
Abstract
Background
The apparent lack of additional missense mutations data on mixed-phenotype leukemia is noteworthy. Single amino acid substitution by these non-synonymous single nucleotide variations can be related to many pathological conditions and may influence susceptibility to disease. This case-control study aimed to unravel whether the ZAP70 missense variant (rs104893674 (C > A)) underpinning mixed-phenotype leukemia.
Methods
The rs104893674 was genotyped in clients who were mixed-phenotype acute leukemia-, acute lymphoblastic leukemia- and acute myeloid leukemia-positive and matched healthy controls, which have been referred to all major urban hospitals from multiple provinces of country- wide, IRAN, from February 11’ 2019 to June 10’ 2023, by amplification refractory mutation system-polymerase chain reaction method. Direct sequencing for rs104893674 of the ZAP70 gene was performed in a 3130 Genetic Analyzer.
Results
We found that the AC genotype of individuals with A allele at this polymorphic site (heterozygous variant-type) contribute to the genetic susceptibility to acute leukemia of both forms, acute myeloid leukemia and acute lymphoblastic leukemia as well as with a mixed phenotype. In other words, the ZAP70 missense variant (rs104893674 (C > A)) increases susceptibility of distinct cell populations of different (myeloid and lymphoid) lineages to exhibiting cancer phenotype. The results were all consistent with genotype data obtained using a direct DNA sequencing technique.
Conclusion
Of special interest are pathogenic missense mutations, since they generate variants that cause specific molecular phenotypes through protein destabilization. Overall, we discovered that the rs104893674 (C > A) variant chance in causing mixed-phenotype leukemia is relatively high.
Similar content being viewed by others


Background
Acute leukemia is one of the most common types of hematological malignancies which affects all blood cell lineages and is associated with increase in the growth rate and disorganized differentiation of hematopoietic cells [1, 2]. Although acute leukemia usually presents in the myeloid or lymphoid lineages, in rare cases some with no clear evidence of differentiation along a single lineage exhibit two distinct affected cell populations of different lineages [2, 3]. In cases of bi-lineal acute leukemia classified as mixed-phenotype acute leukemia (MPAL), the outcome is worse than both acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) [4] and patients with MPAL have poorer survival as compared with age-matched patients with a single phenotype of acute leukemia thereby MPAL prognosis is largely dependent on age and is likely can give rise by aberrant mechanisms at a genomic scale [5]. Protein tyrosine phosphorylation is a substantial signaling mechanism that controls important cellular processes including cell growth, differentiation, metabolism, and motility by activation of downstream pathways. Aberrant routine tyrosine kinases signaling promotes carcinogenesis, an aberrant which has been observed in association with mutations that permit inhibition of the normal function of protein kinases [6, 7]. Most often, missense mutation is the predominant one which results in amino acid changes in the polypeptide chain [8]. Missense mutations or nonsynonymous substitutions which make single nucleotide variants (SNVs) that manifest as protein variants with a single amino acid variation (SAV), are of particular interest in view of the fact that notwithstanding a single amino acid substitution may cause dramatic structural changes by which protein stability is compromised, or a perturbation that destabilizes binding interfaces in protein to the point of impairing its function [9]. The ζ-associated protein of 70 kDa (ZAP-70) is a cytoplasmic tyrosine kinase that has a role in differentiation by regulating the JAK–STAT3–MYC signaling axis. Additionally, ZAP70 was shown to promote migration and invasion of prostate cancer cells as well as identified as a prognostic marker in colorectal cancer [10,11,12]. Currently, 19 nonsynonymous single-nucleotide polymorphisms (nsSNPs) in the Single Nucleotide Polymorphism (dbSNP) database have been reported to cause nonsense or missense changes in ZAP70. It is believed that various causes underlie the different types of leukemia, while a genetic factor might reveal common molecular traits that contribute to the development of AML and ALL simultaneously [13, 14]. In this study, from these nsSNPs, we selected rs104893674 with DNA substitution of 1554 C > A and Ser518Arg amino-acid substitution to identify likely common genetic traits between AML and ALL which would help to uncover the points of susceptibility for MPAL.
Methods
Using a case-control study design, male and female subjects who were diagnosed (depended on complete blood count, peripheral blood film, and flow cytometric immune-phenotyping) with acute myeloid leukemia (n = 94) and acute lymphoblastic leukemia (n = 101), were studied from July 20’ 2021 to June 10’ 2023. The controls for both AML (n = 99) and ALL (n = 101) groups were selected randomly among the age- and sex-matched general population without neoplastic diseases or patient’s companions at Fars and Isfahan (Iran) urban hospitals. Cases from each subtypes of AML or ALL with secondary leukemia, other hematological disease, accompanied with other systemic malignancies, pregnant and lactating women, subjects with uncontrollable systemic infection were excluded.
Forty-seven blood samples were obtained from cases of nonobvious bi-lineage MPAL presentation that microscopic evaluation of their specimens had revealed the coexistence of two different lineages making up the blast cell population, referred to hospitals across the country (Fars, Isfahan, Tehran, Qom, Khuzestan, Bushehr and Yazd), over a 4-years period (2019–2023). Fifteen samples from patients with MPAL were included (cases expressed mixed phenotypic markers) and thirty-two samples (which unequivocally assigned to a myeloid, B- or T-lymphoid lineage) were excluded from this study. Afterwards, enrollment was stopped before reaching the larger sample size because of poor recruitment (as MPAL is rare and giving an incidence of 0.35 cases/1,000,000 person-years) [15]. Therefore, a minimum ratio of 30% (15 individuals) for this population under 50 ensured representativeness of the sample for quantitative analysis [16]. Also, a total of 23 blood samples were obtained from matched healthy subjects, referring to the same hospitals.
A volume of 10 mL of whole blood of each study participant was collected in blood collection tubes containing EDTA for hematologic analysis. Complete blood count was scored in the Dirui Hematology Analyzer (China). Cytomorphologic abnormalities were recorded using Romanowsky-stained blood film.
Immuno-phenotyping assay was performed through flow cytometry analysis (FACSCalibur flow cytometer, BD Biosciences), following a previously described protocol [17], for identification of leukemia cell subsets by their marker profile. The major markers (either cell surface or intracellular) including B-cell lineage markers (CD19, cCD22 and cCD79a) and T-cell-related markers (CD5, sCD3 and CD4 and CD8) for ALL and myeloid markers (CD13 and MPO) for AML were utilized for the distinction of leukemia cell subsets. Markers of multiple lineages including CD19 and CD79a (B -lymphoid lineage), CD7 (T -lymphoid lineage) and CD13, CD33, and MPO (myeloid lineage) were used to make an accurate diagnosis in MPAL ambiguous cases. FlowJo software (FlowJo LLC) was used to analyze flow cytometry data.
We evaluated the Hardy–Weinberg equilibrium (HWE) by computing the observed genotype frequencies versus expected genotype values for polymorphic locus to check whether the population was in Hardy–Weinberg equilibrium.
We used the windows-based software program QUANTO (Version 1.2.4; University of Southern California, Los Angeles, CA, USA, http://biostats.usc.edu/Quanto.html) to estimate the statistical power of our study for detecting an association of rs104893674 with acute leukemia.
ZAP70 genetic polymorphism (rs104893674 (C > A)) was assessed by amplification refractory mutation system–polymerase chain reaction (ARMS-PCR), aka Allele-specific polymerase chain reaction (AS-PCR), after peripheral blood samples were collected in EDTA vacutainer tubes and extraction genomic DNA following a salting out technique standard protocol. We used a pair of internal control primers (Beta-actin was used as an internal control) specific for the normal DNA sequence (A pair of control primers which could not amplify mutant DNA at a given locus was used to confirm that the genomic DNA is, in principle, amplifiable) and allele-specific primers designed using Oligo7 software (version 7.54, Molecular Biology Insights Inc., Cascade, CO, USA). (The designed primer sequences reported in Table 1).
Polymerase chain reaction (PCR) for target DNA amplification was achieved using a final reaction volume of 22 µL composed of, 1 µL template DNA, 11µL of 2× Master Mix Red (Ampliqon), 1 µL of each primer (10 µM), and 5 µL DNase-free water. The PCR cycling conditions were 5 min at 94◦C, followed by 32 cycles of 40 s at 94◦C, 40 s at 58◦C, and 40 s at 72◦C, with a final step at 72◦C for 5 min. PCR products were verified on a 2% agarose gel for 10 min and visualized on UV transilluminator. Direct sequencing of PCR amplification products recovered by the GEL/PCR Purification Kit (Favorgen Biotech Corp., Taiwan, China) was analyzed on a 3130 Genetic Analyzer sequencing machine (Applied Biosystems) according to protocol previously described by Ameri et al. [18] and Parhoudeh et al. [16]. Sanger Sequencing PCR reaction components and thermal cycling steps outlined in Tables 2 and 3, respectively. Sequences were analyzed with the CodonCode Aligner V.5.1.5 software (CodonCode Corporation, Centerville, MA, USA).
To analyze the difference between genotype and allele frequencies in two groups, Chi-square testing and logistic regression analyses were used by SPSS software (Version 22.0, SPSS, Inc, Chicago, IL, USA). P-values less than 0.05 were considered statistically significant. Bonferroni corrections were applied to correct for multiple comparisons, and the threshold for statistical significance was set at ≤ 0.05.
Results
According to immuno-phenotyping findings, common Pre-B ALL was diagnosed in 75% of study’s cases from ALL group who expressed CD19, cytoplasmic- CD22 (cCD22) and CD79a (cCD79a) markers. A total of 25% of cases of this group were represented as T-cell ALL which characteristically expressed CD5, surface CD3 (sCD3) and CD4/CD8 dichotomy markers (Fig. 1). The leukemic cells from all cases in the AML group expressed myeloid markers including CD13 and MoAb anti myeloperoxidase (MPO) (Fig. 2). Immuno-phenotyping results showed that bi-phenotypic cases co-expressed myeloid, B- and T-lymphoid antigens (Fig. 3).

Immuno-phenotyping profile of ALL cases; (A) FSC and SSC, morphologic characteristic of blood cells; (B) negative expression of acute myeloid leukemia specific markers; (C) B-cell lineage acute lymphoblastic leukemia positive for the following markers, cCD79a, CD19, and cCD22 (histograms display a single measurement parameter (Positive dataset)); (D) T-cell lineage acute lymphoblastic leukemia positive for the following markers, CD5, sCD3 and co-express of CD4 and CD8 simultaneously (cells double positive or CD4+\CD8+)

Immuno-phenotyping profile of AML cases; (A) FSC and SSC, morphologic characteristic of blood cells; (B) myeloid leukemia positive for the MPO and CD13 markers (histograms display a single measurement parameter (Positive dataset)); (C) negative expression of acute lymphoblastic leukemia specific markers

Immuno-phenotyping profile of MPAL cases; (A) FSC and SSC, morphologic characteristic of blood cells; (B) Positive markers for multiple lineages including, CD19 and CD79a (B -lymphoid lineage), CD7 (T -lymphoid lineage) and CD13, CD33, and MPO (myeloid lineage); (C) Histograms display positive datasets
We calculated the genotype frequencies expected versus observed genotype values under traditional HWE test for polymorphic locus to determine whether the population was in Hardy–Weinberg equilibrium. As the deviation from HWE in the polymorphic locus was not significant, the results indicated the presence of HWE in in question population at polymorphic rs104893674 site (Table 4).
According to the QUANTO program, the available sample size of cases and controls was adequate and reached the 80% threshold at the significance level of 0.05. The power of the study was calculated as 0.84 at an alpha of 0.05 (Fig. 4). This finding suggests that the rs104893674 polymorphism may contribute to the genetic predisposition of acute leukemia subtypes.

Power analysis for 1554 C > A (Ser518Arg) polymorphism with α = 0.05
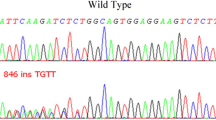
The PCR products in Fig. 5 (Supplementary file) shown a 237 bp (rs104893674) amplification product corresponding to the allele -specific primers and a 169 bp amplification product with internal control primers when the PCR have run on a 1% agarose gel. The accuracy and specificity of our established AS-PCR was further validated by direct sequencing of PCR products.

The pattern of observed bands after agarose-gel electrophoresis and result of DNA sequencing
In our study, the frequencies of the AC (FALL= 89%, OR (95% CI) = 1.57(1.37–1.85), p < 0.004) (FAML= 97.1%, OR (95% CI) = 0.6(0.4–0.9), p = 0.031) (F MPAL = 57.7%, OR (95% CI) = 0.84(0.4–1.2), p < 0.003) and AA (FALL= 11%, OR (95% CI) = 1, p < 0.004) (FAML= 6.1, OR (95% CI) = 0.2(0.1–0.3), p = 0.031) (F MPAL = 26.9%, OR (95% CI) = 0.46(0.15–0.3), p < 0.003) genotypes at rs104893674 and the A (FALL= 50%, OR (95% CI) = 2.19(1.44–3.32), p < 0.006) (FAML= 54.5%, OR (95% CI) = 0.9 (0.6–0.8), p = 0.074) (F MPAL = 96.2%, OR (95% CI) = 0.99(0.4–3.1), p < 0.002) allele were significantly higher in the ALL, AML and MPAL patients groups compared with controls (Table 5). Individuals with AC or AA genotypes or with the A allele at this locus were associated with an increased risk for exhibiting cancer phenotype with both myeloid and lymphoid lineages. These findings indicate the potential biological role of SNP at rs104893674 with the AC or AA genotypes and the A allele, as a common variant involved for the development of both ALL and AML. This is explained by the corresponding amino acid substitution at rs104893674 locus and would help to response to this question that; what underlying molecular features do the patients have when they observed both AML and ALL (patients who have AML and simultaneously have ALL or patients with ALL and mixed with AML)?
Discussion
The biology of co-emergence of mixed different hematopoietic lineages in MPAL has been poorly understood, and is always a matter of debate whether ALL and AML might have similar genetic traits considering their similar symptoms [14, 19]. In this study we understood the contribution of ZAP70 gene missense mutation (rs104893674 (C > A)) in development of both lymphoid and myeloid leukemia and verified the possibility of using the rs104893674 (C > A) variant as an indicator for predicting both ALL and AML as well as mixed lineage leukemia. Identification of this common genetic factor between ALL and AML would help to uncover the points of susceptibility for MPAL and will lead to early diagnosis. With the implementation of ZAP70 gene missense mutation (rs104893674 (C > A)) analysis in the clinical setting, a more molecularly guided precision diagnosis approach could improve the prediction accuracy for likelihood of MPAL developing. In reviewing the literature, no data was found on the association between ZAP70 gene and/or rs104893674 SNP and mixed leukemia phenotype for providing supporting or opposing ideas, nevertheless, several lines of evidence in the literature indicate other genes that have relation to both ALL and AML. These include genes that are both mutated in the two types of leukemia [14]. Some notable are, EP300 gene which encodes E1A binding protein p300 as a histone acetyltransferase. This protein regulates the transcription activity of many genes and plays an important role in differentiation and proliferation [20, 21]. TRAF2 gene which is involved in the signal transduction of the TNF receptor superfamily and plays a significant role in survival and apoptosis of hematopoietic cells [22]. JAK2 tyrosine kinase and STAT1 both are highly associated with leukemia [23]. Mutation in SELL as an adhesion/homing receptor in lymphocyte–endothelial cell interactions is also responsible for the movement of blasts from bone marrow to the circulation [24]. SELPLG gene as a cell adhesion molecule, tethering white blood cells to the inner surface of blood vessels [25]. Generally speaking our results offer an interpretation of common genetic traits between ALL and AML. Considering a larger sample size, and likewise further studies on patients in a variety of ethnic populations as well as if an adequate population of rare MPAL patients is found, will resolve the present study limitation.
Conclusion
MPAL often poses a diagnostic challenge owing to its rarity and underlying lineage plasticity. Identification of common genetic traits between ALL and AML would enhance our knowledge of the molecular and genetic complexity associated with MPAL to propose a novel diagnosis prediction framework.
Data availability
The datasets generated and/or analysed during the current study are available in the [dbSNP] repository [http://www.ncbi.nlm.nih.gov/SNP] and SNP can be searched for using the dbSNP ID (rs104893674).
Abbreviations
- WHO:
-
World Health Organization
- MPAL:
-
Mixed-Phenotype Acute Leukemia
- ALL:
-
Acute Lymphoblastic Leukemia
- AML:
-
Acute Myeloid Leukemia
- SNV:
-
Single Nucleotide Variants
- SAV:
-
Single Amino Acid Variation
- ZAP-70:
-
ζ-Associated Protein Of 70 kDa
- nsSNPs:
-
Nonsynonymous Single-Nucleotide
- ARMS-PCR:
-
Polymorphisms Amplification Refractory Mutation System–Polymerase Chain Reaction
- AS-PCR:
-
Allele-Specific Polymerase Chain Reaction
- PCR:
-
Polymerase Chain Reaction
- HWE:
-
Hardy-Weinberg Equilibrium
- cCD22:
-
Cytoplasmic CD22
- cCD79a:
-
Cytoplasmic CD79a
- sCD3:
-
Surface CD3
- MPO:
-
MoAb Antimyeloperoxidase
References
Bhalodia JN, Patel MM. Profile of Pediatric Malignancy: A three-year study. Nat J Community Med. 2011;2(1):24–7.
George B, Gonzalez A, Rios A. Inside the Biology of Acute Leukemias of Ambiguous Lineage: Diagnostic Work-Up, genomic and clinical characterization. J Cancer Sci Clin. 2022;6(3):276–80.
Eckstein OS, Wang L, Punia JN, Kornblau SM, Andreeff M, Wheeler DA. Mixed-phenotype acute leukemia (MPAL) exhibits frequent mutations in DNMT3A and activated signaling genes. Exp Hematol. 2016;44(8):740–4.
Khan M, Siddiqi R, Naqvi K. An update on classification, genetics, and clinical approach to mixed phenotype acute leukemia (MPAL). Ann Hematol. 2018;97:945–53.
Alexander TB, Orgel E. Mixed phenotype Acute Leukemia: current approaches to diagnosis and treatment. Curr Oncol Rep. 2021;23(2):22.
Kim M, Baek M, Kim DJ. Protein Tyrosine Signaling and its potential therapeutic implications in Carcinogenesis. Curr Pharm Des. 2017;23(29):4226–46.
Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–32.
Crozet P, Margalha L, Confraria A, Rodrigues A, Martinho C, Adamo M, et al. Mechanisms of regulation of SNF1/AMPK/SnRK1 protein kinases. Front Plant Sci. 2014;5:190.
Petrosino M, Novak L, Pasquo A, Chiaraluce R, Turina P, Capriotti E, et al. Analysis and interpretation of the impact of missense variants in Cancer. Int J Mol Sci. 2021;22(11):5416.
Fu D, Liu B, Zang LE, Jiang H. MiR-631/ZAP70: a novel axis in the migration and invasion of prostate cancer cells. Biochem Biophys Res Commun. 2016;469(3):345–51.
Cha Y, Moon BH, Lee MO, Ahn HJ, Lee HJ, Lee KA, et al. Zap70 functions to maintain stemness of mouse embryonic stem cells by negatively regulating Jak1/Stat3/c-Myc signaling. Stem Cells. 2010;28(9):1476–86.
Huang MY, Wang JY, Chang HJ, Kuo CW, Tok TS, Lin SR. CDC25A, VAV1, TP73, BRCA1 and ZAP70 gene overexpression correlates with radiation response in colorectal cancer. Oncol Rep. 2011;25(5):1297–306.
Park D, Jeong HO, Kim BC, Ha YM, Young Chung H. Computational approach to identify enzymes that are potential therapeutic candidates for psoriasis. Enzyme Res. 2011;2011:826784.
Chen J, Huang C, Zhu Y, Dong L, Cao W, Sun L, et al. Identification of similarities and differences between myeloid and lymphoid acute leukemias using a gene-gene interaction network. Pathol Res Pract. 2015;211(10):789–96.
Panagopoulos I, Andersen K, Johannsdottir IMR, Tandsæther MR, Micci F, Heim S. Genetic characterization of Pediatric mixed phenotype Acute Leukemia (MPAL). Cancer Genomics Proteom. 2024;21(1):1–11.
Parhoudeh S, Saadaty A, Khashei Varnamkhasti K, Khashei Varnamkhasti S, Naeimi L, Naeimi S. Highlighting allelic variations at the interleukin-19 locus in term of preeclampsia predisposing factors and access to an accurate diagnostic/screening option. BMC Pregnancy Childbirth. 2023;23(1):839.
Alves GV, Fernandes AL, Freire JM, Paiva Ade S, Vasconcelos RC, Sales VS, et al. Flow cytometry immunophenotyping evaluation in acute lymphoblastic leukemia: correlation to factors affecting clinic outcome. J Clin Lab Anal. 2012;26(6):431–40.
Ameri A, Khashei Varnamkhasti K, Parhoudeh S, Khashei Varnamkhasti S, Naeimi L, Naeimi S. Spontaneous miscarriage driven by maternal genetic mutation at position of PAI-1-844G/A: shed light on a race-specific genetic polymorphism. BMC Res Notes. 2023;16(1):360.
Béné MC, Porwit A. Mixed Phenotype/Lineage leukemia: has anything changed for 2021 on diagnosis, classification, and treatment? Curr Oncol Rep. 2022;24(8):1015–22.
Lynch JT, Somerville TD, Spencer GJ, Huang X, Somervaille TC. TTC5 is required to prevent apoptosis of acute myeloid leukemia stem cells. Cell Death Dis. 2013;4(4):573.
Shigeno K, Yoshida H, Pan L, Luo JM, Fujisawa S, Naito K, et al. Disease-related potential of mutations in transcriptional cofactors CREB-binding protein and p300 in leukemias. Cancer Lett. 2004;213(1):11–20.
Schnetzke U, Fischer M, Spies-Weisshart B, Zirm E, Hochhaus A, Müller JP, et al. The E3 ubiquitin ligase TRAF2 can contribute to TNF-α resistance in FLT3-ITD-positive AML cells. Leuk Res. 2013;37(11):1557–64.
Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annu Rev Immunol. 1998;16:293–322.
Graf M, Reif S, Hecht K, Pelka-Fleischer R, Pfister K, Nuessler V, et al. Low L-selectin (CD62L) expression in acute myeloid leukemia correlates with a bad cytogenetic risk. Eur J Haematol. 2003;71(5):366–76.
Zhang J, Xiang Y, Ding L, Keen-Circle K, Borlawsky TB, Ozer HG, et al. Using gene co-expression network analysis to predict biomarkers for chronic lymphocytic leukemia. BMC Bioinformatics. 2010;11(9):5.
Acknowledgements
Special thanks to the hospitals healthcare workers and all other staffs.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
N.S. designed the study and critically reviewed the manuscript. K.V.K, K.V.S, SH.A, R.M and N.L. performed formal analysis. N.S and N.B administrated project. K.V.K wrote the manuscript. The final manuscript has been approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Islamic Azad University- Kazerun Branch Ethics Committee (IR.IAU.KAU.REC.1398.051). All methods were performed in accordance with the guidelines and regulations of the Islamic Azad University- Kazerun Branch. Written informed consent was provided by all the participants before entering the study groups.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khashei Varnamkhasti, K., Khashei Varnamkhasti, S., Shahrouzian, A. et al. Genetic evidence for predisposition to acute leukemias due to a missense mutation (p.Ser518Arg) in ZAP70 kinase: a case-control study. BMC Med Genomics 17, 200 (2024). https://doi.org/10.1186/s12920-024-01961-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01961-0