
Abstract
The poor prognosis of hepatocellular carcinoma (HCC) is still an urgent challenge to be solved worldwide. Hence, assembling drugs and targeted short peptides together to construct a novel medicine delivery strategy is crucial for targeted and synergy therapy of HCC. Herein, a high-efficiency nanomedicine delivery strategy has been constructed by combining graphdiyne oxide (GDYO) as a drug-loaded platform, specific peptide (SP94-PEG) as a spear to target HCC cells, sorafenib, doxorubicin-Fe2+ (DOX-Fe2+), and siRNA (SLC7A11-i) as weapons to exert a three-path synergistic attack against HCC cells. In this work, SP94-PEG and GDYO form nanosheets with HCC-targeting properties, the chemotherapeutic drug DOX linked to ferrous ions increases the free iron pool in HCC cells and synergizes with sorafenib to induce cell ferroptosis. As a key gene of ferroptosis, interference with the expression of SLC7A11 makes the ferroptosis effect in HCC cells easier, stronger, and more durable. Through gene interference, drug synergy, and short peptide targeting, the toxic side effects of chemotherapy drugs are reduced. The multifunctional nanomedicine GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i (MNMG) possesses the advantages of strong targeting, good stability, the ability to continuously induce tumor cell ferroptosis and has potential clinical application value, which is different from traditional drugs.
Graphical Abstract

Similar content being viewed by others


Introduction
Liver cancer is a global public health problem and hepatocellular carcinoma (HCC) is the most common pathological type of primary liver cancer [1,2,3]. At present, postoperative chemotherapy is the main method of clinical treatment for HCC, which can greatly improve the treatment effect and relative survival rate of patients [4,5,6]. However, due to large postoperative trauma, easy wound infection, and non-specific killing of tumor cells by conventional chemotherapy, patients have obvious toxic reactions and poor recovery ability [7, 8]. Although the new immunotherapy, atezolizumab combined with bevacizumab, can maximize the survival of HCC patients and has an efficacy advantage, the cost of immunotherapy remains unaffordable in developing countries with high population densities and low incomes [9,10,11]. Hence, sorafenib is still the most common and cost-effective systemic treatment for patients with advanced HCC [12,13,14]. However, the long-term use of sorafenib and other chemotherapy drug monotherapy is prone to obvious drug resistance and also faces serious challenges such as poor efficacy, non-targeting, short half-life, and drug toxicity [15,16,17]. Therefore, in order to solve these issues simultaneously, it is an urgent clinical demand to explore a new strategy for HCC chemotherapy with the combined action of several therapies and the advantages of stability, targeting and good biocompatibility.
Ferroptosis, as different from other cell death modes, is triggered by the abnormal accumulation of lipid peroxides caused by excess iron [18,19,20,21]. Iron metabolism, SLC7A11-mediated cysteine transport, NRF2-Keap1, glutathione-GPX4, FSP1-COQ10, and other signal transduction pathways constitute the core molecular mechanism of ferroptosis [22,23,24,25]. Recently, increasing evidence has discovered that some small molecules such as disulfiram, auranofin, iFSP, zalcitabine, and withaferin could act as a promising anticancer therapeutic effect by inducing ferroptotic death [26,27,28,29,30]. As mentioned above, sorafenib, a multi-targeted tyrosine kinase inhibitor, is not only recognized as gold standard first-line therapy for advanced HCC but also identified as a ferroptosis inducer, by inhibiting the expression of the cystine transporter SLC7A11 and reducing the intracellular glutathione (GSH) content [31,32,33,34]. However, monotherapeutic administration strategies often lead to drug resistance and the activation of compensatory mechanisms to escape cell death. Moreover, the synergistic regulation of multiple drugs on different metabolic pathways often ignores their multidrug resistance and toxic side effects from the nonspecific localization, which are important reasons leading to the failure of chemotherapy. Thus, optimizing the combination of sorafenib-based treatment with other therapeutic strategies is the key to solving the problem. In our previous study, we demonstrated that disulfiram/Cu could facilitate sorafenib-induced ferroptosis via targeting different mechanisms and reducing the treatment dose [35]. Therefore, the development of a new drug delivery system for sorafenib-based therapy with properties of the absence of toxic side effects, target-specificity, and ease of drug visualization and assembly is highly desirable.
Graphdiyne oxide (GDYO) is a two-dimensional carbon nanosheet material composed of sp and sp2 hybrid carbon atoms, which is derived from graphdiyne (GDY) through the oxidation of alkyne groups by concentrated nitric acid and sulfuric acid [36,37,38,39]. The surface of GDYO composes of ordered carbon-oxygen and carbon-hydroxyl groups that have a high affinity for proteins driven by the formation of hydrogen bonds and salt bridges [40,41,42]. The depressions formed on their surfaces offer great possibilities for loaded small-molecule drugs and genes, which makes GDYO an ideal delivery carrier for anti-cancer drugs, genes, peptides, and other substances [43,44,45]. For example, the biomimetic GDYO@i-RBM nanosheets designed based on GDYO can target tumor therapy and enhance tumor reoxygenation capacity, and the constructed multifunctional nanomedicine GCDM can be used for photochemical synergistic tumor therapy [46, 47]. Thus, based on the superior physical and chemical properties of GDYO, combining the nanocarrier GDYO with ferroptosis-inducing genes, sorafenib, and DOX-Fe2+ chemotherapeutic drugs to construct a novel nanomedicine may provide a new strategy for HCC chemotherapy.
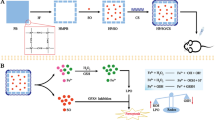
Herein, we developed a multifunctional nanomedicine GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i (MNMG) as a nanomedicine delivery strategy, in which SP94 with the amino acid sequence of NH2-CGGSFSIIHTPILPL-COOH can specifically targeting HCC cells via binding the receptor on the HCC [48,49,50]. After the receptor is mediated, the nanosheets enter the HCC cells through endocytosis and cleave in the acidic lysosomal environment to release sorafenib, DOX, ferrous ion, and SLC7A11-i, which induce HCC cells into the ferroptosis mechanism. As shown in Scheme 1, SP94-PEG has been synthesized by chemically coupling PEG to SP94, and then linked with GDYO to form a nanosheet structure (GDYO@SP94) targeting HCC cells. The synthetic GDYO@SP94 has good dispersion and stability, providing a large specific surface area for loading drugs and genes, and can effectively enrich the drug at the site of HCC, thereby reducing the concentration of drugs in other parts of the body and reducing the toxic and side effects of chemotherapeutic drugs. The combination of targeted therapy and the enhanced permeability and retention (EPR) effect [51,52,53] can enhance the endocytosis of nanocarriers by HCC cells and fulfill the effective aggregation of nanomedicines at the site of HCC. Next, the coordination compound DOX-Fe2+ and sorafenib were loaded onto the GDYO@SP94 nanosheet via π-π stacking. DOX-Fe2+ is a coordination compound formed by the chelation of the traditional chemotherapeutic drug DOX with ferrous ions [54,55,56]. The ferrous ion can preferentially replace the phenolic hydrogen position in doxorubicin (DOX) to form the DOX-Fe2+ complex, which in turn increases the unstable iron pool in tumor cells, induce excessive production of ROS in mitochondria, further amplify the ferroptosis effect of Fe2+ through H2O2 generated by DOX [57,58,59]. Meanwhile, the addition of SLC7A11-i inhibited cystine transport and lowered the synthesized GSH [60,61,62]. Finally, SLC7A11-i has adsorbed to the surface of GDYO@SP94/DOX-Fe2+/sorafenib through the electrostatic interaction of hetero-electric attraction, forming a highly efficient nano-complex MNMG with short peptide targeting, drug synergy, and gene interference.
In this study, the drug-loaded strategy MNMG can effectively inhibit the proliferation, migration, and invasion of hepatocellular carcinoma cells, and has a strong therapeutic effect. Noteworthy, the MNMG disrupted mitochondrial homeostasis and dramatically increased reactive oxygen species and lipid peroxide levels, which severely affected the normal production of GSH in tumor cells and ultimately led to ferroptotic cell death. Moreover, MNMG can effectively aggravate and target the tumor site with minimal systemic side effects in vivo, and effectually suppress HCC through inducing ferroptosis in situ. Meaningfully, the construction of the nanomedicine provides a new strategy for HCC chemotherapy and also provides a novel concept for designing a targeted, efficient, stable, and biocompatible nanomedicine, which may have potential clinical application value.

(A) Schematic diagram of the synthesis of the multifunctional nanomedicine GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i (MNMG). (B) The mechanism of MNMG inducing ferroptosis in tumor cells to treat tumors
Results and discussion
Characterization and validation
The multifunctional nanomedicine GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i was stepwise engineered as illustrated in Fig. 1. Graphdiyne is oxidized to GDYO under the action of concentrated nitric acid and sulfuric acid [63,64,65]. Under the joint action of carbodiimide (EDC) and N-hydroxysuccinimide (NHS), the carboxyl group in GDYO is activated and promotes its reaction with the amino group of SP94-PEG to generate stable amide bonds. Through the π-π stacking interaction between aromatic rings, DOX-Fe2+ and sorafenib were coloaded onto SP94-PEG modified nanosheets, and then the electrostatic interaction between positive and negative charges was used to adsorb siRNA, resulting in the synthesis of GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i (MNMG).

The synthesis schematic of GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i
GDYO is characterized by its high dispersibility, good stability, and sensitivity as a nano enzyme [66,67,68,69]. The most powerful of these properties is its stable loading capacity and sustained release [70,71,72]. To demonstrate the differences between each functionalization step and the morphology change with molecular attachment, we characterized the morphology and elemental composition of different loaded nanosheets using Field Emission Scanning Electron Microscope (FESEM) and Transmission Electron Microscope (TEM) elemental mapping techniques. FESEM and TEM observations indicated that GDY and GDYO had a filmy, uniform lamellar morphology and are ultrathin layered structures (Fig. 2A and B). After decorating with SP94-PEG, DOX-Fe2+, sorafenib and siRNA, it was clearly seen that the ultra-layered morphology of GDYO surface is gradually thickened, forming obvious irregular folds. The Atomic Force microscopy (AFM) images further illustrate the ultrathin layer structure of GDYO with an average thickness of 3 nm (Figure S1). GDYO has a special 2D large-plane all-carbon structure and surface depressions, which provide sites for electrostatic adsorption and π - π stacking of small molecular drugs [73, 74]. In addition, the Energy Dispersive Spectrometer (EDS) elemental mapping of the MNMG was used to identify the corresponding distribution of the C, O, N, Fe, F and Cl elements, indicating that the surface of GDYO was uniformly dispersed DOX-Fe2+ (C27H29NO11Fe) and sorafenib (C21H16ClF3N4O3) (Fig. 2C). Meanwhile, we showed the hydrodynamic particle size based on the number percentage and zeta potential of the GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG (Fig. 2D). The particle size of the corresponding nanosheets increases step by step after each synthesis step.
To demonstrate the successful assembly of loaded drugs and genes on the surface of GDYO, the UV-Vis absorption spectrum of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG were monitored in Fig. 2E. MNMG has an absorption peak at 214 nm to 228 nm. Compared with the UV-Vis of GDYO@SP94 and the calibration curve of SP94, it can be seen that this absorption peak is the characteristic peak of SP94. In the calibration curve, the characteristic absorption peak of sorafenib was 270 nm and that of siRNA was 259 nm (Figure S2). In addition, according to the calibration curve of DOX-Fe2+, the characteristic peak of DOX was at 485 nm and that of Fe2+ was around 268 nm (Figure S3A).The obvious absorption peak of MNMG at 500 nm corresponded to the characteristic peak of DOX, while the absorption peak at 265 nm was very close to the characteristic peak of sorafenib and Fe2+. Although siRNA was reflected in the calibration curve, the intensity of its UV-Vis absorption peak was too weak to show a significant peak in MNMG, which can be seen by comparing the UV-Vis absorption spectra of GDYO@SP94/DOX-Fe2+/sorafenib. The range of calibration curve is consistent with previous research [75,76,77].

Characterization and validation of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG. (A) FESEM images of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG. Scale bar: 1 μm. (B) TEM images of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG. Scale bar: 50 nm. (C) HAADF-STEM images and element mapping results of MNMG. Scale bar: 100 nm. (D) Zeta potential measured changes in the hydrodynamic particle size of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib and MNMG. (E) UV-Vis absorption spectrum of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG
XPS was used to analyze the chemical bindings, electronic states and elemental composition of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG. Figure 3A displayed the XPS full survey spectra of the above nanosheets to show the differences after each functionalization process from pristine GDY. Comparing the changes in C 1s and O 1s of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib, and MNMG, the photoelectron intensity of C = O and C-O significantly increased, which was related to the drug loading (Fig. 3B). And the detection of C 1s and F 1s at GDYO@SP94/DOX-Fe2+/sorafenib yielded the presence of C-F, which reflected the loading of sorafenib on the GDYO nanosheets. In addition, C-NH- and NH4 + were detected in GDYO@SP94 and GDYO@SP94/DOX-Fe2+/sorafenib, and the intensity of NH4 + increased gradually, indicating that the concentrations of SP94 and DOX increased on the surface of GDYO nanosheets. In the high resolution spectrum of S 2p at GDYO@SP94, peaks located at 163 eV and 164.5 eV are observed corresponding to S 2p3/2 and S 2p1/2, and peaks at 168.2 eV and 169.3 eV corresponding to SO42− 2p3/2 and SO42− 2p1/2. Meanwhile, in GDYO@SP94/DOX-Fe2+/sorafenib, the peaks of 167.8 eV and 169.1 eV correspond to SO32− 2p3/2 and SO32− 2p1/2, as well as the peaks located at 169.1 eV and 170.4 eV correspond to SO42− 2p3/2 and SO42− 2p1/2. Since S 2p existed only in SP94, the intensity of S 2p manifested in GDYO@SP94/DOX-Fe2+/sorafenib was reduced after drug loading. Fe 2p3/2 and Fe 2p1/2 corresponding to 710.5 eV and 724 eV can be seen in the Fe 2p spectrum, accompanied by two satellite peaks, indicating that the formation of GDYO-loaded DOX-Fe2+ can protect Fe2+ from environmental oxidation and maintain its reactivity. The binding energies existed at 197.8 and 199.4 eV was corresponding to Cl− 2p3/2 and Cl− 2p1/2, and the other energies of 200.8 and 202.5 eV was attributed to -C-Cl 2p3/2 and -C-Cl 2p1/2. The existence of carbon-chloride covalent bonds and free chloride ions further indicated that GDYO successfully loaded sorafenib. These results indicate that we successfully achieved the ligation of SP94 short peptide and the loading of sorafenib and DOX-Fe2 + on GDYO nanosheets. However, siRNA was composed of 19 base pairs, and it was difficult to detect valence changes between MNMG and GDYO@SP94/DOX-Fe2+/sorafenib due to its small volume.
DOX chelated ferrous ions to form a complex because Fe2 + preferentially chelated on a phenol group of DOX to replace the position of H and formed Fe-O bond. Therefore, we performed characterization analysis of DOX-Fe2+. XPS was also used to analyze the changes of DOX after chelation with Fe2+ (Figure S3B). Compared with DOX and DOX-Fe2+, the major changes occurred in O1s (Figure S3C). The appearance of a binding energy peak corresponding to Fe-O at 530.7 eV confirmed the formation of Fe-O bonds by DOX chelating Fe2+, which was consistent with previous studies. In addition, the coupling between SP94 and PEG occured through the activation of EDC and NHS, where the carboxyl end of the SP94 short peptide chain condensed with the amino group of PEG to form the amide bond. The particle size changes of SP94 and SP94-PEG were detected by zeta potential, and it was found that the particle size of SP94 increased by about 65 nm after coupling PEG (Figure S4A). To demonstrate the changes of SP94 after coupling with PEG, the UV-Vis absorption spectra of SP94, PEG and SP94-PEG were detected in Figure S4B. We found that the peak value of SP94 was slightly shifted under the influence of PEG, which was attributed to the coupling reaction between the carboxyl group of SP94 and the amino group in PEG through the activation group.
To verify particle stability at different time points, we needed to conduct a dispersion study for 5 days, and GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib and MNMG was examined by Dynamic light scattering (DLS) and zeta potential measurements (Table S1). The zeta potential difference of GDY was generally low over the 5 days examined, favoring the condensed state. This was consistent with our observation that black suspended solids were still seen in GDY dispersion after ultrasonic treatment, and some GDY precipitated after standing. It showed that GDY has been in a dispersive system with poor stability. On the contrary, GDYO obtained by oxidation of GDY was in a relatively stable state during these five days, and the nanosheets were evenly distributed in the system. On the first and second days, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib and MNMG were in a relatively stable dispersion. However, with the passage of time, the value of zeta potential gradually decreased, while the particle size showed an increasing trend. This indicates that after GDYO was loaded with drugs and genes, the attraction between the nanosheets gradually exceeded the repulsion over time, and the state tended to condense.

XPS spectra of GDY, GDYO, GDYO@SP94, GDYO@SP94/DOX-Fe2+/sorafenib. (A) Full survey spectra of XPS. (B) High-resolution spectra of XPS
Sequence selection of siRNA, biological toxicity of nanosheet backbone and validation of cellular uptake effect
SLC7A11, one of the core subunits of the systemXc- receptor, is located upstream of the signaling axis in the ferroptosis mechanism and plays a decisive role in GSH synthesis [78, 79]. Due to the elevated SLC7A11 expression accompanied with increased GSH content in HCC cells [80, 81], GDYO nanosheets were loaded with small interfering RNA sequences targeting SLC7A11 to increase the therapeutic effect. Therefore, we designed two interfering sequences targeting SLC7A11 according to different sequences for the inhibition of SLC7A11 expression (Fig. 4A). The interference effect was verified by Western blotting, and GAPDH was used as the internal control. Finally, we selected the SLC7A11-i-1 sequence, the one with more excellent interference efficiency, as the gene fragment loaded on the GDYO nanosheets (Fig. 4B) to hinder the transport function of systemXc- receptor and initiates the process of cells toward ferroptosis.
In order to evaluate the toxic damage, we first tested whether the synthetic backbone of the nanomedicines would cause toxic damage to cells. The proliferation curve results indicated that GDYO nanosheets and GDYO@SP94 at the concentrations of 50, 100, and 150 µg/ml produced negligible cytotoxicity on HCC cell proliferation (Fig. 4C). GDYO is a lamellar, thin-film nanostructure whose surface harbors C = O groups which have a high affinity for proteins binding via hydrogen bonding and salt bridge formation [82, 83]. Therefore, GDYO can be used to target HCC after linking with SP94-PEG. Moreover, the GDYO and GDYO@SP94 framework has no obvious toxicity in vitro, showing special biocompatibility and safety.
To verify whether the synthesized MNMG was able to be uptake by HCC cells successfully, the process of endocytosis was monitored by confocal microscopy. The fluorescence of DOX-Fe2+ and FAM-tagged SLC7A11-i (SLC7A11-iFAM) was used as fluorescent probes for drug tracking and diagnosis. We found that DOX-Fe2+ and SLC7A11-i-1 were successfully assembled on GDYO and could be taken up by cells (Fig. 4D).

Coload verification of MNMG. (A) Target sequence and antisense strand of SLC7A11. (B) Western blotting was performed to detect the expression of SLC7A11 proteins in the control group and HCC cells after adding siRNA interference. (C) Human hepatocellular carcinoma cell lines Huh7 and SMMC-7721 cells were treated with high concentrations of GDYO nanosheets and GDYO@SP94, and the cell proliferation ability was detected by CCK8 assay. (D) Confocal microscopy images of the MNMG endocytosed into cells (Hoechst: blue fluorescence, DOX-Fe2+: red fluorescence, SLC7A11-i: green fluorescence). Scale bar: 5 μm
Stability and antitumor effects of MNMG
To determine the cytotoxic effect of MNMG on HCC cells, we first treated different concentrations of MNMG with Huh7 and SMMC-7721 for 24 h, and cell viability was assessed by CCK-8 assay. In addition to MNMG, we also synthesized nano-drugs lacking various components into the following groups to verify whether these components affect the drug effect. These semi-manufactured nanomedicines respectively are (I) GDYO, (II) GDYO@SP94/DOX-Fe2+/SLC7A11-i, (III) GDYO@SP94/sorafenib/SLC7A11-i, (IV) GDYO@SP94/DOX/sorafenib/SLC7A11-i, (V) GDYO@SP94/DOX-Fe2+/sorafenib, and (VI) GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i. The results revealed that the MNMG was preferentially cytotoxicity to HCC cells and selectively reduced their viability in a dose-dependent manner. Except for group I as control, the toxicity of groups II, III, IV, and V to hepatocellular carcinoma cells at the same concentration was lower than that of group VI (Fig. 5A and B). In addition, we verified the interference effect of SLC7A11-i in each group and found that the interference effect increased with the concentration of MNMG, and compared with other semi-finished product groups, the MNMG group had the most obvious knockdown effect (Fig. 5C). Next, Calcein-AM/PI staining was used to further elucidate the cytotoxic effect of the MNMG. We observed a similar phenomenon that the MNMG can indeed increase the percentage of dead cells and reduce the number of live cells determined by both confocal microscopy and flow cytometry analysis (Fig. 5D and E). In addition, we also tested the stability of the MNMG solution (solvent DMSO) for 21 days and found that MNMG had stable cytotoxicity against HCC cells at -80 °C, 4 °C, and 25 °C (Fig. 5F). Sorafenib is known to be a lipid-soluble chemotherapy drug that is insoluble in water, while GDYO, as a hydrophilic nanomaterial, has good dispersion in water [84, 85]. Therefore, we wonder whether GDYO is capable of improving the water solubility of sorafenib. Accordingly, we found that MNMG also has good dispersibility in water and has similar drug effects to DMSO solvent group (Fig. 5G and H). These results suggest that MNMG has a stable, well-distributed, and effective therapeutic effect as a new candidate for HCC treatment.

Antitumor effect of MNMG. (A) SMMC-7721 and Huh7 were treated with six various concentrations of nanomedicines (0–50 µg/ml) for 24 h. Cell viability was determined by CCK-8 assay. (I. GDYO, II. GDYO@SP94/DOX-Fe2+/SLC7A11-i, III. GDYO@SP94/sorafenib/SLC7A11-i, IV. GDYO@SP94/DOX/sorafenib/SLC7A11-i, V. GDYO@SP94/DOX-Fe2+/sorafenib, and VI. GDYO@SP94/DOX-Fe2+/sorafenib/SLC7A11-i.). (B) Corresponding to the dispersion of six groups of nanomedicines in Figure A. (C) Western blotting ting was used to detect the protein expression levels of SLC7A11 in Huh7 cells treated with six groups of nanomedicines and different concentrations of MNMG. GAPDH was used as the internal reference. (D) Huh7 cells were incubated with 50 µg/ml of GDYO, 20 µg/ml, and 50 µg/ml of MNMG for 24 h, respectively, and the fluorescence of Calcein-AM/PI staining was captured by confocal laser microscopy. (Calcein-AM: live cells, PI: dead cells). Scale bar: 50 μm. On the right is the fluorescence quantification of Calcein-AM/PI staining. (E) The proportion of dead cells treated with MNMG was detected by flow cytometry. On the right is the corresponding statistical graph. (F) Effect stability test of MNMG at different drug storage temperatures. Cell viability was determined by CCK-8 assay. (G) Water solubility test of MNMG compared to sorafenib. (H) The effect of the MNMG dissolved in water was detected by the CCK-8 assay. Values represented mean ± SD. *P < 0.05, **P < 0.01 versus control group
The MNMG inhibits cell viability, invasion, and migration of HCC cells
The most concerning aspect of cancer is its ability to proliferate and migrate. Based on the above observations, we performed a colony formation assay to determine the long-term cell viability and colony-forming ability of HCC cells under low-concentration administration of the MNMG. Low concentrations of MNMG significantly inhibited cell viability for a long time and decreased colony formation with increasing concentrations of MNMG (Fig. 6A and B). In addition, transwell invasion assay and adherent cell wound healing assay were employed to evaluate the effect of MNMG on the invasive and migratory abilities of HCC cells. Clearly, transwell analysis indicated that MNMG could reduce the number of invasive cells in HCC cells (Fig. 6C), and the statistical results revealed that the relative invasion rate was significantly decreased (Fig. 6D). As indicated by the scratch test in Fig. 6E, the wound healing of HCC cells was significantly hindered, and the cell migration ability was weakened after the administration of the low concentration of the MNMG. Histogram statistics showed the wound healing rates of Huh7 and SMMC-7721 cells were significantly reduced (Fig. 6F). Collectively, these results together revealed that the MNMG inhibited cell viability, invasion, and migration capabilities of HCC cells.

MNMG inhibits the proliferation and migration ability of HCC cells. (A) Colonies of Huh7 and SMMC-7721 cells were cultured for 14 days under the GDYO (5 µg/ml) and MNMG (2, 5 µg/ml) treatment. Colonies were recorded with an inverted microscope. (B) Below the colony result graph is the statistical relative colony formation rate. (C) Two HCC cells were treated with a low concentration of the MNMG for 24 h, and cell invasion was detected by transwell assay. (D) Histogram of the number of invasive cells. (E) The migration capacity of the control group and the MNMG-treated cells was detected by wound healing assay. (F) Corresponding quantitative histogram of wound healing experiments. Values represented mean ± SD. *P < 0.05, **P < 0.01 versus control group
The MNMG results in the disturbance of the mitochondrial homeostasis
Ferroptosis is a form of cell death due to the catastrophic accumulation of free iron and unrestrained lipid peroxidation [86,87,88]. Multiple signal transduction pathways such as mitochondrial energy metabolism, cysteine deficiency, and iron-sulfur cluster assembly constitute the core molecular mechanism of ferroptosis [89,90,91]. Considering that mitochondrion plays a vital role in cysteine depletion-induced ferroptosis [92, 93], we then tested whether MNMG would influence mitochondrial dysfunction in hepatoma carcinoma cells. We first focused on mitochondrial morphology, which was visualized by MitoTracker Red fluorescent staining. Compared with the normal morphology of mitochondria in Huh7 and SMMC-7721 cells in the control group, after different concentrations of MNMG delivery, mitochondria appeared fragmented and vacuolated, became disorganized, and aggregated around the nucleus (Fig. 7A). Once mitochondrial dysfunction occurs, mitochondrial oxidative phosphorylation may be affected. So, we used the Seahorse XFe 24 Extracellular Flux Analyzer to explore the effect of the MNMG on mitochondrial respiration capacity. The findings indicated that MNMG was able to inhibit the oxygen consumption rate (OCR) of mitochondrial basal respiration, maximal respiration, and spare respiration, with decreased ATP production (Fig. 7B). Increasing evidence has revealed that there is a close link between the occurrence of ferroptosis and the production of mitochondrial ROS [94]. We further detected mitochondrial ROS production, which originates from the disruption of the electron transport chain, leading to the acceleration of cellular oxidative stress and ferroptosis. The data of mitoSOX staining revealed that the accumulation of mitochondrial ROS was the most significant in cells treated with MNMG (Fig. 7C). Moreover, even in the absence of sorafenib, GDYO@SP94/DOX-Fe2+/SLC7A11-i was able to increase the mitochondrial ROS, a level lower than other synthesized nanomedicines. In addition, the results of flow cytometry also showed that the generation of mitochondrial superoxide also increased with the assembly of different drug components, demonstrating that the different components of the nanotherapeutics exhibited synergistic toxicity effects (Fig. 7D). Meanwhile, flow cytometry confirmed that mitochondrial ROS increased with the increase in MNMG concentration (Fig. 7E).
Ferroptosis and mitochondrial-related protein changes were further detected. Analysis by western blotting, due to the destruction of mitochondrial morphology, the expression of outer mitochondrial membrane protein porin 1 (VDAC1) and mitochondrial transcription factor A (Tfam) was downregulated, which directly led to the blockage of the TCA cycle and inhibited the functions of NADH dehydrogenase flavoprotein 2 (NDUFV2). In addition, the accumulation of ferrous ions increased the intracellular active iron pool, affected the iron homeostasis in the mitochondrial environment, and produced excessive ROS, which led to the block of glycolysis and the inhibition of sirtuin 6 (SIRT6) and lactate dehydrogenase A (LDHA) expression (Fig. 7F and G). The presence or absence of mitochondrial dysfunction is inextricably linked to the occurrence and development of ferroptosis. In summary, the MNMG can lead to a dramatic increase in ROS, inhibit the mitochondrial respiratory chain, and impair ATP metabolism in the presence of massive disruption of mitochondrial morphology.

MNMG disrupts mitochondrial homeostasis and leads to mitochondria damage. (A) Mito Tracker-Red probe-labeled mitochondria of Huh7 and SMMC-7721 cells were observed by confocal microscopy after the MNMG treatment for 6 h. Hoechst was used for nuclear staining. Scale bar: 10 μm. (B) Real-time analysis of OCR using a Seahorse analyzer 45 min after the MNMG treatment of HCC cells. (C) MitoSox fluorescent dye was added to the six groups of nanomedicine-treated Huh7 cells to label mitochondrial ROS, and the fluorescence intensity was observed by confocal microscopy. Below is the corresponding statistical analysis of fluorescence quantification. Scale bar: 25 μm. (D, E) Superoxide dismutase produced by mitochondria was detected by flow cytometry. The corresponding statistical histogram is shown on the right. (F, G) Western blotting analysis of Huh7 cells treated with different concentrations of the MNMG and the same concentration of the six nanomedicines for 24 h. GAPDH was used as an internal control. Values represented mean ± SD. *P < 0.05, **P < 0.01 compared with control group
The MNMG triggers ferroptosis with lipid peroxide accumulation and glutathione deficiency
The production of lipid peroxides due to the exhaustion of GSH is one of the important characteristics of ferroptosis [95,96,97]. We then tested the formation of lipid peroxides by BODIPY staining. Similarly, different groups of synthetic nanomedicines were examined for their effects on lipid peroxide production. Confocal microscopy results showed that after 8 h of MNMG treatment, Huh7 cells showed the most significant lipid peroxide formation with red fluorescence of the BODIPY shifted to green fluorescence (Fig. 8A). In addition, flow cytometry results also confirmed that the production of lipid peroxides was accentuated with the increasing concentration of MNMG, and the green fluorescence detected in the MNMG group was the strongest among the six administration groups (Fig. 8B and C). The results of the western blotting indicated that due to the interference effect of the SLC7A11-i and sorafenib loaded in MNMG, the function of glutamate-cystine antiporter was inhibited, leading to the deficiency of GSH. However, reduced glutathione directly leads to the inactivation of glutathione peroxidase 4 (GPX4), which further increases the sensitivity of HCC cells to oxidative stress and weakens the antioxidant capacity driven by the PERK-NRF2 signal pathway [98,99,100]. The expression of Heat Shock Protein (HSPA5) was decreased during iron death due to the unfolded protein reaction of the protective mechanism was not activated. The inactivation of GPX4 and the Fenton reaction triggered by excess free iron simultaneously promote the lipid peroxidation of polyunsaturated fatty acids (Fig. 8D and E).
To further characterize whether the MNMG induces ferroptosis, Huh7 cells were co-treated with several ferroptosis inhibitors in the presence of the MNMG. The outcomes showed that ferrostatin-1 (ferroptosis inhibitor), DFO (iron chelator), and GSH (glutathione) significantly attenuated the damaging effect of the MNMG on Huh7 cells and the recovery of GSH was the strongest (Fig. 8F). Therefore, we further investigated the effect of MNMG on GSH in Huh7 cells. The mBCL fluorescent probe was used to determine the cellular level of GSH, and it was observed that the fluorescence intensity decreased with the treatment of MNMG and recovered after the addition of GSH (Fig. 8G). Similarly, the results of Calcein-AM/PI staining demonstrated that the addition of GSH increased the number of live cells and decreased the number of dead cells compared with the drug-added group (Fig. 8H). And the results of western blotting also confirmed that GSH restored the expression of Heat Shock Protein Family A (HSP70), LDHA, NRF2, SIRT6, NDUFV2, and Tfam ferroptosis-related genes (Fig. 8I). Collectively, these results concluded that MNMG treatment can hinder GSH production, causing unrestricted lipid peroxidation. Importantly, the ferroptosis inhibitors like ferrostatin-1, iron chelators, and glutathione could attenuate the cell death effect caused by the MNMG.

MNMG causes increased lipid peroxide levels and glutathione deficiency. (A) Huh7 cells were treated with the six groups of nanomedicine for 8 h, and the changes of intracellular total lipid peroxides were observed by Bodipy staining. Nuclear localization was performed using Hoechst. Below is the statistical plot of Bodipy staining that produces red and green fluorescence. Scale bar: 30 μm. (B, C) Flow cytometry was performed to measure intracellular lipid peroxide by the Bodipy probe. The corresponding quantitative histograms are shown on the right. (D, E) Protein levels of PERK, NRF2, HSPA5, and GPX4 were assayed by western blotting in Huh7 cells under exposure to the nanomedicines for 24 h. GAPDH was used as an internal control. (F) Huh7 cells were incubated with the MNMG for 24 h in the presence of 3 ferroptosis inhibitors. (G) mBCL staining of Huh7 cells pretreated with the MNMG for 8 h. Fluorescence quantitative statistics of mBCL staining results on the right. Scale bar: 25 μm. (H) Calcein-AM/PI staining was performed on HCC cells after treatment with the MNMG for 24 h in the presence or absence of GSH. Fluorescence quantitative statistics of Calcin-AM/PI staining results on the left. Scale bar: 50 μm. (I) The levels of related proteins in Huh7 cells treated with MNMG for 24 h in the presence or absence of GSH were determined by western blotting. GAPDH was used as an internal control. Values represented mean ± SD. *P < 0.05, **P < 0.01 compared with control group
The MNMG treatment inhibits xenograft tumors in vivo growth
To verify the efficacy, safety, and targeting ability of this multifunctional nanomedicine in vivo, Huh7 tumor-bearing nude mice were randomly divided into two groups and under the treatment of MNMG for 28 days (Fig. 9A). To determine the ability of MNMG aggregation at the tumor site, Cy5.5 labeled MNMG (MNMG@ Cy5.5) was injected into nude mice and through the tail vein, and animal fluorescence imaging was monitored at different time points. This result showed that significant Cy5.5 fluorescence was detected at the tumor site and increased steadily over time (Fig. 9B). Fluorescence quantification histograms for Cy5.5 showed that MNMG reached a maximum accumulation after 24 h. This effect suggests that the MNMG can prolong the circulatory half-life of drug release and enhance the passive aggregation of the MNMG in tumor tissues through the EPR effect. We wondered whether MNMG exerts excellent anticancer therapeutic potential in xenograft tumors by targeting the multiple mechanisms of ferroptosis. Compared with the control group, the tumors of the MNMG administration group had an obvious antitumor effect (Fig. 9C and D). Through monitoring the tumor volume, we found that the tumor volume in the MNMG group shrank obviously, with almost completely inhibited tumor growth (Fig. 9E and F). Western blotting analysis showed that the expression levels of Glutamate Dehydrogenase 1 (GLUD1), PERK, NRF2, Tfam, and SLC7A11 and other ferroptosis-related genes in tumor tissues of the MNMG administration group were significantly inhibited, indicating that the complex can also activate ferroptosis in vivo, which was also consistent with the in vitro experimental outcomes (Fig. 9G). The above findings support our hypothesis that MNMG can exhibit positive antitumor efficacy in vivo by inducing the activation of the ferroptosis pathway and targeting HCC tissues.
In addition, the weight of tumor-bearing nude mice was measured during the administration period, and the effect of MNMG administration on weight reduction was not obvious, showing high safety (Fig. 9H). Meanwhile, the blood biochemical indexes of the two groups also showed that MNMG had minimal side effects on the tumor-bearing mice (Fig. 9I). Importantly, the results of H&E staining of heart, liver, spleen, and kidney tissues showed that there was almost no difference between the vehicle group and the MNMG group, indicating that the MNMG drug showed minimal toxic side effects in the main organs of the tumor-bearing mice (Fig. 9J). We also extracted tumor tissue for H&E staining, and the MNMG-administered group displayed an increased necrotic area of tumor tissue. Immunohistochemical results showed that the MNMG could decrease the expression of Ki67 and PCNA, thereby inhibiting the proliferation of tumor tissue (Fig. 9K).

Anticancer effect of MNMG in vivo. (A) Schematic diagram of the time node for in vivo treatment by intravenous injection of the MNMG. (B) In vivo fluorescence imaging of Huh7 tumor-bearing nude mice 2, 8, 24, and 48 h after intravenous injection of the MNMG. The red circle points to the location of the tumor in the nude mice. On the right is the square statistical chart of ROI counts. Color scale: 5000–50,000. (C, D) Visual comparison of tumors after 28 days of treatment. (E) Changes in tumor volume after treatment. (F) Changes in the mean tumor weight of each group after 28 days of treatment. (G) Western blotting analysis of key ferroptosis genes expression in two groups of Huh7 tumor-bearing mice. β-Actin and GAPDH were used as the internal control. (H) Changes in body weight of Huh7 tumor-bearing nude mice after treatment. (I) The blood biochemical data of the vehicle group and the MNMG group. (J) H&E staining of tumor tissue and key organ samples after different treatments. Scale bar: 50 μm. (K) Immunohistochemical staining of tumor tissue sections showing changes in the expression of Ki67 and PCNA. Scale bar: 25 μm. Values represented mean ± SD. *P < 0.05, **P < 0.01 compared with control group
Conclusion
Our findings provide valid experimental evidence that the nanomedicine delivery strategy MNMG has significant superiority for targeted and precision treatment in HCC. Based on the GDYO ultra-thin nanosheet with SP94-PEG linked and loaded with sorafenib, DOX-Fe2+, and SLC7A11-i, this multifunctional nanomedicine MNMG can be used to induce ferroptosis in HCC cells. When iron metabolism is abnormal in liver cancer cells, iron accumulation was occurring, which is suitable for targeted therapy by inducing ferroptosis. The induction of ferroptosis by MNMG can enhance the targeted aggregation of drugs at the tumor site and improve the treatment efficiency of chemotherapy. In conclusion, the current research data provide strong evidence that the MNMG can effectively increase drug enrichment at the site of HCC, improve the efficiency of antitumor therapy, and actively target the treatment of HCC. Notably, the MNMG has special advantages and good dispersibility, which solves the problem of toxic side effects caused by traditional chemotherapeutic drugs and the problem of water solubility of sorafenib. It is a promising nanomedicine that preferentially targets HCC cells through ferroptosis. Our study provides experimental evidence that the successfully synthesized MNMG could induce ferroptosis and enable a significant increase in the efficiency of antitumor therapy both in vitro and in vivo, which may be a promising therapeutic strategy.
Data availability
No datasets were generated or analysed during the current study.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
Wege H, Schulze K, von Felden J, Calderaro J, Reig M. Rare liver tumors working group of the European Reference Network on Hepatological D. Rare variants of primary liver cancer: Fibrolamellar, combined, and sarcomatoid hepatocellular carcinomas. Eur J Med Genet. 2021;64(11):104313.
Yaojie F, Luca M, Xin Wei W, Tim FG, Bin G. Alcohol-associated liver cancer. Hepatology. 2024(0).
Jonggi C, Chanyoung J, Young-Suk L. Tenofovir Versus Entecavir on recurrence of Hepatitis B Virus-Related Hepatocellular Carcinoma after Surgical Resection. Hepatology. 2020;73(2).
Xiu-Ping Z, Xiang-Jun C, Bo-Zhao L, Shuai X, Zhou-Liang W, Ming-Gen H et al. Active targeted Janus nanoparticles enable anti-angiogenic drug combining chemotherapy agent to prevent postoperative hepatocellular carcinoma recurrence. Biomaterials. 2022;281(0).
Abin Y, Manqing C, Zhigui G, Bingfeng Z, Junrong G, Hongyuan Z et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J Hematol Oncol. 2016;9(0).
Zeng ZM, Mo N, Zeng J, Ma FC, Jiang YF, Huang HS, et al. Advances in postoperative adjuvant therapy for primary liver cancer. World J Gastrointest Oncol. 2022;14(9):1604–21.
Zhang W, Zhang B, Chen XP. Adjuvant treatment strategy after curative resection for hepatocellular carcinoma. Front Med. 2021;15(2):155–69.
Rumgay H, Arnold M, Ferlay J, Lesi O, Cabasag CJ, Vignat J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77(6):1598–606.
Liu D, Song T. Changes in and challenges regarding the surgical treatment of hepatocellular carcinoma in China. Biosci Trends. 2021;15(3):142–7.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905.
Ding X, Sun W, Li W, Shen Y, Guo X, Teng Y, et al. Transarterial chemoembolization plus lenvatinib versus transarterial chemoembolization plus sorafenib as first-line treatment for hepatocellular carcinoma with portal vein tumor thrombus: a prospective randomized study. Cancer. 2021;127(20):3782–93.
da Fonseca LG, Marta GN, Braghiroli M, Chagas AL, Carrilho FJ, Hoff PM, et al. Safety and efficacy of cytotoxic chemotherapy in hepatocellular carcinoma after first-line treatment with sorafenib. BMC Cancer. 2018;18(1):1250.
Qiang-Qiang F, Huan T, Jiang-Xue C, Jun-Bo Z, Fei L, Jia-Xin Q et al. Research progress of sorafenib drug delivery system in the treatment of hepatocellular carcinoma: an update. Biomed Pharmacother. 2024;177(0).
Yim HJ, Suh SJ, Um SH. Current management of hepatocellular carcinoma: an eastern perspective. World J Gastroenterol. 2015;21(13):3826–42.
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5(1):87.
Hongxing Z, Yuhang L, Jie H, Jinling D, Qinliang M, Yao W et al. Potential targets and therapeutics for cancer stem cell-based therapy against drug resistance in hepatocellular carcinoma. Drug Resist Updat. 2024;74(0).
Hu W, Zhou C, Jing Q, Li Y, Yang J, Yang C, et al. FTH promotes the proliferation and renders the HCC cells specifically resist to ferroptosis by maintaining iron homeostasis. Cancer Cell Int. 2021;21(1):709.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Hannah KCC, Chia-Chou W, Yi-Chen L, Sheng-Hong C. Emergence of large-scale cell death through ferroptotic trigger waves. Nature. 2024;631:8021.
Scott JD, James AO. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6).
Li Y, Xu B, Ren X, Wang L, Xu Y, Zhao Y, et al. Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell Mol Biol Lett. 2022;27(1):81.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–8.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–81.
Marcel SW, Christina M, Lars B-L, Jana KS, Sina CR, Artem S et al. STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell. 2024(0).
Li Y, Chen F, Chen J, Chan S, He Y, Liu W et al. Disulfiram/Copper induces Antitumor Activity against both Nasopharyngeal Cancer Cells and Cancer-Associated fibroblasts through ROS/MAPK and ferroptosis pathways. Cancers (Basel). 2020;12(1).
Yang L, Wang H, Yang X, Wu Q, An P, Jin X, et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther. 2020;5(1):138.
Jo A, Bae JH, Yoon YJ, Chung TH, Lee EW, Kim YH, et al. Plasma-activated medium induces ferroptosis by depleting FSP1 in human lung cancer cells. Cell Death Dis. 2022;13(3):212.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17(4):948–60.
Zhou ZX, Cui Q, Zhang YM, Yang JX, Xiang WJ, Tian N, et al. Withaferin A inhibits ferroptosis and protects against intracerebral hemorrhage. Neural Regen Res. 2023;18(6):1308–15.
Saidak Z, Giacobbi A-S, Louandre C, Sauzay C, Mammeri Y, Galmiche A. Mathematical modelling unveils the essential role of cellular phosphatases in the inhibition of RAF-MEK-ERK signalling by sorafenib in hepatocellular carcinoma cells. Cancer Lett. 2017;392:1–8.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–65.
Zhanwei Z, Huan L, Ruoxi Y, Ying Y, Jingwen D, Yongxiang D et al. Glutathione Depletion-Induced activation of Dimersomes for potentiating the ferroptosis and immunotherapy of Cold Tumor. Angew Chem Int Ed Engl. 2022;61(22).
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
Zhu Q, Yuan Y, Yan B, Zhou J, Zuo J, Bai L. A new biomimetic nanozyme of hemin/graphdiyne oxide with superior peroxidase-like activity for colorimetric bioassays. Analyst. 2021;146(23):7284–93.
Wang C, Yu P, Guo S, Mao L, Liu H, Li Y. Graphdiyne oxide as a platform for fluorescence sensing. Chem Commun (Camb). 2016;52(32):5629–32.
Dinh Phuc D, Viet QB, Minh Chien N, Sohyeon S, Van Dam D, Joosung K et al. Insight into Facile Ion Diffusion in Resistive switching medium toward low operating voltage memory. Nano Lett. 2024;24(26).
Weiqi L, Cong X, Tianyi X, Yanan J, Wenjie M, Ping Y et al. Giant Water Uptake enabled Ultrahigh Proton Conductivity of Graphdiyne Oxide. Angew Chem Int Ed Engl. 2022;62(4).
Tao W, Mingsheng L, Li Y, Wenlong Y, Yuliang L. Controlled growth Lateral/Vertical Heterostructure Interface for Lithium Storage. Adv Mater. 2024(0).
Jian Y, Pengfei Y, Zongwei C, Wei L, Zhaoxi L, Zijian M et al. Interfacial Bonding Induced Charge transfer in two-dimensional amorphous MoO(3-x)/Graphdiyne oxide Non-van Der Waals Heterostructures for Dominant SERS Enhancement. Chemistry. 2024;30(29).
Qianli H, Xue L, Weiwei C, Lin C, Xuefei M. A novel functionalized graphdiyne oxide membrane for efficient removal and rapid detection of mercury in water. J Hazard Mater. 2024;467(0).
Ma W, Xue Y, Guo S, Jiang Y, Wu F, Yu P, et al. Graphdiyne oxide: a new carbon nanozyme. Chem Commun (Camb). 2020;56(38):5115–8.
Yu L, Jiawen W, Yeyu W, Rongxian M, Yuyi Z, Jinyue S et al. An all-graphdiyne electrochemiluminescence biosensor for the ultrasensitive detection of microRNA-21 based on target recycling with DNA cascade reaction for signal amplification. Analyst. 2023;148(6).
Qiwei W, Ying L, Hui W, Penglei J, Wenchang Q, Min Y et al. Graphdiyne oxide nanosheets display selective anti-leukemia efficacy against DNMT3A-mutant AML cells. Nat Commun. 2022;13(1).
Jiang W, Zhang Z, Wang Q, Dou J, Zhao Y, Ma Y, et al. Tumor reoxygenation and blood perfusion enhanced photodynamic therapy using ultrathin Graphdiyne Oxide nanosheets. Nano Lett. 2019;19(6):4060–7.
Xing E, Du Y, Yin J, Chen M, Zhu M, Wen X, et al. Multi-functional Nanodrug based on a three-dimensional Framework for targeted photo-chemo Synergetic Cancer Therapy. Adv Healthc Mater. 2021;10(8):e2001874.
Jiang B, Zhang R, Zhang J, Hou Y, Chen X, Zhou M, et al. GRP78-targeted ferritin nanocaged ultra-high dose of doxorubicin for hepatocellular carcinoma therapy. Theranostics. 2019;9(8):2167–82.
Jian Z, Xiuxiu W, Liang C, Jiandong Y, Zhiyuan Z. SP94 peptide mediating highly specific and efficacious delivery of polymersomal doxorubicin hydrochloride to hepatocellular carcinoma in vivo. Colloids Surf B Biointerfaces. 2020;197(0).
Lei H, Zhaozhao L, Danjie S, Haichen D, Kuo Z, Wangqian Z, et al. Tumor Microenvironment-Responsive Nanocapsule Delivery CRISPR/Cas9 to Reprogram the Immunosuppressive Microenvironment in Hepatoma Carcinoma. Adv Sci (Weinh). 2024;11:26.
Shi Y, van der Meel R, Chen X, Lammers T. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–4.
Mukaddes I, Christy M, Bella BM, Stefaan JS. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem Rev. 2021;121(3).
Jie Kai T, Li Xian Y, Eveline Sheau T, Supawan S, Arun P, Pu Chun K et al. Nanoparticles’ interactions with vasculature in diseases. Chem Soc Rev. 2019;48(21).
Zhang Y, Gu Z, Yun S, Luo K, Bi J, Jiao Y, et al. Green synthesis of DOX-loaded hollow MIL-100 (fe) nanoparticles for anticancer treatment by targeting mitochondria. Nanotechnology. 2022;33:34.
Wei T, Xiaojun X, Peilong C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed Pharmacother. 2020;121:109139.
Xuan W, Yuanyuan Z, Yan H, Yang F, Youbo Z, Chencheng X, et al. Activatable Biomineralized Nanoplatform remodels the intracellular environment of Multidrug-Resistant Tumors for Enhanced Ferroptosis/Apoptosis Therapy. Small. 2021;17:47.
Munnier E, Cohen-Jonathan S, Linassier C, Douziech-Eyrolles L, Marchais H, Souce M, et al. Novel method of doxorubicin-SPION reversible association for magnetic drug targeting. Int J Pharm. 2008;363(1–2):170–6.
Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5(9).
Chen X, Meng L, Yang Z, Jun Z, Yan H, Kai C, et al. Tumor microenvironment-activatable Fe-doxorubicin preloaded amorphous CaCO3 nanoformulation triggers ferroptosis in target tumor cells. Sci Adv. 2020;eaax1346:1.
Chen L, Li X, Liu L, YU B, XUE Y, LIU Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep. 2015;33.
Hideaki O, Kazuaki T, Mariko S, Takafumi K, Hiroshi Y, Reiko W et al. Targeting the vulnerability of glutathione metabolism in ARID1A-Deficient cancers. Cancer Cell. 2019;35(2).
Joanne LP, Justin CD, Dimitrios K, Gabriel K, Philip CB, Susan ML et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc(). Nat Commun. 2021;12(1).
Ma W, Xue Y, Guo S, Jiang Y, Wu F, Yu P, et al. Graphdiyne oxide: a new carbon nanozyme. Chem Commun. 2020;56(38):5115–8.
Zhang Y, Liu W, Li Y, Yang YW, Dong A, Li Y. 2D Graphdiyne Oxide serves as a Superior New Generation of Antibacterial agents. iScience. 2019;19:662–75.
Bai Q, Zhang C, Li L, Zhu Z, Wang L, Jiang F, et al. Subsequent monitoring of ferric ion and ascorbic acid using graphdiyne quantum dots-based optical sensors. Mikrochim Acta. 2020;187(12):657.
Hetong Q, Yuxi T, Mengyue Z, Xuemei W, Ling Y. Boron-doped and Ketonic Carbonyl Group-Enriched Graphdiyne as a dual-site Carbon Nanozyme with enhanced peroxidase-like activity. Anal Chem. 2022;94(49).
Xihong G, Huan H, Rongli C, Dongmei W, Jiali L, Dan W et al. Graphdiyne Oxide Quantum dots: the enhancement of peroxidase-like activity and their applications in sensing H(2)O(2) and cysteine. ACS Appl Bio Mater. 2022;5(7).
Qiqi Z, Yonghua Y, Bin Y, Jing Z, Jianli Z, Lijuan B. A new biomimetic nanozyme of hemin/graphdiyne oxide with superior peroxidase-like activity for colorimetric bioassays. Analyst. 2021;146(23).
Wenjie M, Yifei X, Shuyue G, Yanan J, Fei W, Ping Y et al. Graphdiyne oxide: a new carbon nanozyme. Chem Commun (Camb). 2020;56(38).
Zhu Z, Bai Q, Li S, Li S, Liu M, Du F, et al. Antibacterial activity of Graphdiyne and Graphdiyne Oxide. Small. 2020;16(34):e2001440.
Lizhen W, Yang Z, Linhao L, Xuezheng G, Dandan D, Lu Y et al. Graphdiyne oxide elicits a minor foreign-body response and generates quantum dots due to fast degradation. J Hazard Mater. 2022;445(0).
Kaisong Y, Miguel Ángel L, Beatriz J-S, Alberto E. Janus Micromotors Coated with 2D nanomaterials as dynamic interfaces for (Bio)-Sensing. ACS Appl Mater Interfaces. 2020;12(41).
Hetong Q, Ping Y, Yuexiang W, Guangchao H, Huibiao L, Yuanping Y et al. Graphdiyne oxides as excellent substrate for electroless deposition of pd clusters with high catalytic activity. J Am Chem Soc. 2015;137(16).
Shuyue G, Hailong Y, Fei W, Lijun Z, Ping Y, Huibiao L et al. Graphdiyne as Electrode Material: tuning Electronic State and Surface Chemistry for Improved Electrode Reactivity. Anal Chem. 2017;89(23).
Ma K, Li W, Zhu G, Chi H, Yin Y, Li Y, et al. PEGylated DOX-coated nano graphene oxide as pH-responsive multifunctional nanocarrier for targeted drug delivery. J Drug Target. 2021;29(8):884–91.
Wang X, Zhao Y, Hu Y, Fei Y, Zhao Y, Xue C, et al. Activatable Biomineralized Nanoplatform remodels the intracellular environment of Multidrug-Resistant Tumors for Enhanced Ferroptosis/Apoptosis Therapy. Small. 2021;17(47):e2102269.
Abdelgalil AA, Alkahtani HM, Al-Jenoobi FI. Sorafenib Profiles Drug Subst Excip Relat Methodol. 2019;44:239–66.
Kong R, Wang N, Han W, Bao W, Lu J. IFNgamma-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J Leukoc Biol. 2021;110(2):301–14.
Reem A, Jos vdV, Nicholas H, Zhihua P, Maximilian M, Cuixia E et al. Glutaredoxin attenuates glutathione levels via deglutathionylation of Otub1 and subsequent destabilization of system x(C)(). Sci Adv. 2023;9(37).
Zhang VX, Sze KM, Chan LK, Ho DW, Tsui YM, Chiu YT, et al. Antioxidant supplements promote tumor formation and growth and confer drug resistance in hepatocellular carcinoma by reducing intracellular ROS and induction of TMBIM1. Cell Biosci. 2021;11(1):217.
He Q, Liu M, Huang W, Chen X, Zhang B, Zhang T, et al. IL-1beta-Induced Elevation of Solute Carrier Family 7 Member 11 promotes Hepatocellular Carcinoma Metastasis through Up-regulating programmed death Ligand 1 and colony-stimulating factor 1. Hepatology. 2021;74(6):3174–93.
Guo M, Zhao L, Liu J, Wang X, Yao H, Chang X, et al. The underlying function and Structural Organization of the Intracellular Protein Corona on Graphdiyne Oxide Nanosheet for local Immunomodulation. Nano Lett. 2021;21(14):6005–13.
Guo X, Huang H, Cui R, Wang D, Liu J, Wang D, et al. Graphdiyne Oxide Quantum dots: the enhancement of peroxidase-like activity and their applications in sensing H(2)O(2) and cysteine. ACS Appl Bio Mater. 2022;5(7):3418–27.
Yan H, Guo S, Wu F, Yu P, Liu H, Li Y, et al. Carbon Atom hybridization matters: Ultrafast Humidity response of Graphdiyne Oxides. Angew Chem Int Ed Engl. 2018;57(15):3922–6.
Chen F, Fang Y, Chen X, Deng R, Zhang Y, Shao J. Recent advances of sorafenib nanoformulations for cancer therapy: Smart nanosystem and combination therapy. Asian J Pharm Sci. 2021;16(3):318–36.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Baiyu Q, Fereshteh Z, Carla TB, Eduard R, Rajesh Kumar S, Wei G et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell. 2024;187(5).
Tao X, Yingchao C, Qiang P, Shuai L, Saran L, Mingle L et al. Lipid droplet targeting type I photosensitizer for ferroptosis via lipid peroxidation Accumulation. Adv Mater. 2023;36(4).
Du J, Wang X, Li Y, Ren X, Zhou Y, Hu W, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 2021;12(7):705.
Li Y, Wang X, Huang Z, Zhou Y, Xia J, Hu W, et al. CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 2021;12(9):839.
Erdem MT, Vladislav OS, Samantha WA, Gabrielle CW, Richard P. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7(22).
Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. 2020;99(1).
Nathan PW, Sang Jun Y, Tyce F, Amanda MS, Maddison AO, Juliana M et al. Mitochondrial respiratory function is preserved under cysteine starvation via glutathione catabolism in NSCLC. Nat Commun. 2024;15(1).
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Mengyao C, Xiaohan T, Yanting S, Chunyan D, Chen L, Chunhui W et al. A ferroptosis amplifier based on triple-enhanced lipid peroxides accumulation strategy for effective pancreatic cancer therapy. Biomaterials. 2024;309(0).
Suleixin Y, Yi W, Wenzhao Z, Ruie C, Meilin W, Meiwan C. GSH/pH dual activatable cross-linked and fluorinated PEI for Cancer Gene Therapy through Endogenous Iron De-hijacking and in situ ROS amplification. Adv Mater. 2023;36(2).
Jiawei Z, Xiaorui W, Yan S, Jinjun S, Xuejiao S, Wenjun W et al. Multifunctional nanolocks with GSH as the key for synergistic ferroptosis and anti-chemotherapeutic resistance. Biomaterials. 2022;288(0).
Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17(11):2703–17.
Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, et al. The unfolded protein response Mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING Signaling. Immunity. 2020;52(4):668–82. e7.
Yilei Z, Robert VS, Litong N, Xiaoguang L, Chao W, Hyemin L et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1).
Acknowledgements
Not applicable.
Funding
This research was supported by National Natural Science Foundation of China (No. 82102938); Zhejiang Provincial Natural Science Foundation (No. LQ21H200005, LR21H090001); Zhejiang Provincial Health Bureau (No. 2023RC126); Zhejiang Provincial Administration of Traditional Chinese Medicine (No. 2023ZF001); major projects of Hangzhou health science and technology (No. Z20220024).
Author information
Authors and Affiliations
Contributions
Luyang Wang, Le Tong and Zecheng Xiong: Experiment, Methodology, Writing - Original Draft. Yi Chen, Ping Zhang, Yan Gao and Jing Liu: Methodology, Investigation. Lei Yang, Chunqi Huang and Gaoqi Ye: Validation. Jing Du: Writing - Review & Editing, Funding acquisition. Huibiao Liu: Data Curation, Supervision. Wei Yang: Conceptualization, Writing - Review & Editing, Funding acquisition. Ying Wang: Supervision, Project administration, Funding acquisition. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Tong, L., Xiong, Z. et al. Ferroptosis-inducing nanomedicine and targeted short peptide for synergistic treatment of hepatocellular carcinoma. J Nanobiotechnol 22, 533 (2024). https://doi.org/10.1186/s12951-024-02808-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-024-02808-7