
Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) protein is an ion channel found in numerous epithelia and controls the flow of water and salt across the epithelium. The aim of our study to find natural compounds that can improve lung function for people with cystic fibrosis (CF) caused by the p.Gly628Arg (rs397508316) mutation of CFTR protein. The sequence of CFTR protein as a target structure was retrieved from UniProt and PDB database. The ligands that included Armepavine, Osthole, Curcumin, Plumbagine, Quercetin, and one Trikafta (R*) reference drug were screened out from PubChem database. Autodock vina software carried out docking, and binding energies between the drug and the target were included using docking-score. The following tools examined binding energy, interaction, stability, toxicity, and visualize protein-ligand complexes. The compounds having binding energies of −6.4, −5.1, −6.6, −5.1, and − 6.5 kcal/mol for Armepavine, Osthole, Curcumin, Plumbagine, Quercetin, and R*-drug, respectively with mutated CFTR (Gly628Arg) structure were chosen as the most promising ligands. The ligands bind to the mutated CFTR protein structure active sites in hydrophobic bonds, hydrogen bonds, and electrostatic interactions. According to ADMET analyses, the ligands Armepavine and Quercetin also displayed good pharmacokinetic and toxicity characteristics. An MD simulation for 200 ns was also established to ensure that Armepavine and Quercetin ligands attached to the target protein favorably and dynamically, and that protein-ligand complex stability was maintained. It is concluded that Armepavine and Quercetin have stronger capacity to inhibit the effect of mutated CFTR protein through improved trafficking and restoration of original function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most prevalent and potentially fatal autosomal recessive disorders is cystic fibrosis (CF). Mutations in the CFTR gene, which codes for the Cystic Fibrosis Transmembrane-conductance Receptor anion channel, cause the hereditary disease known as cystic fibrosis (CF) (Ashraf et al. 2023a). It mostly affects the reproductive, digestive, and respiratory systems. The gene encoding the transmembrane conductance regulator CFTR for cystic fibrosis is located in the 7q31 region on the chromosome’s long arm (q arm). To control the movement of chloride ions across cell membranes, this gene encodes a protein that acts as a chloride channel on the cell surface (Fonseca et al. 2020; López-Valdez et al. 2021; Ghani et al. 2022; Ullah et al. 2023).
The cystic fibrosis transmembrane conductance regulator CFTR protein has five structural domains required to function as a chloride ion channel. This complex mechanism is based on CFTR’s unique structure, which includes two transmembrane domains (TMDs), two cytoplasmic nucleotide-binding domains (NBDs), and a regulatory domain with serine/threonine residues that are phosphorylated by PKA (Hwang et al. 2018; Kleizen et al. 2021; Ullah et al. 2024). Once phosphorylated, the CFTR gating process begins with ATP-induced NBD dimerization and hydrolysis-triggered NBD dimer separation. CFTR gene mutations can alter ATP binding or hydrolysis, reducing channel function. Many CF-causing mutations result in faulty CFTR channels that cannot open properly, resulting in a lack of chloride ion transport and poor mucus clearance (George Priya Doss et al. 2008; Laskar et al. 2023). This results in the classic CF symptoms, such as persistent lung infections and breathing difficulties (Shanthi et al. 2014; McDonald et al. 2022; GOHAR et al. 2023; Sheema et al. 2024).
The development of CFTR potentiators, correctors, amplifiers, and small molecules aimed at rectifying the CFTR gating defect has been the subject of significant research (Cutting 2015). Notably, the approval of these by the U.S. Food and Drug Administration in 2012 marked a substantial milestone in CF treatment (Ghazi et al. 2023). The rational design of potent compounds with improved pharmacological properties and helpful in disease management enhanced the life expectancy of CF patients. The cellular phenotype and complexity of the CF mutations determine which belong to different classes I, II, III, IV, V, and VI. Ivacaftor alone did not improve outcomes in trials conducted on patients with homozygous F508del mutation, the most common type of CF mutation involving protein processing defects. Consequently, in 2015, a potentiator-corrector double-combination therapy, i.e., Ivacaftor-Lumacaftor, was approved to treat patients homozygous for F508del mutation. Then, in 2018, a combination medication called Ivacaftor-Tezacaftor (corrector) was licensed, demonstrating better tolerability than Lumacaftor. Trikfta, a triple combination therapy, was introduced in 2019. It consists of two correctors (Tezacaftor and Elexacaftor) and one potentiator (Ivacaftor). It is widely believed to function better in homozygous people for the F508del mutation because it raises CFTR activity. Research and clinical trials demonstrate that Trikafta outperforms other treatments regarding respiratory symptoms, lung functions, and overall quality of life (AMIN et al. 2023; Ashraf et al. 2023a; Safdar et al. 2023; Awan et al. 2024).
List of 177 CFTR gene mutations that Trikafta was used to treat: Combining the correctors Elexacaftor and Tezacaftor, which aid in repairing CFTR protein defects so that it can take on the proper shape and traffic to the cell surface, results in the CFTR modulator Trikafta (Becq et al. 2022).
The fact that substances derived from nature and used in traditional medicine will generally be thoroughly evaluated for their effects on the body long before they ever get passed to consumers, is also very helpful. The strategy might increase the chances of finding compounds with different modes of action as better candidates for drugs. The diversity of possibilities allows for a robust evaluation of the multitude and possibly hundreds or more, which may someday reverse, or at least mitigate some aspect of mutation Gly628Arg (Lopes-Pacheco et al. 2021).
The function of natural compounds in the context of cystic fibrosis is still limited. According to recent studies, natural substances may affect cystic fibrosis (Fiedorczuk and Chen 2022). In addition to their antioxidant properties, flavonoids and phenolic compounds have a potent anti-inflammatory action. The anti-inflammatory effects of bioactive extracts and their natural compounds have demonstrated that these agents function biologically through the inhibition of two major signalling pathways, NF-κB and mitogen-activated protein kinases (MAPKs), which play a major role in the production of proinflammatory mediators (Arulselvan et al. 2016; Akbari et al. 2022). The current study aims to check whether the selected natural compounds can improve lung function for people with severe cystic fibrosis (CF) having the (p.Gly628Arg) mutation.
Methodology
Dataset of target protein and preparation
It has been confirmed that the experimental data collection complied with relevant institutional, national, and international guidelines and legislation with appropriate permissions from authorities of the Institute of Molecular Biology and Biotechnology, The University of Lahore, Lahore, Pakistan. Sequences and information about CFTR protein were retrieved from UniProt ID: P13569.3 and protein databank database PDB ID: 8UBR.
Dataset of multiple ligands (MLPs)
A total of 65 natural compounds from the compound library were used for virtual screening with ADMET prediction. Further we have selected five different natural compounds and one reference drug Trikafta (R*) for docking and downloaded from PubChem (Kim et al. 2023) including Armepavine Compound CID: (442169), Osthole Compound CID: (10228), Curcumin Compound CID: (969516), Plumbagin Compound CID (1025), Quercetin Compound CID (5280343) and Trikafta Compound CID: (165363555). The structures of the compounds were downloaded in SDF format from PubChem.
Mutagenesis by PyMOL software
PyMOL was selected for mutagenesis given its vast structural biology functionalities and molecular modeling-oriented utilities. The software is ideal for fine-tuning operations of mutagenesis that requires insertion into a specific residue number and the selection from one or more suitable amino acids to replace another at this position such as creating P.GLY628ARG mutation in CFTR protein. PYMOL has a graphical user-interface, which makes certain operations like mutagenesis much easier. Most of the molecular graphics was performed using PyMOL due to its rich functionalities, easy-to-use interface and integration abilities. To open PyMOL, either double-click the shortcut, open the PyMOL app, or type “Pymol” into a terminal. First, open the protein structure that you wish to modify by navigating to file open Choose a PDB file. Then, Wizard mutagenesis was selected to start the mutagenesis process. Next, a residue from the structure of the desired mutation was selected. The right panel shows the current residue. Click on this to select the amino acid to be changed. The structure of the mutated amino acid is shown in the lower-right corner. To see the rotamers, arrow keys are used to cycle through them. Steric clashes are represented by the red structures surrounding the residue. Use the arrow keys to now cycle through the rotamers. The steric clashes were the red structures surrounding the residue. Navigate through the rotamers to see the console. Once a rotamer has been chosen, click apply and then done (Schrodinger 2010).
Structure validation of protein
The quality of the structure was validated using RAMACHANDRAN plot and ERRAT score evaluation (Colovos and Yeates 1993).
Sequence based predictions
SIFT
Sorting Intolerant from Tolerant (SIFT) is a web-based server that analyzes and predicts the effects of amino acid substitutions on protein functions. These effects are calculated based on sequence similarity, identity, and physicochemical changes between the substituted amino acids. This tool is useful for determining whether an amino acid substitution is likely to affect the protein function (Ng and Henikoff 2003).
PolyPhen 2
PolyPhen2 is a computational tool that assesses the impact of amino acid changes on protein structure and function by utilizing sequence homology, Pfam annotations, and 3D protein structures. It employs a set of classifier models to score the most detrimental or least impactful variants, providing a qualitative assessment of the effects of the change (Adzhubei et al. 2013).
PROVEAN
A web service called Protein Variation Effect Analyzer (PROVEAN) employs a quick set of computer methods to forecast how an amino acid change will affect a specific protein sequence. Initially, PROVEAN uses an alignment method to examine the protein sequence and determine the score. Then, each value derived from the sequence cluster is computed to provide the score (Choi and Chan 2015).
Structure based predictions
CUPSAT
The impact of substituting amino acids on protein stability is analyzed and forecasted by the web-based program CUPSAT. The software program determines the torsional potential between atoms by taking use of structural and conformational peculiarities. Furthermore, in order to determine the impact, this software predicts energy scores, more precisely the variation in the free energy of folding and unfolding structures between wild-type and mutant (Parthiban et al. 2007).
mCSM
A computational program called mCSM uses the environment of protein residues to train prediction algorithms by encoding different patterns between their atoms. This allows the program to understand how mutations impact the structural consistency and stability of interactions between proteins and nucleic acids. The frequency of each residue in a particular data set, for both wild-type (WT) and mutational variants, is analyzed by the program (Pires et al. 2014).
DUET
Using an integrated computational approach, missense mutations in proteins can be studied using the DUET online service (http://biosig.unimelb.edu.au/duet/). In order to produce a consensus prediction, DUET integrates the results of two complimentary approaches (mCSM and SDM) into an improved predictor that uses Support Vector Machines (SVM) (Worth et al. 2011).
Molecular docking
Computational techniques such as AutoDock Vina facilitate detection and development of candidate CFTR modulators. Example of a well known open source molecular docking program: AutoDock Vina, by which interactions and binding poses of small molecules or ligands to their target proteins could be predicted. It is project to foretell docking plus scoring utilisation towars the approval energy of ligand within active sites upon protein target, in-silico virtual screening compound libraries along with catering perimeter creation used for synthetic logical novel pharmaceutical. The prepared protein was then exported to Auto Dock Vina, where the missing atoms were examined and mended, Kollman’s charges were inserted, and polar hydrogen was dissolved. During docking, Kollman charges (partial atomic charges ) are given to protein atoms in order to mimic the electrostatic interactions as close as possible. While the latter is restated by everything in printed circuit designing, it takes a posh data set to foresee bonding situations and affinities of ligands; those charges are gotten from quantum-mechanical reproduction. Here it is applied Kollman charges to the CFTR protein model in order to ensure proper electrostatic interactions between the protein and selected natural chemicals. The final result was saved in the PDBQT file format.
Ligands preparation
The ligands were exported into the PyMOL and convert into PDB format. The three-dimensional structures of the compounds were taken from PubChem and subsequently exported to PyMOL to convert into PDB format. Subsequently, the enhanced ligands were saved in the PDBQT file format after being exported to AutoDock Vina, where they underwent further preparation, including the removal of heteroatoms from the ligand preparation, energy minimization, and the addition of hydrogen atoms.
Drug likeness, pre-clinical testing and ADMET evaluation of the multiple ligands (MPLs)
The evaluation of a therapeutic compound’s drug-likeness is crucial for determining its oral bioavailability, as many medications are taken orally. Lipinski’s rule of five was used to evaluate the most distinct natural ligands for this essential parameter. The ADMET 2.0 web server, a free resource, was utilized for in silico ADMET screening and drug-likeness examination of multiple ligands. Compounds with the highest ranked binding energy scores were included in this phase of the screening. The ligands’ smiles were entered into ADMET’s search bar, and various physical and chemical properties were calculated, such as molecular weight (MW), LogP, LogS, HBA, and HBD. The Lipinski screening guidelines were used to evaluate the drug-likeness of the compounds. The absorption, distribution, metabolism, excretion, and toxicity of drugs all play a role in biological systems. The ADMET 2.0 online server was used to evaluate the identified bioactive substances (Daina et al. 2017; Fatima et al. 2023).
MD simulation
The Desmond program, a molecular dynamics tool (version 3.1, Desmond Molecular Dynamics System, Schrodinger), which uses fixed optimal potentials for liquid simulations (OPLS-2005) force field, was employed to carry out simulations for the optimal dock protein-ligand combination. A simple point charge (SPC) model was used as the solvent in an orthorhombic box with periodic boundary conditions, with the box’s size and shape set at 10 Å x 10 Å x 10 Å to prepare the system for simulation. Using the system builder option, the simulation’s desired electrically neutral system was constructed with 0.15 M NaCl (the physiological concentration of monovalent ions) in 10 Å buffer areas between the box sides and protein atoms. Steepest Decline and short-term memory. A hybrid algorithm of the Broyden-Fletcher Goldfarb-Shanno algorithms was used to obtain the system’s relaxation (Unnisa et al. 2023). The Schrödinger LLC Desmond software was used for the 200-nanosecond molecular dynamics simulation. Prior to the MD simulation, the protein and ligand docking was performed, which served as a critical step in predicting the static view of the molecule’s binding position at the protein’s active site. By incorporating Newton’s classical equation of motion, MD simulations generally simulate atomic movements over time and predict ligand-binding status in a physiological environment. The ligand-receptor complex was preprocessed using Maestro’s Protein Preparation Wizard, which included optimization, minimization, and filling in missing residues if required, as well as the system created using the System Builder tool. The TIP3P (Intermolecular Interaction Potential 3 Points Transferable) solvent model was employed, which is based on an orthorhombic box with a temperature of 300 K, a pressure of 1 atm, and the OPLS_2005 force field. Counter ions and 0.15 M sodium chloride were used to neutralize the models and simulate physiological conditions, respectively (Ferreira et al. 2015).
MMGBSA binding free energy calculation
The MM/GBSA method was used to determine the binding free energy of the entire complex, which integrates the ideas of molecular mechanics and the generalized Born surface area model. The Glide posture viewer file was utilized to calculate the overall binding free energy during the main MM-GBSA simulation, and these values were inputted into the energy minimization processes for the protein-ligand complexes (E-complex), free proteins (Eprotein), and free ligands (Eligand). The equation ΔGbind was employed to determine the binding free energy (Hildebrand et al. 2019a).
ΔGbind = E-complex (minimized)- E-ligand (minimized)- E-receptor (minimized)
The relative binding affinity of ligands to the receptor (given in kcal/mol) is estimated using the MM-GBSA calculations. Since the binding energies of MM-GBSA are approximations of free energies of binding, a larger negative number denotes stronger binding.
PCA analysis
In order to gain a deeper understanding of the movement of macromolecules, we utilized an unsupervised learning approach known as Principal Component Analysis (PCA) to analyze the motion of molecular dynamics (MD) trajectories. This analysis focused specifically on the internal motion of the protein. To carry out the PCA, we employed the R package Bio3D, and a custom R script was used to compute the PCA and extract valuable information from the simulation data. This research was greatly aided by the script, which allowed us to gain a deeper understanding of the molecular dynamics simulation (Palma and Pierdominici-Sottile 2023).
Results
In silico validation of Sequence based and Structure based analysis
Using sequence-based predictors, the retrieved missense mutations of the CFTR (Gly628Arg) protein were first processed, and the desired outcomes were obtained. Sequence-based predictors' results indicated which substitutions were harmful and acceptable; SIFT (0), PolyPhen (1), and PROVEAN (−5.327) were found to be responsible for harmful substitutions. The webservers CUPSAT (58.9%), mCSM-123 (−0.105 kcal/mol), and DUET (−0.033 kcal/mol) were used to test changes that were anticipated to destabilize the protein structure in the structure-based predictors in Table 2, 3 (Supplementary Material tables).
Results
Structural validation
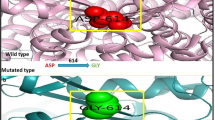
The wild type and mutated structure of CFTR (Gly628Arg) were shown in Fig. 1a, b. The Ramachandran plot shown in Fig. 1(c) shows that mutated CFTR (Gly628Arg) structure 84% residues were in allowed regions, and 0.6% are in the disallowed region, with an ERRAT score of 81.059 and quality pass by VERIFY 3D and PROCHECK in Table 1 (Supplementary Material tables).

a–c: Cartoon represents wild type and mutated type Human CFTR protein structure: a Wild type CFTR represent glycine residue red color area. b Mutated CFTR represent Arginine residue in Blue color area. The strain value of CFTR (Gly628Arg) mutated was 23.74. c Ramachandran plot shows most of the residues are in most favored allowed region
In silico experimental validation of molecular docking
Molecular Docking is the most effective method for identifying protein-ligand interactions in molecular models. The site finder successfully predicted the active site of the muted CFTR (Gly628Arg) structure. To compare our chosen natural compounds, such as Armepavine, Osthol, Curcumin, Plumbagin, and Quercetin, in Table 1 (Supplementary Material tables), with mutated CFTR (Gly628Arg) structure, computation-based mutations were introduced in the CFTR wild-type structure at chain A: Glycine 628 by replacing it with position A: Arginine 628. The docking scores for these ligands against mutant human CFTR (Gly628Arg) are − 6.3 for Armepavine, −5.1 for Osthole, −6.5 for Curcumin, −3.9 for Plumbagin, −5.1 for Quercetin, and − 6.5 for the reference drug Trikafta, see in Table 3 (Supplementary Material tables).
Analyses of the protein-ligand interface were conducted to investigate the interactions between ligands and crucial residues of the muted CFTR (Gly628Arg). The ligands Armepavine, Glu-92, Trp-1145 from chain A are likely significant residues of mutated CFTR (Gly628Arg), as they are implicated in strong hydrogen bonding and are situated inside the binding pocket. The binding pocket surrounding essential muted CFTR amino acids such as Arg-352, Trp-1145, Ser-1141, leu-138, Lys-95, Glu-92, His-139, Ile-142, Arg-134, Gln-1144, Met-348, and Trp-356 from chain A is involved, according to the 2D diagram shown in Fig. 2a with Armepavine. Armepavine fits inside the binding pocket of human CFTR protein and is shown to interact in Fig. 2b−c. This suggests that Armepavine has strong potential to improve CFTR (Gly628Arg) activity.

a–r: Mutated CFTR (Gly628Arg) structure: a–c) CFTR mutated protein-Armpavin complex 2D, 3D, interacting residues of complex. d–f CFTR mutated protein- Osthole complex 2D, 3D, interacting residues of complex. g–iCFTR mutated protein- Curcumin complex 2D, 3D, interacting residues of complex. j–l CFTR mutated protein- Plumbagin complex 2D, 3D, interacting residues of complex. m–o CFTR mutated protein- Quercetin complex 2D, 3D, interacting residues of complex. p−r CFTR mutated protein- Trikafta complex 2D, 3D, interacting residues of complex
In silico study of protein-ligand interactions with key residues in the structure of the mutated CFTR. According to the 2D diagram, Ser-307 from chain A, Fig. 2d is a key amino acid for interaction and contributes to hydrogen bonding with Osthole in mutated-type human CFTR. Chain A’s Trp 356, Asn 1148, Arg 352, Phe 311, Leu 997, Ser 1149, Asp 1152, and Thr 990 have also been shown to interact with the Osthole residues close to the binding pocket. Osthole showed the best fit within the muted CFTR (Gly628Arg) binding pocket, indicating a strong potential for inhibitory interactions to stop the activity of the binding site, as shown in Fig. 2e, f.
According to the 2D diagram, Arg-1078 and Glu-193 from chain A, Fig. 2g, are key amino acids for interaction and contribute to hydrogen bonding with curcumin in mutated-type human CFTR. Chain A’s Phe-311, Trp-366, Gln-1100, ser-207, asp-993, Arg-1097, leu-997, Trp-1145, Arg-352, His-139 have also been shown to interact with the Curcumin close to the binding pocket. Curcumin showed the best fit within the mutated CFTR (Gly628Arg) binding pocket, indicating a strong potential for inhibitory interactions to stop the activity of the binding site Fig. 2h, i.
A protein-ligand contact 2D diagram with critical residues of the muted CFTR structure was examined. Asp-993 and Trp-356 from chain A are key amino acids of the mutated-type human CFTR (Gly628Arg) for interaction and contribute to hydrogen bonding with Plumbagin Fig. 2j. The surrounding binding pocket residue of mutated CFTR (Gly628Arg) involves Leu-989, Asn-306, Phe-310, Ser-307, Gln-237, Thr-351, and Arg-352 from chain A, which also interacted with Plumbagin. The mutated CFTR (Gly628Arg) protein binding pocket is that the ligands fit best, showing strong potential for inhibitory interactions to stop the activity of the binding site in Fig. 2k, l.
Analyses were performed on protein-ligand interactions. Gln-1144 from chain A is a direct residue of muted CFTR (Gly628Arg) involved in the hydrogen bonding observed with Quercetin Fig. 2 (m). The surrounding binding pocket contained essential mutated CFTR amino acids such as Leu-1096, His-145, Arg-1097, Asn-1148, Glu-193, Ileu-142, Leu-197, and Trp-356 from chain A, indicating a high inhibitory interaction potential to block muted CFTR (Gly628Arg) activity, in Fig. 2(n, o).
The protein-ligand contact 2D diagram with critical residues of the mutated CFTR structure was investigated. The interaction plot indicates that Arg-352 and Asp-993 from chain A are key amino acids of the mutated-type human CFTR for interaction and contribute to hydrogen bonding with Trikafta. The surrounding binding pocket residues involved Asp-1152, Arg- 352, Phe-311 Leu-989, Ser-307, Arg-1097, leu-997, Asn-306, which also interacted with R*. The ligands best fit within the binding pocket of the mutated CFTR (Gly628Arg), indicating a high inhibitory interaction, in Fig. 2p, q, r.
From AutoDock Vina, we can say that Armepavine, Curcumin may tractably reach a thermodynamic minimum with the CFTR p.Gly628Arg mutant protein. Evidence of interactions with key residues provide insight into how they might be able to rescue the defective CFTR protein function seen in connection with p.Gly628Arg mutation and could potentially serve as a therapeutic recourse for patients carrying this genetic variant.
In silico experimental validation of ADMET analysis
Drug-likeliness, oral bioavailability, results from a drug’s physicochemical composition. To display the pharmacokinetic and toxicity profiles of different ligands in Tables 5, 6 and 7 (Supplementary Material tables).
A total of sixty five compounds were analyzed as a suitable drug candidate. Best five compounds were filtered and processed further. The two online servers that were chosen for this section are publically accessible, have been well validated using experimental data, and provide dependable computational techniques for estimating a worldwide assessment of the pharmacokinetics and toxicity of small compounds. For the initial stages of drug design and evaluation, ADME. 2.0 were utilized to forecast the pharmacokinetics and toxicity characteristics of substances. The following characteristics of natural substances were calculated: drug-likeness, absorption, distribution, metabolism, excretion and toxicity, see Fig. 3a, f.

a−f Silicos -IT LogSW, BBB and P-gp substrate, Human intestinal absorption and Cytochromes CYPs, excretion and toxicity of compounds. The prediction was done with Swiss ADMET.2
Drug-likeness
It is important to note that all natural compounds have a molecular weight of less than 500 g/mol, an HBA of less than 10, and an HDB of less than 5, and they all adhere to Lipinski’s Rule of 5.
Absorption
The process by which a drug permeates the systemic circulation from an extravascular site during administration is known as absorption. It can be represented using various property listed below.
Solubility
ADME has selected Silicos-IT LogSw as the descriptor. SILICOS-IT developed it as a predictor for solubility. High negative results in Silicos-IT LogSw point to the existence of poorly soluble chemicals. Armepavine, Osthole, Curcumin, Plumbagine, Quercetin, and Trikafta compounds have the lowest LogS, indicating that overall, it contains more water-soluble compounds, with the majority of values falling between − 10 and 0. The mean value of Trikafta is −3.45. Conversely, the lowest values for Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin, which were − 1.991, −5.01, −4.45, −2.85, and − 3.24. It means that all natural compounds were lowest mean value of LogS which indicate significantly increase solubility of compounds and may effect on mutated CFTR activity.
Lipophilicity
In this work, we were used the descriptor LogPo/w. The mean value of LogP was 2.92, 3.34, 3.03, 1.72, and 1.23 for Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin, whereas Trikafta has a cLogP value of 7.33. These compounds are highly lipophilic and permeable across biological membranes. Furthermore, all natural compounds and the reference drug demonstrated significantly higher intestinal absorption in the human body and may effect on CFTR corrector. The results of consensus cLog Po/w and water solubility demonstrated that all of these compounds appeared to have good water solubility, which could provide a reference for the formulation design of pharmaceutical preparations. When lipophilicity is too low, it may be reduced, but very hydrophilic substances often cannot diffuse passively through them.
P-gp substrate
The percentages of compounds predicted to inhibit P-gp substrate, Armepavine and Trikafta the reference drug did not inhibit P-gp substrate but Osthole, Curcumin, Plumbagine, Quercetin, have shown the inhibition of P-gp substrate in the CFTR corrector. Armepavine compounds 0.75%, have not inhibit the P-gp substrate can stop from attaching to the P-gp protein or stop P-gp from pumping in an ATP-dependent manner like Trikafta 0.95% may affect the CFTR activity. The substrate that P-gp would typically pump out of the cell instead gathers inside the cell as a result. In particular, the discipline of pharmacology will be greatly impacted by this interplay between P-gp substrates and inhibitors.
Human intestinal absorption (HIA)
All natural compounds and the reference drug demonstrated significantly higher intestinal absorption in the human body. The results also indicate that the distributions of Armepavine, Osthole, Curcumin, Quercetin, and Trikafta have to exhibit to increase intestinal absorption ≥ 30%. The HIA descriptor was calculated in this work using pkCSM pharmacokinetics.
Distribution
The distributions of Osthole, Armepavine, Curcumin, Quercetin, and Trikafta are comparable, according to the results. Additionally, it suggests that all of these organic substances exhibited increased intestinal absorption.
Blood brain barrier (BBB) and plasma protein binding (PPB)
The percentages of compounds predicted to cross the BBB, Armepavine has been shown to cross the BBB then Osthole, Curcumin, Plumbagine, Quercetin and Trikafta the reference drug. The biological significance that Armepavine succeeded in crossing blood-brain barrier (BBB) was significant. Armepavine properties could also fit for treating CNS symptoms or consequences of cystic fibrosis, notably CFTR linked to neuroinflammation and cognitive deficits. In this way it has an extended therapeutic opportunity beyond the tissue circulation. R* drug and other natural substances are unable to pass across the BBB. PPB stands for Plasma Protein Binding, a crucial concept in pharmacokinetics. All natural compounds had excellent outcomes such as Armepavin, Osthole, Curcumin, Quercetin except for the Plumbagin and R* drug, which had a PPB of over 90%.
Metabolism
Swiss ADME, a binary form (yes/no) of primary cytochrome inhibition was anticipated. Drug development heavily relies on metabolism, therefore it was important to fully understand how it affects PK, pharmacodynamics (PD), and safety.
For predicting the percentage of isoenzyme inhibition, ADMET was utilized. Armepavine compound had non-inhibitory action of four CYP P450 isoenzymes, as per the data and may effect on mutated CFTR activity. Osthole compounds have been demonstrated to inhibit CYP-1A2, CYP2C19, CYP2C9, and CYP2D6 except for CYP-3A4 (0.865%). The curcumin inhibits the activity of all other cytochrome isoforms but does not affect that of CYP-2D6 (0.138%) or CYP-2C19 (0.171%). All isoenzymes of cytochromes CYP-1A2 (0.975%), CYP2C19 (0.583), CYP2C9 (0.666%), CYP2D6 (0.885%), and CYP-3A4 (0.716%) were demonstrated to be inhibited by plumbagin. The percentage of curcumin molecule that inhibits CYP2C9 (0.596%) and CYP3A4 (0.865%). Furthermore, it demonstrates that CYP2C19 and CYP3A4 are anticipated to be substantially inhibited by Curcumin, and Plumbagine Cytochrome P450 (CYP) 3 A was responsible for the metabolism of xenobiotic, including as drugs, hormones, carcinogens, and eicosanoids, in accordance with its maximum abundance in humans.
Excretion (total clearance)
This is an important feature since it influences the drug’s half-life and bioavailability, which in turn influences the dosage and amount of the medication. Armepavine’s high significant mean value of 12.46 is similar to Osthole’s 13.583 and Curcumin’s 12.8. Between Armepavine and Osthole, the mean value of Quercetin was 8.28%. It suggests that the greatest values demonstrated a large overall drug elimination from the body. Trikafta’s lowest total clearance value (3.54) is comparable to Plumbagine’s (4.94), suggesting that drug accumulation can lead to toxicity.
Toxicity
The ability of a substance to damage an organism or any of its organs, as in the case of a failed late-stage drug development, is known as its toxicity. Hepatoxicity (0.1%, 0.2%), AMES toxicity (0.09%, 0.057%), carcinogenicity (0.084%, 0.05%), and hERG (0.2%, 0.09%) were the lowest percentages of Armepavine and Quercetin compound. Osthole and Trikafta exhibited the highest percentage of hepatoxicity (0.95%, 0.98%), carcinogenicity (0.79%, 0.81%), and hERG (0.02%, 0.21%) after AMES toxicity (0.20%). The sole data set with the highest percentage of substances with AMES toxicity test predictions is Plumbagine and Trikafta (0.908%, 0.71%). The compounds with the highest proportion of carcinogenicity (60%) were found to be Osthole, Curcumin, Plumbagin, and Trikafta. The majority of these compounds—98%—were non-hERG inhibitors.
In silico experimental validation of MD simulation
Armepavine-CFTR complex MD simulation
We used the Desmond program to carry out MD simulations in order to validate the ligand binding modes and the stability of the protein-ligand complexes. The simulations were conducted for 200 ns on the top dock scorer, which included Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin in conjunction with the CFTR (Gly628Arg) complexes. RMSD plot displayed the root mean standard deviation of the protein-ligand complex’s C-α backbone throughout the simulation. The C-α backbone of the CFTR protein in the Armepavine complex was simulated using MD for 200 ns. The RMSD plot revealed that the protein-ligand complex remained stable, with only slight fluctuations observed until 80 ns. The RMSD of the C-α backbone remained within a light blue color, while the ligand was represented in pink. The maximum RMSD of the CFTR protein C-α backbone was 3.5 Å, indicating that the protein persisted throughout the simulation. Additionally, the RMSF was used to evaluate the mobility of amino acid residues by comparing the protein’s RMSFs to a reference structure. The average value of all atomic fluctuations for each amino acid residue was calculated using the root mean square fluctuation (RMSF). The RMSF of the simulation results revealed that the residues of Val-452, Leu-574, and Ala-400 fluctuated above 6.4 Å. These protein residues were not associated with a ligand during the simulation. Armepavine-CFTR complex forms stable hydrogen bonds with water-mediated interactions with residues such as LYS-464, GLN-493, HIS-1375, and ARG-1403, as shown in the protein’s crystal structure, see Fig. 4a−c. Additionally, Armepavine interacts with VAL-580 and ALA-1374 through hydrophobic bonding with water-mediated interactions, and other interactions were detected with various amino residues throughout the simulation. However, the interaction with LYS-464, GLN-493, VAL-580, and ALA-1374 residues of the receptor was persistent and stable throughout the entire simulation process. The stability of the Armepavine-CFTR (Gly628Arg) complex was assessed using the RGyr parameter, which was calculated from the entire trajectory of the molecular dynamics simulation. A steady RGyr value of 3.9 Å indicates that the protein remained stable throughout the simulation. The RGyr parameter was utilized to illustrate the structural compactness and variations in folding of Armepavine with its protein complexes during the 200-ns simulation. Importantly, no significant fluctuations were detected in the complex, indicating that the protein-ligand complexes remain stable after binding, and may effect on mutated CFTR activity see Fig. 4d.

a−d. MD simulation analysis of the Armepavine with CFTR (Gly628Arg) complex. a Time dependent-RMSD of protein Cα atoms and the ligand relative to the protein are displayed in blue and red, respectively. The stability of the complex was maintained as evidenced by the RMSD value within 3.8 Å for the maximum value of CFTR protein Cα atoms. Initially, the RMSD values of the ligand until 60 ns and then stabilized with a mean RMSD of 4 Å. b The protein RMSFs represent the individual fluctuations of each amino acid during the simulation. c The protein–ligand contact histogram displays the distribution of contact between the protein and the ligand. d Gyration radii of the complex. The graph depicts the gyration radii of the complexes formed by Armepavine with CFTR protein that were remain maintained after 3.9 Å determined from molecular dynamics simulations
Osthole-CFTR complex MD simulation
The CFTR protein (Gly628Arg)) and Osthole complex underwent a 200 ns molecular dynamics simulation. The RMSD of the protein-ligand complex varied slightly but remained stable until the 20 ns mark, at which point it remained stable for the rest of the simulation. The RMSD of the C-α backbone (shown in light blue) was calculated relative to the reference frame backbone and ligand (pink). The maximum RMSD of the CFTR protein C-α backbone was 3.5 Å, while the RMSD of the ligand was 1.5 Å, indicating that both the protein and ligand were stable throughout the simulation. The CFTR-osthole complex exhibited major fluctuations but remained unstable throughout the simulation. The mutated CFTR protein exhibit similar RMSF trends, suggesting less stability than Armepavine and Quercetin compound. The residues of Arg-600 fluctuated above 5.4 Å and were not linked to a ligand during the simulation. The crystal structure of the Osthole-CFTR complex reveals three hydrogen bond interactions between the protein and ligand. These interactions were stable throughout the 200-ns simulation, as indicated by the RGyr parameter, which remained at 3.24 Å, see Fig. 5a, b. The RGyr parameter was used to illustrate the structural compactness or folding variations of Osthole with the protein complexes during the simulation. Notably, significant fluctuations were observed for the complex, suggesting that the protein-ligand complex remains stable after binding, see Fig. 5d.

a−d. MD simulation analysis of the Osthole with CFTR (Gly628Arg) complex. a Time dependent-RMSD of protein Cα atoms and the ligand relative to the protein are displayed in blue and red, respectively. The stability of the complex was maintained as evidenced by the RMSD value within 3.5 Å for the maximum value of CFTR protein Cα atoms. Initially, the RMSD values of the protein with ligand were not stabilized. b The protein RMSFs represent the individual fluctuations of each amino acid during the simulation. c The protein–ligand contact histogram displays the distribution of contact between the protein and the ligand. d Gyration radii of the complex. The graph depicts the gyration radii of the complexes formed by Armepavine with CFTR protein that were determined 3.25 Å from molecular dynamics simulations
Curcumin-CFTR complex MD simulation
The CFTR (Gly628Arg) protein C-α backbone was simulated using MD for 200 ns, and slight fluctuations in the RMSD of the Curcumin-CFTR complex complex were observed until 150 ns. The maximum RMSD of the CFTR protein C-α backbone was 3.5 Å, and the RMSD of the ligand Curcumin was 1.5 Å, indicating that both the protein and ligand persisted throughout the simulation. In the figure, the Root Mean Square Deviation (RMSD) of the protein is displayed. Throughout the simulation, the mobility of amino acid residues was assessed by comparing the protein’s Root Mean Square Fluctuation (RMSF) to a reference structure. The RMSF of the amino acid residue was used to calculate the average value of all atomic fluctuations for a specific amino acid. RMSF values of Leu-574, Ala-400, and Arg-600 were above 5.6 Å, and no ligand was associated with these residues during the simulation. As shown in the crystal structure of the protein, the Curcumin-CFTR complex forms hydrogen bond interactions with water mediated interactions at THR-460, GLY-463, THR-495, SER-469, TRP-496, and THR-1064, as well as hydrophobic bond interactions at VAL-440, PHE-494, and TRP-1063. LYS-464 was the key interacting residue of the protein with the ligand, and it was observed in every frame of the simulation, indicating a stable interaction throughout the simulation process, see Fig. 6a–c. The stability of the Curcumin-CFTR (Gly628Arg) complex was evaluated using the RGyr parameter, which was calculated from the entire molecular dynamics (MD) simulation trajectory set. A stable RGyr value of 6.0 Å indicates that the protein remained stable during the simulation. The RGyr parameter was used to illustrate the structural compactness or folding variations of Curcumin with protein complexes during the 200-ns simulation. Notably, no significant fluctuations were observed for the complex. These results suggest that protein-ligand complexes remain stable after binding, see Fig. 6d.

a–d. MD simulation analysis of the Curcumin with CFTR (Gly628Arg) complex. a Time dependent-RMSD of protein Cα atoms and the ligand relative to the protein are displayed in blue and red, respectively. The stability of the complex was maintained as evidenced by the RMSD value within 3.2 Å for the maximum value of CFTR protein Cα atoms. Initially, the RMSD values of the protein ligand until 150 ns and then stabilized with a mean RMSD of 4 Å. b The protein RMSFs represent the individual fluctuations of each amino acid during the simulation. c The protein–ligand contact histogram displays the distribution of contact between the protein and the ligand. (d) Gyration radii of the complex. The graph depicts the gyration radii of the complexes formed by Curcumin with CFTR protein that were determined 6.0 Å from molecular dynamics simulations
Plumbagine -CFTR complex MD simulation
The RMSD of the CFTR (Gly628Arg) protein C-α backbone in the Plumbagine complex was simulated using MD for 200 ns. The protein-ligand complex exhibited major fluctuations but remained unstable throughout the simulation. The residues of Arg-600 fluctuated above 5.4 Å and were not linked to a ligand during the simulation. The crystal structure of the protein reveals that the Plumbagine-CFTR complex forms hydrogen bond interactions with water-mediated interactions at SER-271 and SER-1225, and hydrophobic bond interactions with water-mediated interactions at LLE-546. These interactions was observed in each frame of the simulation. To evaluate the compactness of the complex, the RGyr parameter was used, which was calculated from the entire MD simulation trajectory set, see Fig. 7a, c. A stable RGyr value was 2.57 Å indicates that the protein remained stable during the simulation. This parameter was also used to illustrate the structural compactness or folding variations of Plumbagine with protein complexes during the 200-ns simulation. Notably, significant fluctuations were observed for the complex, suggesting that protein-ligand complexes remain stable after binding see Fig. 7d.

a−d. MD simulation analysis of the Plumbagine with CFTR (Gly628Arg) complex. (a) Time dependent-RMSD of protein Cα atoms and the ligand relative to the protein are displayed in blue and red, respectively. The stability of the complex was maintained as evidenced by the RMSD value within 3.2 Å for the maximum value of CFTR protein Cα atoms. Initially, the RMSD values of the ligand were not stabilized. b The protein RMSFs represent the individual fluctuations of each amino acid during the simulation. cThe protein–ligand contact histogram displays the distribution of contact between the protein and the ligand. d Gyration radii of the complex. The graph depicts the gyration radii of the complexes formed by Plumbagine with CFTR protein that were determined 2.57 Å from molecular dynamics simulations
Moreover, the ADMET results provide a detailed insight into natural compounds as CFTR modulators besides our docking findings. It possesses a great ADMET profile and shows high oral bioavailability which suggests that the compounds could be safe as well as useful if applied to clinical settings. This latter observation is biologically significant, as it potentially represents a mechanism for the therapeutic efficacy of Armepavin and Querectin in patients bearing disease-conferring P.GLY628ARG mutation in CF. ArmePavine and Quercetin may enhance the quality of life, reduce lung infections frequency, and ameliorate CF symptoms by promoting functional stabilization of the CTFR protein.
Quercetin -CFTR complex MD simulation
The CFTR (Gly628Arg) protein C-α backbone of Quercetin complex was simulated using MD for 200 ns. The RMSD of the protein-ligand complex showed slight fluctuations but remained stable. The maximum RMSD of the CFTR protein C-α backbone was 3.1 Å until 75 ns, and the RMSD of the ligand was 3.5 Å, indicating that both the protein and ligand persisted throughout the simulation. In the figure, the root mean square deviation (RMSD) of the protein is depicted. This value is computed by averaging all atomic fluctuations for each amino acid residue’s root mean square fluctuation (RMSF). Residues such as Arg-600, which fluctuated at 5.4 Å and were not linked to a ligand during the simulation, are displayed. Moreover, as shown in the crystal structure of the protein, the Quercetin-CFTR complex forms hydrogen bond interactions with water. These interactions involve residues such as SER-573, GLU-583, GLU-1405, SER-1442, ASP-1445, which are stable during each frame of the simulation. The stability of the Quercetin-CFTR (Gly628Arg) complex was assessed using the RGyr parameter, which was calculated from the entire MD simulation trajectory set, see Fig. 8a, c. A stable RGyr value at 3.7 Å indicates that the protein remained stable during the simulation. The RGyr parameter was used to demonstrate the structural compactness or folding variations of Quercetin with protein complexes during the 200-ns simulation. Notably, no significant fluctuations were observed for the complex. These findings suggest that protein-ligand complexes remain stable after binding, see Fig. 8d.

a−d. MD simulation analysis of the Quercetin with CFTR (Gly628Arg) complex. a Time dependent-RMSD of protein Cα atoms and the ligand relative to the protein are displayed in blue and red, respectively. The stability of the maximum value of CFTR protein Cα atoms was maintained as evidenced by the RMSD value within 3.6 Å after 75 ns. Initially, the RMSD values of the ligand until 60 ns and then stabilized with a mean RMSD of 4 Å. b The protein RMSFs represent the individual fluctuations of each amino acid during the simulation. c The protein–ligand contact histogram displays the distribution of contact between the protein and the ligand. d Gyration radii of the complex. The graph depicts the gyration radii of the complexes formed by Quercetin with CFTR protein that were determined 3.75 Å from molecular dynamics simulations
MM-GBSA binding free energy calculation
The ΔGbind values, which were calculated using the MM-GBSA method after the simulation, indicate the strength of binding between the protein and ligand complexes. The average ΔGbind values for the complexes formed between CFTR and Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin were − 65.7650, −15.6751, −45.3145, −50.1859, and − 52.7872, respectively. The ΔGbind values for the complexes formed between CFTR with Armepavine was − 65.7650 greater than other natural compounds. A value that is highly negative suggests a stronger binding affinity between the two, see Table 7 (Supplementary Material tables).
In silico experimental validation of principle component analysis
The use of MDS parameters in conjunction with principal component analysis (PCA) allows for the identification of cluster conformations in the lead molecule and the assessment of optimal ligand stability. During the MDS process, PCA results showed that Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin were the most stable ligands among the chosen molecules. PCA eigenvalue plots show the percentage of variance for each component, which is presented in three distinct sections. For example, the varying areas for Armepavine with CFTR protein variations in PC1, PC2, and PC3 account for 55.96%, 5.32%, and 5% of the total variance, respectively. Similarly, the varying areas for Osthole-CFTR protein in variations in PC1, PC2, and PC3 account for 40.5%, 184%, and 10.38% of the total variance, respectively. The PCA results for Curcumin, Plumbagine, and Quercetin are also provided, with varying areas accounting for 28.3%, 15.44%, and 7.27% of the total variance for Curcumin; 32.85%, 15.36%, and 7.47% for Plumbagine; and 49.35%, 22.3%, and 7.38% for Quercetin, a visual representation of these results see Fig. 9a–e.

a−e. Principal Component Analysis. a PCA of Armepavine with CFTR (G628A) complex. b PCA of Osthole with CFTR (G628A) complex c PCA of Curcumin with CFTR (G628A) complex. d PCA of Plumbagine with CFTR (G628A) complex. e PCA of Quercetin with CFTR (G628A) complex
Discussion
Cystic fibrosis is caused by mutations in the CFTR protein, which misfolded or improperly configure protein, which may lose its capacity to facilitate the transport of chloride ions and water effectively across cell membranes. This disruption in NBD1’s function significantly contributes to the hallmark clinical manifestations of cystic fibrosis marked by the accumulation of thick and viscid mucus within the respiratory and digestive system (Sosnay et al. 2013). A buildup of thick mucus results from CFTR gene mutations that cause the CFTR protein to fail or not be produced at all, which in turn causes recurring lung infections, pancreatic damage, and complications in other organs (McKee et al. 2023), (Shan et al. 2024).
An efficient strategy might be to predict the binding regions on the mutated CFTR (Gly628Arg) protein surface using docking, bio-viability, and toxicity analysis and then verify the predictions using MD simulation trajectory. Virtual screening of the investigated compounds using molecular docking against mutated CFTR (Gly628Arg) showed that the ligands have high binding affinities to the target’s active sites. The mutated CFTR (Gly628Arg) protein structure was present different amino acid of the active binding site included, Gln-237, Thr-351, Arg-352, His-145, Arg-1097, Asn-1148, Glu-193, Ileu-142, Leu-197, Trp-356, Asp-1152, Arg- 352, Phe-311 Leu-989, Ser-307 in Table 4 (Supplementary Material tables), see Fig. 1 (a-c).
In this study, we have analyzed the detail in silico analysis of five anti-mutated CFTR protein ligands i.e. Armepavin, Osthole, Curcumin, Plumbagin, and Quercetin as inhibitors of mutated CFTR (Gly628Arg) protein structure. The docking-score of the multiple ligands included the Armepavine compound with a binding affinity of s-score − 6.5 kcal/mol against the target mutated CFTR (Gly628Arg) structure. According to the analysis of the 2D diagram, the target protein’s active sites were bound by the Armepavine ligand via the formation of two hydrogen bonds with Glu-92, Trp-1145 with bond distances of 3.23 Å and 3.64 Å. It is important to note that the combination of ligand with the mutated CFTR (Gly628Arg) contained hydrophobic, electrostatic, and hydrogen bond interactions in Table 4 (Supplementary Material tables), Fig. 2a, c.
The osthole ligand interacted with the amino acid residues in the target CFTR (Gly628Arg) mutated structure in the ways described below, with a binding energy − 5.1 kcal/mol. According to the 2D diagram, the target site formed one hydrogen bond with Ser-307 with a bond distance of 2.98 Å to stabilize the interaction in Table 4; Fig. 2 (d, f). Further, the interaction of ligand Curcumin with the active sites of the mutated CFTR (Gly628Arg) has an s-score of -6.6 kcal/mol in Table 4 (Supplementary Material tables), Fig. 2g, i. In this complex were formed two hydrogen bonds with Arg-1078, and Glu-193 with bond distance 2.92 Å, 2.9 Å (Hassan et al. 2021).
The ligand Plumbagin docking against the target mutated CFTR (Gly628Arg), displayed the following interactions with the amino acid residues with a binding energy of -3.9 kcal/mol. The 2D diagram indicates that the ligand formed two hydrogen bonds with Asp-993, Trp-356 with bond distances 3.17 Å, and 3.11 Å. These interactions were stabilized in the ligand/protein complex in Table 4; Fig. 2(j, k, l). Furthermore, the Quercetin ligand formed a complex with the active sites of muted CFTR (Gly628Arg) with an s-score − 5.1 kcal/mol. The interactions found in this complex were hydrogen bond interactions with Gln-1144, with a bond distance of 2.8 Å. Similar to all ligands, hydrophobic, electrostatic, and hydrogen bond interactions were also found in the complex of the ligand with mutated CFTR (Gly628Arg), as shown in Table 4 (Supplementary Material tables), Fig. 2m, n, o. In addition, R* (Reference drug Trikafta) is bound to mutated CFTR (Gly628Arg) with an S-score of -6.4 kcal/mol. The following interactions were found between the ligand and the active sites of the protein target: formation of three conventional hydrogen bonds with Arg-352, Asp-993, Trp-1145 with a bond distance of 3.94, 2.3, 3.5 Å see Table 4 (Supplementary Material tables), Fig. 2(p, q, r). This is because strong hydrogen bonds and good docking-scores are formed Armepavine, Curcumin with mutated CFTR (Gly628Arg) protein interacting residues compared with the R* drug. According to the docking data, Armepavin and Curcumin are more effective than other ligands at binding CFTR (Gly628Arg) in mutant states, hence stopping the process of mutated CFTR protein structure that leads to cystic fibrosis. These ligands should enhance the activity of CFTR protein through improved trafficking and restoration of original function. Through different mechanisms, CFTR modulator medications improve or even repair the expression, stability, and function of a faulty CFTR. This is because two strong hydrogen bonds are formed between residues of mutated CFTR protein structure. Through different mechanisms, CFTR modulator medications improve or even repair the expression, stability, and function of a faulty CFTR (Bergbower et al. 2018; Lopes-Pacheco 2020; Sakhawat et al. 2023).
According to our results to find that, the screening for the multiple ligands (MLP) and R*- drug are shown. Lipinski’s rule states that ligands with MW < 500 and HBA < 10 may have high oral absorption. But in case of reference drug R* did not have passed Lipinski’s rule. Pharmacokinetics examines a medicinal substance in a biological system, focusing on its absorption, distribution, metabolism, and excretion (Brylinski and design, 2018). Multiple ligands’ ADME and toxicity (T) profiles are shown in Table 5 (Supplementary Material tables). The percentage of medicinal chemical impact entering the circulatory system is called oral bioavailability. The bioavailability scores of the various ligands ranged from 0.55 to 0.56, indicating that they had good oral bioavailability according to Lipinski’s rule of five (Chen et al. 2020).
In Table 6 show that, the important feature of oral medications is their (absorption) solubility in intestinal fluid since inadequate solubility may prevent the drug from being absorbed through the portal vein system and having a therapeutic impact when systemic effects are appropriate. Armepavine, Osthole, Curcumin, Plumbagine, and Quercetin, which indicate that all natural compounds were lowest Log S which indicate that increase solubility of compounds. All these compounds are highly' lipophilicity (in the lipid phase) and easily permeable across membrane.
The fact that Armepavine was able to cross blood-brain barrier (BBB) is also significant in terms of its biological importance. Thus, properties of Armepavine could be used for the treatment of CNS symptoms or complications associated with cystic fibrosis (CF), such as CFTR-related neuroinflammation and cognitive impairments. This enables it to act as a depot and allows for an extended therapeutic time frame outside of the tissue circulation. R*-drug and natural substances do not pass the BBB. These are characteristics that minimise central nervous system side-affects and ensure therapeutic effect is focussed on peripheral tissue. Compared with all natural compounds higher PPBs was observed except Armepavin indicating that an additional part of the proportion is bound to plasma proteins. This might mean a more enduring effect, although then you need to balance dosage carefully against toxicity.
A substance’s human intestinal absorption (HIA) is often expressed as the percentage of the administered dose that has passed through the portal vein and to increased intestinal absorption in the body. All ligands, as well as the R*-drug, were found to have gastrointestinal absorption potential. Curcumin, Quercetin, and the reference drug R*, cannot enter the BBB, which may positively affect the central nervous system (Bors and Erdő 2019). Membrane transporters called P-glycoproteins (P-gps) restrict cellular absorption of their substrates from blood circulation into the brain by efflux activity. All ligands except Armepavine were discovered to be non-substrates of P-gps and hence may be unaffected by P-gp efflux activity, resulting in reduced serum concentration and a failure therapeutic effect and may also effect CFTR activity. P-gp inhibition may raise Armepavine intracellular concentration and improve its effectiveness as a CFTR corrector. In contrast, Armepavine and reference drug R*, a substrate of the protein P-gp, may have significantly increase serum concentrations, leading to eliminate from the body which show positive effect (Zhou et al. 2021), see Fig. 3(a, f). Table 6 (Supplementary Material tables). Screening for potential drug-drug interactions is critical in managing CF patients to determine the effect of cytochrome P450 (CYP450) induction or inhibition on drug bioavailability. CYP450 enzymes metabolize clinically relevant drug interactions enhancing its efficacy as a CFTR corrector. Inhibitors either prohibit CYP450 enzymes from functioning or limit the pace of enzyme-catalyzed processes (Ameji et al. 2023). As a result, drug metabolism in the body is reduced, and the risk of toxicity increases. The cytochrome P450 (CYP450) monooxygenase family is critical in this process. The metabolism of natural compounds and R*drug effect was against four CYP450 isoforms, CYP1A2, CYP2C19, CYP2C9, and CYP3A4. Tricaffta and other ligands’ pharmacokinetics can be better understood, including how they are absorbed, distributed, metabolized, and removed. However, in silico analysis of the inhibition of CYP450 enzymes by therapeutic ligands, such as Armepavine, and Quercetin did not reveal a significant effect on toxicity profiles. Furthermore, when the toxicity of the ligands was tested using mutagenicity, tumorigenic effect, reproductive impact, and irritating effect as endpoints, it was discovered that none of the numerous ligands showed any of the aforementioned tendencies and hence may not offer a serious toxicity risk. Cytochrome P450 (CYP) 3 A is responsible for the metabolism of xenobiotics, including as drugs, hormones, carcinogens, and eicosanoids, in accordance with its maximum abundance in humans. A prevalent indicator of numerous ailments, inflammation plays a role in the etiology of infections, cancer, diabetes, rheumatoid arthritis, and inflammatory bowel disease in addition to age-related phenomena like normal aging and metabolic abnormalities. Exposure to Armepavine, Quercetin and Trikafta is significantly increase the use of strong CYP3A inducers, which may increase the therapeutic effectiveness (Tsai et al. 2020). Through different mechanisms, CFTR modulator medications improve or even repair the expression, stability, and function of a faulty CFTR. Armepavine, Osthole and Curcumin were the greatest and good to eliminate large drug from the body as compared to the Trikafta’s have poor excretion or clearance which cause toxicity. The effect of compounds to damage an organism or any of its organs, as in the case of a failed late-stage medication development, cause hepatotoxicity, AMES toxicity, carcinogenicity and eye irritation. The natural ligands like Armepavine, Quercetin Osthole and Curcumin have non toxicity profile (Ernst et al. 2021), see Table 6 (Supplementary Material tables).
A useful technique for studying protein-ligand interactions is Molecular Dynamics, which may also be used to the binding efficacy of pharmaceuticals. Virtual screening using MD simulations, illnesses caused by misfolding of proteins can be addressed by determining the kinetics of ligand binding and the stability of protein-ligand complexes. With the CFTR (P.GLY628ARG) mutated protein, MDS equilibration could be completed in 200 ns. The RMSD graph of CFTR (P.GLY628ARG) indicated that the minor changes were noted, the system was fully stabilized with an RMSD of 3.5–4.8 Å, and the minimal fluctuations were seen in each complexes such as Armepavine and Quercetin, see Figs. 4, 5, 6, 7 and 8 (a). The trajectory residue-based root-mean-square fluctuation (RMSF) was calculated to examine the flexibility of the peptide structure (Hildebrand et al. 2019a). The middle residue of amino acids generally shows a higher RMSF value than other amino acid residues. In protein-ligand complexes, the amino acids showed RMSF values lower than 3Å, and these RMSF results indicate a stabilized protein structure (Kumar et al. 2014), see Figs. 4, 5, 6, 7 and 8b. The protein-ligand contacts, Armepavin, and Quercetin ligand, interacted with many amino acids and does not diverge significantly from its initial structure. This can only occur if the ligand changes its position within the active binding site. Using an MD simulation trajectory, our study examined the interactions between the mutated CFTR structure and five natural compounds. These complexes reached an equilibrium state in 200 ns during simulation, see Figs. 4, 5, 6, 7 and 8d. Armepavin, and Quercetin suppressed the mutated structure of CFTR (Gly628Arg) activity because to their safety and rapid onset of action than other natural compounds. It is necessary to comprehend the computed energy components and that contribute to the total free energy change in a molecular system in order to interpret MM-GBSA results was showing higher ΔGbind in Armepavine − 65.5 and Quercetin − 52.5 values than other natural compounds. This is frequently the case when analyzing protein-ligand binding. All natural ligands in our in silico research were shown higher negative binding energy by a favorable binding, it means that binding is thermodynamically beneficial, see Table 8 (Supplementary Material tables) (Li et al. 2020; Alameen et al. 2022).
The covariance matrix’s eigenvalues, which the PCA approach used to determine the total combined motion of the C-atoms in the protein ligand complexes. The time-dependent switching between different conformations during the simulation is shown by the continuous red-to-blue color transition, where dots that begin and end with red represent the alignment of each frame (Rasheed et al. 2021). These findings show the best and highest %age of effective PCA analysis of Armepavine and Quercetin is at clear complex conformational dynamics, which can help with the creation of new medicines. In contrast, continuous overlap ligand complexes with the CFTR mutant protein (P.GLY628ARG) show localized fluctuations and more frequent jumps. These results collectively imply that mutations modify the internal dynamics of the complexes and significantly affect their structure (Palma and Pierdominici-Sottile 2023), see Fig. 9a–e. Based on molecular docking, ADMET characteristics, and simulation, then we selected two best natural compounds Armepavine and Quercetin. These compounds were shown to have the best interactions with the mutated CFTR protein active site.
Our study focused on different natural compounds and discovered initially five compounds to be the most promising ligands for blocking the cystic fibrosis mutated CFTR (p.Gly628Arg) structure. Notably, two of the substances Armepavine and Quercetin exhibited a higher docking score, which indicates that these two have stronger capacity to enhance the activity of CFTR protein through improved trafficking and restoration of original function. Armepavine and Curcumin possess favorable pharmacokinetic and toxicity profiles, making them suitable candidates for further investigation. To fully appreciate the medicinal potential of natural substances, it is imperative to understand their pharmacophore and new possibilities for producing long-lasting, efficient drugs that draw inspiration from the wide range of natural substances.
Data availability
The dataset analyzed during the current study are available in the NCBI-database (CFTR protein) (www.ncbi.nlm.nih.gov) and PubChem database. Sr.No#-Identification of Protein-Accession No#. 1-CFTR protein- P13569.3. -Identification Natural compounds-. 2- Armepavine-442169. 3-Osthole-10228. 4- Curcumin-969516. 5. Plumbagin-10205. 6-Quercetin-5280343. 7-R* (Trikafta)-165363555
References
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protocols Hum Genet 76(20):7 21-27.20. 41
Akbari B, Baghaei-Yazdi N, Bahmaie M, Mahdavi Abhari FJB (2022) The role of plant‐derived natural antioxidants in reduction of oxidative stress. 48:611–633
Alameen AA, Abdalla M, Alshibl HM, AlOthman MR, Alkhulaifi MM, Mirgany TO, Elsayim RJJSCS (2022) In-silico studies of glutathione peroxidase4 activators as candidate for multiple sclerosis management. Journal of Saudi Chemical Society. 2022;26(6):101554. 26: 101554
Ameji PJ, Uzairu A, Shallangwa GA, Uba SJJTUMS (2023) Molecular docking-based virtual screening, drug-likeness, and pharmacokinetic profiling of some anti-Salmonella typhimurium cephalosporin derivatives. Journal of Taibah University Medical Sciences. 2023 Jun 9
Amin F, Hassan N, Bashir K, Khan A, Bibi H, Irshad M, Khan S, Nawaz K, Ullah Z (2023) antimicrobial susceptibility profile of various bacteria isolated from respiratory tract infection. Bulletin of Biological and Allied Sciences Research 2023: 48–48
Arulselvan P, Fard MT, Tan WS, Gothai S, Fakurazi S, Norhaizan ME, Kumar SSJO (2016) longevity c Role of antioxidants and natural products in inflammation. Oxidative medicine and cellular longevity. 2016;2016. 2016
Ashraf A, Ghani MU, Khan MU, Rehman HM, ul Hassan M, Mehdi ZJALS (2023) Personalized Medicine; a Potential Therapy for Cystic Fibrosis. Advancements in Life Sciences. 2023;9(4):437– 45. 9: 437–445
Ashraf A, Ghani MU, Khan MU, Rehman HM, ul Hassan M, Mehdi Z (2023a) Personalized medicine; a potential therapy for cystic fibrosis. Advancements Life Sci 9:437–445
Awan SJ, Fatima Z, Kamran S, Khan AS, Fatima T, Imran S, Shabbir M, Nadeem SI (2024) Guar gum in therapeutics: a succinct exploration. Bulletin of Biological and Allied Sciences Research 2024: 60
Bareil C, Bergougnoux A (2020) CFTR gene variants, epidemiology and molecular pathology. Archives de Pédiatrie 27:eS8–eS12
Becq F, Mirval S, Carrez T, Lévêque M, Billet A, Coraux C, Sage E, Cantereau AJERJ (2022) The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. ivacaftor. 59
Bergbower E, Boinot C, Sabirzhanova I, Guggino W, Cebotaru LJCP, Biochemistry (2018) The CFTR-associated ligand arrests the trafficking of the mutant ∆F508 CFTR channel in the ER contributing to cystic fibrosis. Cellular Physiology and Biochemistry. 2018;45(2):639– 55. 45: 639–655
Bors LA, Erdő FJSP (2019) Overcoming the blood–brain barrier. challenges and tricks for CNS drug delivery. Scientia Pharmaceutica. 2019;87(1):6. 87: 6
Brylinski, MJCb (2018) design d Aromatic interactions at the ligand–protein interface: Implications for the development of docking scoring functions. Chemical biology & drug design, 91(2), 380–390. 91: 380–390
Chen X, Li H, Tian L, Li Q, Luo J, Zhang YJJ (2020) Analysis of the physicochemical properties of acaricides based on Lipinski’s rule of five. Journal of computational biology. 2020;27(9):1397– 406. 27: 1397–1406
Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31:2745–2747
Colovos C, Yeates TOJP (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein science. 1993;2(9):1511-9. 2: 1511–1519
Cuevas-Ocaña S, Laselva O, Avolio J, Nenna R (2020) The era of CFTR modulators: improvements made and remaining challenges. Breathe 16
Cutting GRJNRG (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nature Reviews Genetics. 2015;16(1):45–56. 16: 45–56
Daina A, Michielin O, Zoete VJS (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports. 2017;7(1):42717. 7: 42717
Ernst SW, Knight R, Royle J, Stephenson L (2021) Pharmaceutical toxicology. Principles of Translational Science in Medicine. Elsevier, pp 265–279
Fatima S, CHEEMA K, Shafiq M, Manzoor M, Ali Q, Haider M, Shahid M (2023) The genome-wide bioinformatics analysis of 1-aminocyclopropane-1-carboxylate synthase (acs), 1-aminocyclopropane-1-carboxylate oxidase (aco) and ethylene overproducer 1 (eto1) gene family of fragaria vesca (woodland strawberry). Bulletin of Biological and Allied Sciences Research 2023: 38–38
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo ADJM (2015) Molecular docking and structure-based drug design strategies. Molecules 20:13384–13421
Fiedorczuk K, Chen J (2022) Molecular structures reveal synergistic rescue of ∆508 CFTR by Trikafta modulators. Science 378:284–290
Fonseca C, Bicker J, Alves G, Falcão A, Fortuna AJP (2020) Cystic fibrosis: Physiopathology and the latest pharmacological treatments. Pharmacological research. 2020;162:105267. 162: 105267
George Priya Doss C, Rajasekaran R, Sudandiradoss C, Ramanathan K, Purohit R, Sethumadhavan RJG (2008) A novel computational and structural analysis of nsSNPs in CFTR gene. Genomic medicine. 2008;2:23–32. 2: 23–32
Ghani MU, Sabar MF, Awan FI (2022) WS21.03 low-cost chain termination DNA sequencing PCR reaction to diagnose CFTR gene mutations. J Cyst Fibros 21:S41–S42
Ghazi BK, Bangash MH, Razzaq AA, Kiyani M, Girmay S, Chaudhary WR, Zahid U, Hussain U, Mujahid H, Parvaiz UJBRI (2023) In Silico Structural and Functional Analyses of NLRP3 Inflammasomes to Provide Insights for Treating Neurodegenerative Diseases. BioMed Research International. 2023;2023. 2023
Gohar M, Rehman I, Ahmad J, Ahmad F, Bashir K, Ikram S, Hassan N, Khan M, Ullah A (2023) Prevalence of hepatitis b virus and genotypes in the region of khyber pakhtunkhwa pakistan. Bulletin of Biological and Allied Sciences Research 2023: 53–53
Hassan M, Baig AA, Attique SA, Abbas S, Khan F, Zahid S, Ain QU, Usman M, Simbak NB, Kamal MAJDJPS (2021) Molecular docking of alpha-enolase to elucidate the promising candidates against Streptococcus pneumoniae infection. DARU Journal of Pharmaceutical Sciences. 2021;29:73–84. 29: 73–84
Hildebrand PW, Rose AS, Tiemann JKJTBS (2019) Bringing molecular dynamics simulation data into view. 44:902–913
Hildebrand PW, Rose AS, Tiemann JKJTBS (2019a) Bringing molecular dynamics simulation data into view. Trends Biochem Sci 44:902–913
Hwang T-C, Yeh J-T, Zhang J, Yu Y-C, Yeh H-I, Destefano SJJGP (2018) Structural mechanisms of CFTR function and dysfunction. J Gen Physiol 150:539–570
Hwang T-C, Braakman I, van der Sluijs P, Callebaut IJJCF (2023) Structure basis of CFTR folding, function and pharmacology. Journal of Cystic Fibrosis. 2023;22:S5-11. 22: S5-S11
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu BJN (2023) PubChem 2023 update. Nucleic acids research. 2023;51(D1):D1373-80. 51: D1373-D1380
Kleizen B, van Willigen M, Mijnders M, Peters F, Grudniewska M, Hillenaar T, Thomas A, Kooijman L, Peters KW, Frizzell RJJMB (2021) Co-translational folding of the first transmembrane domain of ABC-transporter CFTR is supported by assembly with the first cytosolic domain. Journal of Molecular Biology. 2021;433(13):166955. 433: 166955
Kumar K, Anbarasu A, Ramaiah SJMB (2014) Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Molecular BioSystems. 2014;10(4):891–900. 10: 891–900
Laskar FS, Bappy MNI, Hossain MS, Alam Z, Afrin D, Saha S, Ali Zinnah KMJIJoG (2023) An In silico Approach towards Finding the Cancer-Causing Mutations in Human MET Gene. International Journal of Genomics. 2023;2023. 2023
Li J, Guo Q, Lei X, Zhang L, Su C, Liu Y, Zhou W, Chen H, Wang H, Wang F (2020) Pristimerin induces apoptosis and inhibits proliferation, migration in H1299 Lung Cancer cells. J Cancer 11:6348
Lopes-Pacheco M (2020) CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol 10:1662
Lopes-Pacheco M, Pedemonte N, Veit G (2021) Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin Drug Discov 16:897–913
López-Valdez JA, Aguilar-Alonso LA, Gándara-Quezada V, Ruiz-Rico GE, Ávila-Soledad JM, Reyes AA (2021) Pedroza-Jiménez FDJBmdHIdM (2021) cystic fibrosis: current concepts. Boletín médico Del Hosp Infantil De México 78(6):584–596
McDonald EF, Woods H, Smith ST, Kim M, Schoeder CT, Plate L, Meiler JJB (2022) Structural Comparative Modeling of Multi-Domain F508del CFTR. Biomolecules. 2022;12(3):471. 12: 471
McKee AG, McDonald EF, Penn WD, Kuntz CP, Noguera K, Chamness LM, Roushar FJ, Meiler J, Oliver KE, Plate LJCCB (2023) General trends in the effects of VX-661 and VX-445 on the plasma membrane expression of clinical CFTR variants
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814
Padányi R, Farkas B, Tordai H, Kiss B, Grubmüller H, Soya N, Lukács GL, Kellermayer M, Hegedűs TJC, Journal SB (2022) Nanomechanics combined with HDX reveals allosteric drug binding sites of CFTR NBD1. Computational and Structural Biotechnology Journal. 2022;20:2587-99. 20: 2587–2599
Palma J, Pierdominici-Sottile GJC (2023) On the uses of PCA to Characterise Molecular Dynamics simulations of Biological macromolecules: Basics and Tips for an effective use. 24:e202200491
Parthiban V, Gromiha MM, Hoppe C, Schomburg D (2007) Structural analysis and prediction of protein mutant stability using distance and torsion potentials: role of secondary structure and solvent accessibility. Proteins: Structure, Function, and Bioinformatics 66: 41–52
Pereira SV, Ribeiro JD, Ribeiro AF, Bertuzzo CS, Marson FAL (2019) Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools. Sci Rep 9:6234
Pires DE, Ascher DB, Blundell TL (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30:335–342
Rasheed MA, Iqbal MN, Saddick S, Ali I, Khan FS, Kanwal S, Ahmed D, Ibrahim M, Afzal U, Awais MJL (2021) Identification of lead compounds against scm (fms10) in Enterococcus faecium using computer aided drug designing. Life (Basel) 11:77. https://doi.org/10.3390/life11020077
Safdar A, Ghani MU, Bano I, Rafique H, Sabar MF, Mumtaz A, Akbar A, Shaikh RS (2023) P027 mutational analysis of CFTR gene in Pakistani cystic fibrosis patients. J Cyst Fibros 22:S71–S72
Sakhawat A, Khan MU, Rehman R, Khan S, Shan MA, Batool A, Javed MA, Ali Q (2023) Natural compound targeting BDNF V66M variant: insights from in silico docking and molecular analysis. Amb Express 13:134
Schrodinger LJNY (2010) The PyMOL molecular graphics system, version 1.3 r1 Schrodinger LLC. 30
Shan MA, Khan MU, Ishtiaq W, Rehman R, Khan S, Javed MA, Ali Q (2024) In silico analysis of the Val66Met mutation in BDNF protein: implications for psychological stress. AMB Express 14:11
Shanthi V, Rajasekaran R, Ramanathan, KJCb (2014) biophysics Computational identification of significant missense mutations in AKT1 gene. 70: 957–965
Sheema, Bashir K, Fiaz S, Khan AW, Haqqani S, Bibi A, Nawaz K, Khan MA, Ullah A (2024) Molecular identification of hcv genotypes among injecting drug users having HCV and HIV co-infection. Bulletin of Biological and Allied Sciences Research 2024: 71
Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski JJN (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nature genetics. 2013;45(10):1160-7. 45: 1160–1167
Tsai A, Wu S-P, Haseltine E, Kumar S, Moskowitz SM, Panorchan P, Shah KJP (2020) Physiologically based pharmacokinetic modeling of CFTR modulation in people with cystic fibrosis transitioning from mono or dual regimens to triple-combination elexacaftor/tezacaftor/ivacaftor. Pulmonary therapy. 2020;6:275– 86. 6: 275–286
Ullah I, Ullah A, Rehman S, Ullah S, Ullah H, Haqqni S, Amir M, Gul F, Bashir K (2023) Prevalence and risk factors of helicobacter pylori infection among individuals with tobacco consumption habits in district Peshawar: a cross-sectional study. Bulletin of Biological and Allied Sciences Research 2023: 42–42
Ullah A, Bibi A, Ullah I, Kayani REZ, Asim M, Munawar N, Amjad M, Siraj M, Gohar M, Khan MA (2024) An overview of hepatitis c virus and liver cirrhosis in Pakistan. Bulletin of Biological and Allied Sciences Research 2024: 64
Unnisa A, Jandrajupalli SB, Elamine BA, Mohamed OA, Alreshidi KS, Alshammari AH, shaty Alshammari H, Bandar N, Gangireddy RJC, Biology M (2023) Screening of natural products atlas for identification of lead molecule to treat angina pectoris using bioinformatics approaches. Cellular and Molecular Biology. 2023;69(8):125– 31 69: 125–131
Vankeerberghen A, Wei L, Jaspers M, Cassiman J-J, Nilius B, Cuppens H (1998) Characterization of 19 Disease-Associated missense mutations in the Regulatory Domain of the cystic fibrosis transmembrane Conductance Regulator. Hum Mol Genet 7:1761–1769
Worth CL, Preissner R, Blundell TL (2011) SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res 39:W215–W222
Zhou X, Smith QR, Liu XJWIRN (2021) Brain penetrating peptides and peptide–drug conjugates to overcome the blood–brain barrier and target CNS diseases. Nanomedicine and Nanobiotechnology. 2021;13(4):e1695. 13: e1695
Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP2024R165), King Saud University, Riyadh, Saudi Arabia.
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
AS and RR carried out research work and wrote the initial draft of the manuscript. AA, AHW and MUG conducted the data analysis. QA, and MUK planned and supervised the study and edited the final version of the manuscript. MUK, ZY, DA, MAJ, and QA technically reviewed and finalized the draft. All authors reviewed the final version of the manuscript and approved it for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors have no potential conflict of interest
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khan, M.U., Sakhawat, A., Rehman, R. et al. Identification of novel natural compounds against CFTR p.Gly628Arg pathogenic variant. AMB Expr 14, 99 (2024). https://doi.org/10.1186/s13568-024-01762-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-024-01762-9