
Abstract
Background
Asthma, a prevalent chronic inflammatory disorder, is shaped by a multifaceted interplay between genetic susceptibilities and environmental exposures. Despite strides in deciphering its pathophysiological landscape, the intricate molecular underpinnings of asthma remain elusive. The focus has increasingly shifted toward the metabolic aberrations accompanying asthma, particularly within the domain of pyrimidine metabolism (PyM)—a critical pathway in nucleotide synthesis and degradation. While the therapeutic relevance of PyM has been recognized across various diseases, its specific contributions to asthma pathology are yet underexplored. This study employs sophisticated bioinformatics approaches to delineate and confirm the involvement of PyM genes (PyMGs) in asthma, aiming to bridge this significant gap in knowledge.
Methods
Employing cutting-edge bioinformatics techniques, this research aimed to elucidate the role of PyMGs in asthma. We conducted a detailed examination of 31 PyMGs to assess their differential expression. Through Gene Set Enrichment Analysis (GSEA) and Gene Set Variation Analysis (GSVA), we explored the biological functions and pathways linked to these genes. We utilized Lasso regression and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) to pinpoint critical hub genes and to ascertain the diagnostic accuracy of eight PyMGs in distinguishing asthma, complemented by an extensive correlation study with the clinical features of the disease. Validation of the gene expressions was performed using datasets GSE76262 and GSE147878.
Results
Our analyses revealed that eleven PyMGs—DHODH, UMPS, NME7, NME1, POLR2B, POLR3B, POLR1C, POLE, ENPP3, RRM2B, TK2—are significantly associated with asthma. These genes play crucial roles in essential biological processes such as RNA splicing, anatomical structure maintenance, and metabolic processes involving purine compounds.
Conclusions
This investigation identifies eleven PyMGs at the core of asthma's pathogenesis, establishing them as potential biomarkers for this disease. Our findings enhance the understanding of asthma’s molecular mechanisms and open new avenues for improving diagnostics, monitoring, and progression evaluation. By providing new insights into non-cancerous pathologies, our work introduces a novel perspective and sets the stage for further studies in this field.
Similar content being viewed by others

Introduction
Asthma, a chronic and complex disease, affects over 300 million individuals globally, with projections suggesting an increase to 400 million by 2025 [1]. Characterized by pervasive airway inflammation and intricate cellular dynamics, asthma presents significant global health challenges, as indicated by the Global Asthma Network (GAN) with prevalence rates of 10.5% among children and 4.4% among adults [2]. The disease manifests through symptoms such as airway hyperresponsiveness, eosinophilic infiltration, reversible airflow obstruction, airway remodeling, mucus hypersecretion, and goblet cell hyperplasia [3]. Despite extensive research, the underlying mechanisms of asthma remain poorly understood, a consequence of its multifactorial nature involving genetic, environmental, infectious, immunological, and dietary components. Current therapeutic approaches primarily target symptom control, often leaving a considerable number of patients undermanaged [4]. This gap underscores the critical need for an in-depth investigation into the genetic and molecular bases of asthma, which could revolutionize our understanding of its onset and progression [5]. Advancements in this field promise to shift from merely managing symptoms to altering the disease trajectory, potentially transforming asthma treatment and significantly enhancing patient care worldwide [6].
PyM plays a pivotal role in the synthesis, degradation, and recycling of critical pyrimidine nucleobases, such as cytosine and uracil, which are crucial to nucleic acid structures. Beyond its foundational role in nucleic acid synthesis, PyM is intricately linked to broader aspects of energy metabolism through its biosynthetic and catabolic pathways, encompassing both de novo and salvage routes [7]. These pathways are particularly vital in rapidly dividing cells [8]. Disruptions in PyM pathways can lead to a range of inherited metabolic and autoimmune inflammatory disorders, including asthma. Recent studies highlight the role of microRNAs, notably miR-146a and miR-155, in promoting asthma by downregulating genes that typically suppress cell proliferation [9]. Furthermore, modulation of these microRNAs through TSHR-mediated pathways reveals insights into the fibroproliferative characteristics of asthma. Research by Madera-Salcedo et al. has shown that inflammation-induced hypermethylation of PPP2R2B (B55ß) leads to resistance to apoptosis in the absence of cytokines [10]. Concurrently, Zhu et al. demonstrated how UBE2T exacerbates the progression of hepatocellular carcinoma by influencing PyM [11]. This research aims to dissect the role of PyMGs in asthma immunotherapy, seeking to uncover new therapeutic opportunities by investigating purinosome formation and the glutamine pyrimidine metabolism pathway [12]. Despite significant progress, the impact of PyM on the immunogenic landscape and its critical role in enhancing the efficacy of immunotherapies for asthma remain poorly understood. This study is committed to a comprehensive assessment of PyMGs and their integration with immunotherapeutic strategies in asthma, potentially leading to transformative clinical advancements.
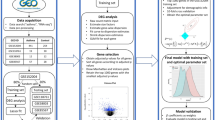
The Asthma Initiative heralds a transformative leap in biomedical research, integrating comprehensive transcriptomic sequencing with detailed clinical annotations to elucidate the transcriptional and molecular intricacies of asthma [13,14,15]. This project employs cutting-edge bioinformatics to navigate vast datasets, aiming to unveil the complex pathophysiological foundations of the disease. Notably, the role of PyMGs in the asthma schema remains underexplored. Our study addresses this oversight by utilizing asthma-specific datasets from the GEO to investigate the impact and relevance of PyMGs on asthma pathogenesis, as illustrated in Fig. 1. Through this focused analysis, we aim to enhance our understanding of asthma’s molecular basis and pave the way for novel therapeutic strategies.

Framework
Materials and methods
The methodologies proposed by Zi-Xuan Wu et al. in 2023 were employed in this study [16].
Raw data
GEO was searched for mRNA expression. Series: GSE76262 and GSE147878. Platform: GPL13158 and GPL6480. Strategy for searching (‘Parkinson’ [MeSH] mRNA [All Fields] and normal) AND (‘Homo sapiens’ [Organism] AND ‘Non-coding RNA profiling by array’ [Filter]). Specifically, this investigation harnessed the datasets GSE76262 and GSE147878, underpinned by the GPL13158 and GPL6480 platform. GSE76262 functioned as the training cohort, while GSE147878 constituted the testing group (Table 1). We also identified PyMGs from the MSigDB (Table S1).
Analysis of differentially expressed genes (DEGs)
Our methodology for extracting precise mRNA profiles involved the utilization of Perl scripts to meticulously match and sort transcriptional data from the GSE76262 dataset. Following normalization procedures, we applied stringent criteria for identifying differential expression among PyMGs: FDR < 0.05 and |log2FC|≥ 1. This rigorous approach enabled the isolation of significantly altered PyMGs for further scrutiny. To elucidate the intricate relationships among these genes, Pearson's correlation coefficient was harnessed, leveraging the corrplot package in R for comprehensive correlation analysis. This step was pivotal in highlighting genes with statistically significant associations within the identified modules.
GO and KEGG analysis
To elucidate the biological implications and pathway involvements of the differentially expressed genes (DEGs), we conducted comprehensive Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. Employing the R programming language, we investigated the impact of differentially expressed PyMGs on biological processes (BP), molecular functions (MF), and cellular components (CC). This analysis aimed to delineate the overarching biological themes and molecular pathways influenced by these genes, thereby enhancing our understanding of their roles in disease pathology and identifying potential therapeutic targets. Through this multi-faceted approach, we not only categorized the DEGs but also shed light on the intricate interplay between purine metabolism and its broader biological and clinical significance.
Model construction and analysis of immune cell infiltration
In our pursuit of a predictive model characterized by unparalleled precision and reliability, we utilized the glmnet package for Lasso regression analysis, complemented by cross-validation, to refine and enhance our model. This methodology effectively mitigated overfitting, thereby boosting the model's predictive capability for complex biological datasets. For further validation, we employed the sophisticated SVM-RFE algorithm using the e1071 package, meticulously constructing a machine learning model. Cross-validation was pivotal in assessing the model's error rates and accuracy, ensuring its robustness and dependability. The Random Forest algorithm, renowned for its efficacy in ensemble learning, was integral to our analysis. By generating multiple decision trees and aggregating their predictions, it minimized overfitting risks and enhanced model generalization. The algorithm's distinctive feature—random feature selection and bootstrap sampling—fostered diversity among decision trees, thus improving the model's overall accuracy. Utilizing the randomForest and ggplot2 packages, we concentrated on analyzing differentially expressed genes, identifying key genes crucial for disease classification. In the final phase, we ranked the significance of these feature genes using an integrated approach that synthesized insights from Lasso regression, Random Forest, and SVM models. This provided a nuanced understanding of their roles in disease pathology. Furthermore, the CIBERSORT algorithm enabled us to analyze the immune cell composition, offering deeper insights into the immune landscape associated with the disease. This comprehensive and rigorous analytical approach not only augmented the accuracy of disease classification but also unveiled new avenues for understanding the molecular basis of the disease.
GSEA and GSVA
To elucidate the functional dynamics and pathway alterations across a spectrum of samples, we employed Gene Set Enrichment Analysis (GSEA) and Gene Set Variation Analysis (GSVA). These powerful methodologies enabled the identification of functionally related gene sets and pathway changes, utilizing quantitative scores and visual representations to highlight active biological processes and pathways within various risk stratifications. Using R, we thoroughly investigated the impact of differentially expressed PyMGs on BP, MF, CC, and pathways, providing a granular understanding of their roles in disease mechanisms. This approach allowed us to uncover the intricate biological themes and molecular pathways influenced by these genes, thereby enhancing our comprehension of their involvement in disease pathology.
Drug-gene interactions in asthma
As bioinformatics in asthma research progresses towards pinpointing viable biomarkers, the construction of biological models and the identification of effective markers for disease diagnostics have become increasingly critical. Grasping the clinical relevance of these biomarkers is essential for crafting targeted therapeutic approaches. Anticipating drug reactions based on these pivotal markers is fundamental for the development of future preventative measures and therapeutic regimens for asthma. In this vein, validated biomarkers stand as crucial reference points. Thus, precise prediction of drug-gene interactions holds paramount significance. In this study, the Drug-Gene Interaction database (DGIdb) (https://dgidb.genome.wustl.edu/) was utilized to forecast interactions between the identified hub genes and prospective therapeutic agents, highlighting the potential for targeted intervention strategies in asthma management.
Investigation of shared miRNAs and lncRNAs in asthma
The regulatory landscape of genetics is profoundly influenced by non-coding RNA transcripts, particularly microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). miRNAs modulate gene expression by enhancing or repressing mRNA degradation and translation, while lncRNAs, consisting of over 200 nucleotides, orchestrate a multitude of cellular processes through mechanisms such as chromosomal modifications, transcriptional activation, and interference. Recent studies have illuminated the extensive interplay between miRNAs and lncRNAs, fostering a competitive binding scenario among miRNAs, lncRNAs, and other regulatory entities. This interaction has led to the concept of competitive endogenous RNAs (ceRNAs), where lncRNAs can regulate gene expression by sequestering miRNAs. In light of these findings, our investigation aims to determine whether specific miRNAs and lncRNAs share regulatory mechanisms and developmental pathways in Asthma, potentially unveiling novel avenues for understanding and treating this complex condition.
Establishment of a common mRNA-miRNA-lncRNA network in asthma
To elucidate the interactive landscape among mRNA, miRNA, and lncRNA entities in Asthma, we sourced target gene information from miRTarBase and PrognoScan, databases renowned for providing empirically validated miRNA-lncRNA-target interactions. By intersecting the target genes of common mRNA-miRNA-lncRNA interactions with Asthma-associated genes, we established a regulated network. This network was visualized using Cytoscape software, providing a graphical representation of the molecular interplay critical to Asthma pathophysiology.
Mendelian randomization analysis
To ensure the independence of exposure and outcome variables in our genome-wide association study (GWAS) summary data, we engaged in an association analysis via the TwoSampleMR package in R. Designating NME7 and POLR2B-related expression as the exposure and Asthma as the outcome, we aimed to explore potential causal relationships. The analysis entailed: 1. Instrumental Variables (IVs) Configuration: NME7 and POLR2B-related expressions were screened with a P-value threshold of < 5 × 10^-8 to identify strongly associated exposures. 2. Independence Configuration: Linkage disequilibrium (LD) between SNPs was calculated using the PLINK clustering method, excluding SNPs with LD coefficient r^2 > 0.001 and within 10,000 kb to ensure SNP independence and reduce pleiotropic biases. 3. Statistical Strength Configuration: The robustness of instrumental variables was assessed using the F-statistic (F = β^2/SE^2), with variables having F < 10 deemed inadequate to mitigate confounding effects.
Leveraging GWAS data, SNPs associated with the instrumental variables were identified, and through the “harmonise_data” function within TwoSampleMR, we aligned allelic directions of exposure and outcome, excluding incompatible SNPs. The inverse variance-weighted (IVW) method served as the cornerstone for causal inference, employing the variance of instrumental variables as weights to determine causal dynamics, thereby advancing our understanding of the genetic architecture underlying disease states.
Active components-targets docking
ADCY4 and PNPT1 were docked to verify the accuracy of principal components and prediction targets. The protein configurations of the core targets were obtained from the Uniprot database by using the minimum resolution (Resolution) and the source (Method) as X-ray as the screening condition, and the crystal structure of these protein configurations were obtained from the RCSB PDB database). 2D structures of 6 active components of core targets were obtained from PubChen database, and these 2D structures were minimized by chem3d software. The binding strength and activity of active components and targets were evaluated by SYBYL2.0 software, and the active components of binding TotalScore greater than 3 were selected for sub-docking. Then imported the crystal into the Pymol 2.4 for dehydration, hydrogenation, and separation of ligands; it then imported AutoDockTools 1.5.6 to construct the docking grid box for each target. Docking was completed by Vina 1.1.2 software, and the molecules with the lowest binding energy in the docking conformation were selected to observe the binding effect by matching with the original ligands and intermolecular interactions (such as hydrophobicity, cation-π, anion-π, π-π stacking, hydrogen bonding, etc.). Finally, the Pymol2.4 software was utilized to visualize the molecular docking.
Results
Identification of DEGs and principal component analysis
Among the 31 examined PyMGs, several exhibited significant differences in expression levels. Furthermore, gene clustering analysis revealed distinct clusters in the treatment and control groups. Notable PyMGs in the treatment group included DHODH, POLE, UCK2, ENTPD1, NT5E, UPB1, ENPP1, TXNRD1, while control group PyMGs comprised PNPT1, POLR3F, PRIM2, POLR2C, POLR2G, POLR1D, POLR2B (Fig. 2a). Correlation analysis was conducted among these PyMGs, and a correlation matrix was generated for visualization (Fig. 2b) (Table S2).

Principal Component Analysis. a Analysis of difference. b Analysis of correlation
Enrichment analysis of PyMGs
GO enrichment analysis identified 290 core target genes, encompassing BP, MF, and CC. The MF category primarily involved nucleoside binding (GO: 0001882), catalytic activity, acting on RNA (GO: 0140098), ribonucleoside binding (GO: 0032549). The CC category was mainly associated with transferase complex, transferring phosphorus-containing groups (GO: 0061695), nuclear chromosome (GO: 0000228), RNA polymerase complex (GO: 0030880). The BP category included RNA splicing (GO: 0008380), anatomical structure homeostasis (GO: 0060249), purine-containing compound metabolic process (GO: 0072521). KEGG enrichment analysis revealed that the upregulated genes were primarily involved in Pyrimidine metabolism (hsa00240), Huntington's disease (hsa05016), Purine metabolism (hsa00230) (Fig. 3 and Table S3a, b).

For PyMGs, GO, and KEGG analyses were performed. a The GO circle illustrates the scatter map of the selected gene’s logFC. b The KEGG barplot and bubble illustrates the scatter map of the logFC of the indicated gene
Model construction
In our study, we meticulously established a gene signature by employing LASSO and Cox regression analysis, judiciously selecting the optimal value, as depicted in Fig. 4a, b. To validate the precision and reliability of our model, we constructed a machine learning model using SVM-RFE. This model demonstrated exceptional accuracy, achieving a score of 0.863, and maintained a minimal error rate of 0.137, as shown in Fig. 4c, d. Some key genes were screened by random forest tree, including NME7, POLR3C, ENTPD4, ENPP1, UCKL1, etc. (Fig. 4e, f). The intersection of the 11 PyMGs identified by LASSO, RF and SVM revealed strong concordance (Fig. 4g). Upon evaluating the model in relation to the 11 hub genes, we observed notably high accuracy rates for each gene: DHODH (AUC = 0.680), UMPS (AUC = 0.823), NME7 (AUC = 0.776), NME1 (AUC = 0.767), POLR2B (AUC = 0.672), POLR3B (AUC = 0.775), POLR1C (AUC = 0.637), POLE (AUC = 0.699), ENPP3 (AUC = 0.679), RRM2B (AUC = 0.744), TK2 (AUC = 0.720) (Fig. 4f). Remarkably, an AUC of 0.934 (95% CI 0.887−0.973) was achieved in dataset GSE76262, underscoring the high accuracy and robustness of our prediction model (Fig. 4g) (Table 1 and S4). In evaluating the performance of our study, particularly with regard to the AUC, a detailed analysis of Fig. 4 demonstrates an impressive AUC value of 0.934, underscoring the high accuracy of our model. Addressing concerns about the lower AUC values observed for certain genes, it is essential to consider the influence of individual genetic variations on these outcomes. Despite these variations, it is important to note that the aggregated AUC values for these genes consistently approximate a significant benchmark of 0.7. This collective result substantiates the overall credibility, precision, and robustness of our predictive model, affirming its utility in both clinical and research contexts.

The development of the PyMGs signature. a Regression of the 11 Asthma-related genes using LASSO. b Cross-validation is used in the LASSO regression to fine-tune parameter selection. c, d Accuracy and error of this model. e, f: RF. g Venn. h AUC of 10 hub genes. i AUC of train group
Gene set enrichment analysis
In this study, the AUC of each gene in the test group, the Rank ranking of each gene, and the results of the test group validation were observed. We found that NME7 and POLR2B may be the most relevant genes. Through literature evaluation and analysis of hub gene sensitivity within the model, it was determined that NME7 and POLR2B may be the most relevant genes to Asthma. In terms of GO analysis, NME7 was found to be associated with CC ribosome, MF cytokine receptor activity, MF structural constituent of ribosome. On the other hand, POLR2B was primarily involved in the CC mitochondrial protein containing complex, CC organellar ribosome, CC ribosomal subunit (Fig. 5a). In KEGG analysis, NME7 was mainly associated with cytokine cytokine receptor interaction, neuroactive ligand receptor interaction, oxidative phosphorylation, while POLR2B was involved in neuroactive ligand receptor interaction, oxidative phosphorylation, parkinsons disease (Fig. 5b) (Table S5).

GSEA of Analysis in NME7 and POLR2B. a GO. b KEGG
Analysis of immune cells
In this investigation, we explore the nuanced role of the immune microenvironment in the onset and progression of asthma, a complex condition shaped by the dynamics of immune cell interactions. Our extensive analysis was designed to unravel the expression patterns and interrelations of these cells, utilizing violin plots to vividly delineate the differential expression profiles of immune cells in control versus asthma-impacted tissues. These graphical representations distinctly showcased an upregulation of activated mast cells, eosinophils, and both resting and activated dendritic cells in the control samples. Conversely, tissues afflicted by asthma exhibited heightened levels of CD8 T cells, follicular helper T cells, and both M0 and M2 macrophages, illuminating the altered or engaged immune response mechanisms in asthma. This endeavor aimed to elucidate the potential genetic interplays with, or influences by, the immune microenvironment in asthma, striving to enrich our comprehension of the disease's pathophysiological underpinnings. The results, showcased in Fig. 6a, reveal a complex symbiosis between immune cells and genetic factors, broadening our understanding of the intricate immune-genetic interplay in asthma. In addition, we added NME7 and POLR2B to the immune infiltration analysis of their respective genes alone.

Expression of Immune cells. a Expression of immune cells in different clusters. b NME7. c POLR2B
GSVA
In the GO analysis, NME7 was primarily associated with CC septin cytoskeleton, BP negative regulation of myoblast proliferation, BP axonemal central apparatus assembly, MF olfactory receptor binding, CC iga immunoglobulin complex. POLR2B was mainly involved in the MF udp xylosyltransferase activity, BP regulation of mirna catabolic process, BP positive regulation of mirna catabolic process, CC fancm mhf complex, MF glycine n acyltransferase activity (Fig. 7a). In terms of KEGG analysis, NME7 was mainly associated with glycosaminoglycan degradation, taurine and hypotaurine metabolism, renin angiotensin system, glycosphingolipid biosynthesis lacto and neolacto series, asthma, sulfur metabolism. POLR2B was involved in glycosaminoglycan biosynthesis heparan sulfate, linoleic acid metabolism, arachidonic acid metabolism, taste transduction, retinol metabolism, nitrogen metabolism (Fig. 7b).

GSVA of Analysis in NME7 and POLR2B. a GO. b KEGG
Drug-gene interactions
Some drugs were predicted to interact with the eleven hub genes, including leflunomide, teriflunomide, vidofludimus, chembl1164954, leucovorin (Table S6) (Fig. 8). The investigation unveiled eleven PyMGs—DHODH, UMPS, NME7, NME1, POLR2B, POLR3B, POLR1C, POLE, ENPP3, RRM2B, TK2—significantly associated with asthma. According to rank, NME7 and POLR2B were selected for molecular docking. To verify the credibility of our results. We performed molecular docking of NME7 and POLR2B with CHEMBL1164954, LEFLUNOMIDE, TERIFLUNOMIDE, ZALCITABINE (Table 2 and Fig. 9).

Drug-gene interactions. Red circles are up-regulated genes, green hexagons are down-regulated genes, and blue squares are associated drugs

Molecular docking results. a UMPS. b DHODH
Identification of common RNAs and construction of miRNA-lncRNA shared genes network
A total of 212 miRNAs and 241 lncRNAs associated with Asthma were identified from three databases (Table S7a, b). Table S7 shows the matching of these genes against the corresponding miRNA database. These databases include miRanda [17], miRDB [18], and TargetScan [19]. When the corresponding database matched the relevant miRNA, the score was marked as 1. It can be seen that when all three databases can be matched, it is 3 points. The miRNA was matched by spongeScan database [20] to obtain the corresponding lncRNA data. The miRNA-lncRNA-gene network was constructed by intersecting these non-coding RNAs with the shared genes obtained through Lasso regression and SVM-RFE. The network consisted of 190 lncRNAs, 182 miRNAs, and some common genes, including the 11 hub genes (NME7, TK2, UMPS, POLE, ENPP3, POLR1C, DHODH, RRM2B, POLR3B, NME1, POLR2B) (Fig. 10).

miRNAs-LncRNAs shared Genes Network. Red circles are mrnas, blue quadrangles are miRNAs, and green triangles are lncRNAs
Validation of hub genes
To enhance the confidence and prediction accuracy of the model, GSE147878 dataset was used for validation. The GSE147878 analysis further confirming their potential relevance to Asthma (Fig. 11).

eleven hub genes were validated
Model verification
The Boxplots depicted the residual expression patterns of these genes in Asthma (Fig. 12a). There are some differences in the proportions of the four different modes (Fig. 12b, c). The PyMGs' diagnostic capacity in distinguishing Asthma from control samples revealed a satisfactory diagnostic value, with an AUC of RF: 0.967; SVM: 0.919; XGB: 0.943; GLM: 0.933 (Fig. 12c). An AUC of 1.000 (95% CI 1.000–1.000) in GSE147878 (Fig. 12d).

Model verification. a Residual expression patterns. b, c Model expression patterns (d) AUC of model. e AUC of test group
Mendelian randomization analysis
In our exploration of the intrinsic connection between NME7 and POLR2B, and asthma forest plots were meticulously employed to visually articulate the associations. For NME7, the all SNPs (rs73078636, rs10178845, rs11071559, rs2460555, rs7734635, rs5743618, etc.) conspicuously positioned itself to the right of the confidence interval, indicating a positive association. (Fig. 13a). In the case of POLR2B, all SNPs (rs10178845, rs11071559, rs2460555, rs7734635, rs5743618, rs61957178, rs4795399, etc.) were all situated to the right of the confidence interva, suggesting a similar trend of association with asthmal (Fig. 13b). Further dissecting the heterogeneity inherent in our analysis, the funnel plot tailored to asthma revealed a deviation from the expected symmetrical distribution, albeit maintaining a general symmetry. This nuanced observation was further scrutinized through sensitivity analysis, employing a “leave-one-out” approach. Remarkably, the omission of any individual SNP from the analysis had a negligible effect on the results of the Inverse Variance Weighted (IVW) analysis, indicating that the remaining SNPs consistently mirrored the outcomes of the aggregate dataset. Substantiating the validity of our findings, the MR-Egger regression analysis was invoked, providing a solid foundation that bolsters both the robustness and authenticity of our results and the methodologies applied. This Mendelian randomization analysis unequivocally confirms the intimate association of NME7 and POLR2B with asthma. Hence, it delineates a potential pathway to modulate the incidence, evolution, and progression of asthma by intervening in the functions of NME7 and POLR2B, presenting a promising avenue for therapeutic intervention and a deeper understanding of the disease mechanism.

Mendelian Randomization Analysis. a NME7. b POLR2B
Discussions
Asthma, a chronic inflammatory disorder of the airways, presents a significant global health challenge, with the Global Asthma Network (GAN) study reporting prevalence rates of 10.5% in children and 4.4% in adults [21]. Characterized by airway inflammation, hyperresponsiveness, mucus hypersecretion, and remodeling, the pathophysiology of asthma is complex, resulting from a multifaceted interplay of genetic and environmental factors [22]. Despite the availability of treatments primarily aimed at symptomatic relief, a substantial proportion of patients remain inadequately managed, highlighting the urgent need for advanced therapeutic interventions [23]. In this context, the identification of asthma biomarkers through comprehensive bioinformatic analysis is a critical endeavor. This approach aims to unravel the complex mechanisms underlying asthma and uncover novel therapeutic targets, potentially shifting asthma management towards early detection and precise disease characterization [24]. PyM is essential for the synthesis, breakdown, and utilization of key pyrimidine nucleobases, such as cytosine and uracil, which are integral to nucleic acid structures and energy metabolism [8]. Dysregulation of PyM pathways is implicated in various inherited metabolic disorders, including autoimmune inflammatory diseases [9]. Recent studies have highlighted the role of microRNAs, such as miR-146a and miR-155, in modulating cell proliferation in asthma by targeting genes that inhibit cell growth, and the TSHR-mediated regulation of these microRNAs in asthma's fibroproliferative pathology [10]. Additionally, research has identified dysregulation in PPP2R2B (B55ß) through inflammation-driven hypermethylation, affecting apoptosis resistance, and the role of UBE2T in exacerbating hepatocellular carcinoma by influencing PyM [12]. This study focuses on PyMGs and their relevance in asthma immunotherapy, exploring new therapeutic avenues through the investigation of purinosome formation and the glutamine pyrimidine metabolism pathway [25]. Despite advancements, the impact of PyM on the immunogenic landscape and its role in modulating immunotherapy efficacy in asthma remains to be fully elucidated. Our research aims to conduct a thorough analysis of PyMGs and their interaction with immunotherapeutic strategies in asthma, paving the way for groundbreaking clinical progress.
In our comprehensive study, we identified a network of 31 DEGs intricately associated with PyMGs in asthma. Utilizing a rigorous analytical framework that combines DEG analysis, Lasso regression, and SVM-RFE, we identified eleven critical PyMGs: DHODH, UMPS, NME7, NME1, POLR2B, POLR3B, POLR1C, POLE, ENPP3, RRM2B, and TK2. These genes have demonstrated significant diagnostic relevance in asthma, a finding further validated by external datasets, underscoring their essential role in the disease's pathogenesis. However, our research also revealed a significant gap in understanding the interaction between these genes and specific transcription factors, especially within the context of purine metabolism. Notably, NME7 and POLR2B emerged as key mediators in the link between asthma and PyMGs. Further investigation into their biological functions indicated their involvement in various immune-related processes, including RNA splicing, anatomical structure homeostasis, and the metabolic processing of purine-containing compounds. This finding suggests that PyMGs may regulate a broad spectrum of biological pathways, particularly those related to immune responses, thereby potentially influencing the pathophysiological progression of asthma. Our results propose that these genes are crucial in understanding asthma’s progression and could unveil new avenues for therapeutic targeting, presenting promising opportunities for future research and the development of treatment strategies. This underscores the paramount importance of PyMGs within the molecular landscape of asthma, indicating a novel direction in the quest to understand and mitigate this complex disease.
The investigation into PyM, a cornerstone of cellular energy balance and proliferation, has revealed its extensive implications across various diseases and metabolic disorders. In the intricate landscape of asthma pathogenesis, recent research has highlighted the critical roles of NME7 and POLR2B in shaping the molecular framework of this disease. Asthma, characterized by chronic airway inflammation, hyperresponsiveness, and reversible airflow obstruction, arises from a complex interplay of genetic, environmental, and immunological factors [26]. Identifying NME7 and POLR2B as key players in this intricate narrative adds a new dimension to our understanding of asthma's origins and potential therapeutic avenues. NME7, associated with intracellular signaling and cellular differentiation pathways, has been significantly linked to asthma [27]. Its role in modulating signal transduction pathways underscores its impact on the inflammatory cascade and airway smooth muscle cell dynamics, which are integral to asthma's pathophysiology [28]. However, the precise mechanisms by which NME7 influences asthma require further elucidation. The gene's association with asthma suggests that any deviation in NME7's expression or functionality could exacerbate the inflammatory environment, leading to increased asthma symptoms and airway structural changes [29]. Similarly, POLR2B, a vital component of the RNA polymerase II complex essential for mRNA synthesis, has been identified as a gene of interest in asthma research. Its fundamental role in gene expression regulation positions POLR2B as a key mediator in asthma by controlling genes involved in immune responses and airway inflammation [30]. Changes in POLR2B expression or function could alter the transcriptional profile of asthma-related genes, influencing disease severity and the effectiveness of therapeutic interventions. Furthermore, analyses of the GSE147878 dataset have highlighted PyM-related markers as emerging prognostic indicators in asthma, representing a dynamic frontier in genomic exploration. These insights pave the way for innovative therapeutic strategies in asthma management, heralding the advent of personalized medicine in contemporary healthcare. This body of research not only deepens our understanding of asthma's molecular etiology but also underscores the potential of targeted genetic and molecular interventions in revolutionizing asthma treatment paradigms.
Asthma, a chronic disorder, is characterized by persistent airway inflammation and increased reactivity, resulting from a complex interplay between host immune mechanisms and environmental factors within the pulmonary environment. The pathogenesis of asthma is marked by an imbalance between the innate and adaptive immune systems in the airways [31]. Environmental triggers—such as allergens, pollutants, and respiratory pathogens—induce airway epithelial cells to initiate an immune response, characterized by the release of pro-inflammatory cytokines and chemokines. This response recruits various innate immune cells, including neutrophils, macrophages, dendritic cells, and innate lymphoid cells, leading to acute inflammation [32]. Simultaneously, dysregulation of adaptive immune responses, particularly within T-helper (Th) cell subsets, plays a crucial role in sustaining chronic inflammation and structural changes characteristic of asthma [33]. A shift towards Th2-dominated responses, marked by the secretion of interleukins (IL)-4, IL-5, and IL-13, promotes eosinophilic inflammation, airway hyperresponsiveness, and excessive mucus production [34]. Additionally, recent studies have highlighted the significant roles of aberrant Th17 and regulatory T cell (Treg) functions in modulating airway inflammation and remodeling, adding complexity to our understanding of asthma’s immunology. Our analysis, utilizing violin plot visualizations, has identified distinct immune cell expression profiles. The control group exhibited higher levels of activated mast cells, eosinophils, and both resting and activated dendritic cells. In contrast, the treatment group showed increased levels of CD8 T cells, follicular helper T cells, and both M0 and M2 macrophages. These findings provide deeper insights into the immune landscape of asthma. These observations underscore the importance of elucidating immune pathways to advance innovative therapeutic strategies. Immunomodulatory interventions, aimed at reducing inflammation and correcting immune dysregulation, present a promising avenue for novel asthma treatments. This research heralds a new era in targeted therapeutic interventions, poised to reshape the future of asthma therapy by emphasizing the critical role of immune pathways in understanding and managing the disease.
The merging of asthma research with metabolic studies marks a groundbreaking phase in medical science, energized by the adoption of advanced bioinformatics techniques. This shift has significantly broadened our understanding of the molecular complexity and diverse pathological expressions of asthma [35,36,37]. The scientific community's collaborative efforts have been crucial in deciphering the molecular foundations and clinical spectrum of this disease. Our research represents a pivotal advancement in this burgeoning field, highlighting the critical role of PyMGs within the asthma paradigm. Utilizing extensive datasets from the GEO, specifically GSE76262 and GSE147878, we have applied a comprehensive suite of analytical tools, including GO, KEGG, and GSEA. These tools have enabled the development of a sophisticated predictive model that illuminates the intricate role of PyMGs in asthma's etiology. Our work establishes a foundational theoretical framework while also opening new pathways for investigating the metabolic imbalances at the heart of asthma and developing targeted therapeutic approaches. However, the necessity for further empirical research to confirm the mechanisms proposed in our findings is paramount. Such validation, achievable through rigorous in vivo and in vitro testing, is essential to deepen our understanding of asthma. These exacting scientific endeavors are vital for progressing towards effective treatment solutions, shaping the future of asthma research and therapeutic development.
Conclusions
Within the complex landscape of asthma, PyMGs play a central role, orchestrating a broad array of biological interactions, pathways, signaling cascades, and regulatory mechanisms. At the core of asthma's molecular framework, PyMGs underlie the synthesis of key biomolecules such as DHODH, UMPS, NME7, NME1, POLR2B, POLR3B, POLR1C, POLE, ENPP3, RRM2B, and TK2, which are crucial in essential physiological processes and metabolic regulation related to asthma. Notably, NME7 and POLR2B are distinguished by their significant regulatory effects on metabolic pathways, profoundly influencing asthma's pathophysiological terrain. Our investigations into PyMGs highlight their vital role in the metabolic imbalances characteristic of asthma, proposing the targeting of these pathways as a viable therapeutic strategy.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the [GEO] repository. https://www.ncbi.nlm.nih.gov/geo/.
Abbreviations
- GO:
-
Gene ontology
- TCM:
-
Traditional Chinese medicine
- MF:
-
Molecular functions
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GEO:
-
Gene expression omnibus
- PyMGs:
-
Pyrimidine metabolism genes
- BP:
-
Biological processes
- CC:
-
Cellular components
- DEGs:
-
Differentially expressed genes
References
Gomez JL. Epigenetics in asthma. Curr Allergy Asthma Rep. 2019;19(12):56.
Lauzon-Joset JF, Marsolais D, Tardif-Pellerin E, Patoine D, Bissonnette EY. CD200 in asthma. Int J Biochem Cell Biol. 2019;112:141–4.
Cormier M, Lemiere C. Occupational asthma. Int J Tuberc Lung Dis. 2020;24(1):8–21.
Pinyochotiwong C, Chirakalwasan N, Collop N. Nocturnal asthma. Asian Pac J Allergy Immunol. 2021;39(2):78–88.
Khan DA, Oppenheimer JJ, Lee GB, Fineman SM. Asthma. Ann Allergy Asthma Immunol. 2019;123(4):416–7.
Katwa U, Kabra SK. Advances in asthma. Indian J Pediatr. 2018;85(8):641–2.
Garavito MF, Narvaez-Ortiz HY, Zimmermann BH. Pyrimidine metabolism: dynamic and versatile pathways in pathogens and cellular development. J Genet Genomics. 2015;42(5):195–205.
El KM. Pyrimidine metabolism in schistosomes: a comparison with other parasites and the search for potential chemotherapeutic targets. Comp Biochem Physiol B Biochem Mol Biol. 2017;213:55–80.
Pfenninger KH. Plasma membrane expansion: a neuron’s Herculean task. Nat Rev Neurosci. 2009;10(4):251–61.
Woeller CF, Roztocil E, Hammond C, Feldon SE. TSHR signaling stimulates proliferation through PI3K/Akt and induction of miR-146a and miR-155 in thyroid eye disease orbital fibroblasts. Invest Ophthalmol Vis Sci. 2019;60(13):4336–45.
Madera-Salcedo IK, Sanchez-Hernandez BE, Svyryd Y, Esquivel-Velazquez M, Rodriguez-Rodriguez N, Trejo-Zambrano MI, Garcia-Gonzalez HB, Hernandez-Molina G, Mutchinick OM, Alcocer-Varela J, et al. PPP2R2B hypermethylation causes acquired apoptosis deficiency in systemic autoimmune diseases. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.126457.
Zhu Z, Cao C, Zhang D, Zhang Z, Liu L, Wu D, Sun J. UBE2T-mediated Akt ubiquitination and Akt/beta-catenin activation promotes hepatocellular carcinoma development by increasing pyrimidine metabolism. Cell Death Dis. 2022;13(2):154.
Yuan Y, Li N, Fu M, Ye M. Identification of critical modules and biomarkers of ulcerative colitis by using WGCNA. J Inflamm Res. 2023;16:1611–28.
Xu M, Kong Y, Chen N, Peng W, Zi R, Jiang M, Zhu J, Wang Y, Yue J, Lv J, et al. Identification of immune-related gene signature and prediction of CeRNA network in active ulcerative colitis. FRONT IMMUNOL. 2022;13:855645.
Wu Y, Liu X, Li G. Integrated bioinformatics and network pharmacology to identify the therapeutic target and molecular mechanisms of Huangqin decoction on ulcerative Colitis. Sci Rep. 2022;12(1):159.
Wu Z, Liu P, Huang B, Deng S, Song Z, Huang X, Yang J, Cheng S. A novel Alzheimer’s disease prognostic signature: identification and analysis of glutamine metabolism genes in immunogenicity and immunotherapy efficacy. Sci Rep. 2023;13(1):6895.
De Carvalho TR, Giaretta AA, Teixeira BF, Martins LB. New bioacoustic and distributional data on Bokermannohyla sapiranga Brandao et al., 2012 (Anura: Hylidae): revisiting its diagnosis in comparison with B. pseudopseudis (Miranda-Ribeiro, 1937). Zootaxa. 2013;3746:383–92.
Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–31.
Mon-Lopez D, Tejero-Gonzalez CM. Validity and reliability of the TargetScan ISSF Pistol & Rifle application for measuring shooting performance. Scand J Med Sci Sports. 2019;29(11):1707–12.
Furio-Tari P, Tarazona S, Gabaldon T, Enright AJ, Conesa A. spongeScan: a web for detecting microRNA binding elements in lncRNA sequences. Nucleic Acids Res. 2016;44(W1):W176–80.
Williams EJ, Berthon BS, Stoodley I, Williams LM, Wood LG. Nutrition in asthma. Semin Respir Crit Care Med. 2022;43(5):646–61.
Guerau-de-Arellano M, Britt RJ. Sterols in asthma. Trends Immunol. 2022;43(10):792–9.
Quirt J, Hildebrand KJ, Mazza J, Noya F, Kim H. Asthma. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):50.
King-Biggs MB. Asthma. Ann Intern Med. 2019;171(7):C49–64.
Liu C, Gu C, Huang W, Sheng X, Du J, Li Y. Targeted UPLC-MS/MS high-throughput metabolomics approach to assess the purine and pyrimidine metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1113:98–106.
Sedova L, Bukova I, Bazantova P, Petrezselyova S, Prochazka J, Skolnikova E, Zudova D, Vcelak J, Makovicky P, Bendlova B, et al. Semi-lethal primary ciliary dyskinesia in rats lacking the Nme7 gene. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22083810.
Sedova L, Prochazka J, Zudova D, Bendlova B, Vcelak J, Sedlacek R, Seda O. Heterozygous Nme7 mutation affects glucose tolerance in male rats. Genes. 2021. https://doi.org/10.3390/genes12071087.
Ren X, Rong Z, Liu X, Gao J, Xu X, Zi Y, Mu Y, Guan Y, Cao Z, Zhang Y, et al. The protein kinase activity of NME7 activates Wnt/beta-catenin signaling to promote one-carbon metabolism in hepatocellular carcinoma. Cancer Res. 2022;82(1):60–74.
Sun Y, Li S, Li H, Yang F, Bai Y, Zhao M, Guo J, Zhao M, Zhou P, Khor CC, et al. TNFRSF10A-LOC389641 rs13278062 but not REST-C4orf14-POLR2B-IGFBP7 rs1713985 was found associated with age-related macular degeneration in a Chinese population. Invest Ophthalmol Vis Sci. 2013;54(13):8199–203.
Li XL, Xie Y, Chen YL, Zhang ZM, Tao YF, Li G, Wu D, Wang HR, Zhuo R, Pan JJ, et al. The RNA polymerase II subunit B (RPB2) functions as a growth regulator in human glioblastoma. Biochem Biophys Res Commun. 2023;674:170–82.
Wang H, Jia Y, Gu J, Chen O, Yue S. Ferroptosis-related genes are involved in asthma and regulate the immune microenvironment. Front Pharmacol. 2023;14:1087557.
Yip W, Hughes MR, Li Y, Cait A, Hirst M, Mohn WW, McNagny KM. Butyrate shapes immune cell fate and function in allergic asthma. Front Immunol. 2021;12:628453.
Shamji MH, Sharif H, Layhadi JA, Zhu R, Kishore U, Renz H. Diverse immune mechanisms of allergen immunotherapy for allergic rhinitis with and without asthma. J Allergy Clin Immunol. 2022;149(3):791–801.
Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Environ Res Public Health. 2018. https://doi.org/10.3390/ijerph15051033.
Huang F, Yu J, Lai T, Luo L, Zhang W. The combination of bioinformatics analysis and untargeted metabolomics reveals potential biomarkers and key metabolic pathways in asthma. Metabolites. 2022. https://doi.org/10.3390/metabo13010025.
Chen ST, Yang N. Constructing ferroptosis-related competing endogenous RNA networks and exploring potential biomarkers correlated with immune infiltration cells in asthma using combinative bioinformatics strategy. BMC Genomics. 2023;24(1):294.
Wu Z, Cai Z, Shi H, Huang X, Cai M, Yuan K, Huang P, Shi G, Yan T, Li Z. Effective biomarkers and therapeutic targets of nerve-immunity interaction in the treatment of depression: an integrated investigation of the miRNA-mRNA regulatory networks. Aging. 2022;14(8):3569–96.
Funding
The Second “Xinglin Scholars-Nursing Youth” Program of Shanghai University of Traditional Chinese Medicine (2023HLXL09); Science and Technology Development Project (Nursing Special Project), Shanghai University of Traditional Chinese Medicine 23HLZX06.
Author information
Authors and Affiliations
Contributions
Dihui Zhang and Xiaowei Pu drafted and revised the manuscript. Dihui Zhang and Man Zheng were in charge of data collection. Dihui Zhang and Xiaowei Pu were in charge of design of frame. Jia Chen and Guanghui Li conceived and designed this article, in charge of syntax modification and revised of the manuscript. All the authors have read and agreed to the final version manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, D., Pu, X., Zheng, M. et al. Employing a synergistic bioinformatics and machine learning framework to elucidate biomarkers associating asthma with pyrimidine metabolism genes. Respir Res 25, 327 (2024). https://doi.org/10.1186/s12931-024-02954-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-024-02954-4