
Abstract
Background
Autism is a neurodevelopmental disorder that is usually diagnosed in early childhood. Timely diagnosis and early initiation of treatments such as behavioral therapy are important in autistic people. Discovering critical genes and regulators in this disorder can lead to early diagnosis. Since the contribution of miRNAs along their targets can lead us to a better understanding of autism, we propose a framework containing two steps for gene and miRNA discovery.
Methods
The first step, called the FA_gene algorithm, finds a small set of genes involved in autism. This algorithm uses the WGCNA package to construct a co-expression network for control samples and seek modules of genes that are not reproducible in the corresponding co-expression network for autistic samples. Then, the protein–protein interaction network is constructed for genes in the non-reproducible modules and a small set of genes that may have potential roles in autism is selected based on this network. The second step, named the DMN_miRNA algorithm, detects the minimum number of miRNAs related to autism. To do this, DMN_miRNA defines an extended Set Cover algorithm over the mRNA–miRNA network, consisting of the selected genes and corresponding miRNA regulators.
Results
In the first step of the framework, the FA_gene algorithm finds a set of important genes; TP53, TNF, MAPK3, ACTB, TLR7, LCK, RAC2, EEF2, CAT, ZAP70, CD19, RPLP0, CDKN1A, CCL2, CDK4, CCL5, CTSD, CD4, RACK1, CD74; using co-expression and protein–protein interaction networks. In the second step, the DMN_miRNA algorithm extracts critical miRNAs, hsa-mir-155-5p, hsa-mir-17-5p, hsa-mir-181a-5p, hsa-mir-18a-5p, and hsa-mir-92a-1-5p, as signature regulators for autism using important genes and mRNA–miRNA network. The importance of these key genes and miRNAs is confirmed by previous studies and enrichment analysis.
Conclusion
This study suggests FA_gene and DMN_miRNA algorithms for biomarker discovery, which lead us to a list of important players in ASD with potential roles in the nervous system or neurological disorders that can be experimentally investigated as candidates for ASD diagnostic tests.
Similar content being viewed by others

Background
Autism spectrum disorder (ASD), as a neurodevelopmental disorder, is characterized by limitations in social interaction and communication alongside repetitive, restricted behavioral, interest, or activity patterns [1]. These symptoms are typically diagnosed in children during their second year of life (12–24 months) [1]. It should be noted that it is essential to diagnose these children early and begin behavioral therapy and drug therapy as soon as possible [2]. Therefore, identifying candidate ASD risk factors for diagnosis and finding appropriate drugs for early treatment of this condition can significantly improve the future life quality of autistic children. Although some studies showed the role of immunological, neurological, and environmental factors in this condition, the main reason for ASD is the genetic basis [3, 4]. The phenotype and pathophysiological mechanisms of ASD are highly heterogeneous and complex [5, 6]. However, the characterization of gene expression profiles of ASD cases is a way to detect genetic causes, the effect of non-genetic risk factors on the gene expressions causes an understanding of the pathophysiological mechanisms in ASD [3]. In this regard, some studies focus on finding genes involved in autism with statistical, data mining, and machine learning approaches. For example, Latkowski and Osowski utilized eight different data mining methods independently, each followed by a genetic algorithm to find the most related genes to autism [7]. Another study detected important genes in autism using GBPSO-SVM, a Geometric Binary Particle Swarm Optimization with a Support Vector Machine as a fitness function [8]. They ran a tenfold GBPSO-SVM independently on three sets of genes having a higher rank in three statistical tests. The important genes are proposed based on the frequency of genes in optimal solutions [8]. Kong and his colleagues ranked genes based on differential expression between sample groups. They repeatedly used cross-validation to select some of the top genes from the ranked list. Then, a set of 55 genes is proposed as important genes [9].
MicroRNAs (miRNAs) are a class of non-coding RNAs that play critical roles in regulating gene expression. They target messenger RNAs (mRNAs), the coding RNAs that encode proteins, and activate or repress the translation of mRNAs [10]. So, changes in miRNA expression can affect protein production and cause various problems, including nervous system diseases and disorders [11]. The difference in miRNA expression is also detected in autistic patients compared to normal individuals [12,13,14]. In this regard, some studies focus on discovering miRNAs involved in autism. Shen and his colleagues used the gene expression profiles of ASD ones to calculate the differentially expressed genes (DEGs) relative to control ones. Then, a sub-network is extracted from an experimentally validated and computationally predicted miRNA-mRNA network by mapping DEGs onto it. Finally, 11 miRNAs are proposed as biomarkers for autism based on three self-defined parameters [15]. Another study constructed a co-expression network for a group of male samples and detected eight dysfunctional modules in autism related functionally to metabolism, immunity, neurodevelopment, and signaling. They also reported miRNAs and transcription factors for each module [16].
As mentioned, some studies try to find a small set of genes involved in autism and some others concentrate on finding relevant miRNAs. Although there is evidence that miRNAs and their corresponding targets might contribute to understanding autism [17], there are few studies that focus on investigating both. Hence, we propose a framework in two steps, one for finding a small set of genes involved in autism and the other for finding a minimum set of miRNAs regulators. In the first step, we offer an algorithm named FA_gene which is based on the gene co-expression network and Protein–Protein Interaction networks. In this way, instead of considering genes individually as in most statistical and machine learning studies, we benefit from a module-based view. In the second step, which is called DMN_miRNA algorithm an mRNA–miRNA is constructed based on the genes obtained from the first step and corresponding miRNA regulators. Then, we use a combinatorial-graph based method by defining a set cover problem over the mRNA–miRNA network. Based on the first step, we find 20 genes as abnormal genes in autism, and the second step announces five miRNAs targeting abnormal genes. Finally, we evaluate the results using previous studies and enrichment analysis on the target genes of the detected miRNAs. Therefore, in this study, a framework consisting of different statistical and systems biology approaches has been proposed to investigate both genes and miRNAs involved in autism.
Methods
This paper aims to reduce genes involved in autism and find a minimum set of miRNAs (as much as possible) that are effectively related to these genes. In this section, we first declare some notations and describe the extracted gene expression dataset for control and autism samples. Then, we represent a computational framework for miRNA discovery in autism.
Notations
Two sets, \(S = \left\{ {s_{1} , \ldots s_{n} } \right\}\) and \(G = \left\{ {g_{1} , \ldots g_{m} } \right\}\), with \(n\) individuals and \(m\) genes, are defined as sample and gene sets, respectively. The gene expression matrix of gene set \(G\) in the sample set \(S\) is shown by \({\mathbb{E}}_{n \times m}^{S,G}\) where
We represent two autistic and control sample sets with \(S^{C}\) and \(S^{A}\), respectively. Gene expression \(g_{j}\) across control and autistic samples is determined by column j of the \({\mathbb{E}}^{{S^{C} ,G}}\) and \({\mathbb{E}}^{{S^{A} ,G}}\) matrices, \({\mathbb{E}}^{{S^{C} ,G}} \left[ j \right]\) and \({\mathbb{E}}^{{S^{A} ,G}} \left[ j \right]\), respectively. A set of miRNAs is represented by \(R = \left\{ {r_{1} , \ldots r_{l} } \right\}\), where \(\left| R \right| = l\) shows the number of miRNAs.
Extracting control and autism gene expression dataset
Peripheral blood samples of ASDs and control ones (from GSE18123 [9]), which are more accessible than brain tissue samples [18, 19], are retrieved from the Gene Expression Omnibus (GEO) database [20]. Gene expression of these samples is achieved using two GPL570 and GPL6244 platforms with 103 and 182 samples, respectively. We use the GPL6244 platform, which has more samples. Based on the diagnosis, samples are grouped into four categories: CONTROL (81 samples), AUTISM (40 samples), PDD-NOS (47 samples), and ASPERGER’S DISORDER (14 samples). The first and second categories show samples without and with autism, respectively. The third represents the samples that do not fully meet autism criteria (called Pervasive Developmental Disorder-Not Otherwise Specified). The last one indicates samples diagnosed with Asperger disorder (typically have stronger verbal language skills and intellectual ability than autistic samples). This study analyzes samples with CONTROL and AUTISM diagnoses. Besides the diagnosis of autism, there are also some other features for each sample, such as developmental or speech disorder situations and infection to other psychiatric disorders or neurological disorders. We extract samples with no neurological, psychiatric, developmental, or speech disorder to remove the effect of any neurological or psychiatric disorder such as Seizures, Landau-Kleffner Syndrome, Bipolar, or Attention Deficit Hyperactivity Disorder. Though, 77 samples remain for the control group and 21 for autism. We also remove drug-treated controls and those who were allergic to have a purer set of control samples, resulting in 60 control samples. Using all remaining control samples, which are three times larger in size than the autistic sample, biases the variance toward the control samples (in the second step of the first algorithm). Hence, using unequal-sized groups affects the power parameter (fourth and fifth steps of the first algorithm), co-expression network, and changes the connectivity pattern and module extracting. To generate a balanced dataset of control and autistic samples, we select 21 out of 60 control samples. Therefore, we have two sample sets \(S^{A}\) and \(S^{C}\) with sizes \(n_{A} = 21\) and \(n_{C} = 21\) for autism and control samples, respectively. Each sample has 32,321 probes in raw files. We use the “Affy” [21] package in R for RMA normalization to construct gene expression matrices from raw files. We also use “annotate” [22] and “hugene10sttranscriptcluster” [23] packages in R to convert probe IDs into gene symbols to achieve a set \(G = \left\{ {g_{1} , \ldots g_{m} } \right\}\) with \(m = 18801\) gene symbols. Finally, we obtain a gene expression profile for control and autistic samples named \({\mathbb{E}}_{21 \times 18801}^{{S^{C} {,}G}}\) and \({\mathbb{E}}_{21 \times 18801}^{{S^{A} {,}G}}\), respectively.
The details of the selected dataset are available in Table 1. The first and second columns indicate the autism and control samples, respectively. The third and fourth columns display the number of females and males in autism and control samples separately. The last column shows the number of genes for each sample.
We also performed differential gene expression analysis using the “Limma” R package [24] to detect genes with different expressions (DEGs) between control and autism. This package performs t-tests to find up-regulated and down-regulated genes. Here, genes with higher absolute log fold-change of than one and adjusted p-value less than 0.05 were selected as DEGs between autism and control.
Proposed framework for predicting involved miRNAs in autism
This paper presents a new framework including two main steps for predicting a minimum set of involved miRNAs in autism as follows:
-
1.
Introducing the FA_gene algorithm to find a small set of genes, \(G^{A} \subseteq G\), involved in autism as an abnormal gene set.
-
2.
Introducing the DMN_miRNA algorithm to detect the minimum number of miRNAs, \(\mathop R\limits^{\prime } \subseteq R\), as regulators covering (targeting) the gene set \(G^{A}\).
Each step of our framework is explained in more detail below.
FA_gene algorithm: finding abnormal genes in autism
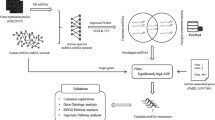
We propose an algorithm for finding a small set of genes, \(G^{A} \subseteq G\), involved in autism named FA_gene (see Fig. 1). In the following, we illustrate this algorithm in more detail.

FA_gene algorithm to find important genes involved in autism
In the first step, the set \(G\) with 18,801 genes and two gene expression matrices \({\mathbb{E}}_{21 \times 18801}^{{S^{A} {,}G}}\) and \({\mathbb{E}}_{21 \times 18801}^{{S^{C} {,}G}}\) are given as inputs to the FA_gene algorithm for finding genes involved in autism. The second step extracts a gene set \(\mathop G\limits^{\prime } \subseteq G\) with 4704 genes, \(\mathop m\limits^{\prime } = 4707\) where \(\mathop m\limits^{\prime } \ll m\). Set \(\mathop G\limits^{\prime }\) includes genes whose expressions are diverse between control and autistic samples. Then, the third step generates \({\mathbb{E}}_{{n_{A} \times \mathop m\limits^{\prime } }}^{{S^{A} ,\mathop G\limits^{\prime } }}\) and \({\mathbb{E}}_{{n_{C} \times \mathop m\limits^{\prime } }}^{{S^{C} ,\mathop G\limits^{\prime } }}\) based on \({\mathbb{E}}_{{n_{A} \times m}}^{{S^{A} ,G}}\) and \({\mathbb{E}}_{{n_{C} \times m}}^{{S^{C} ,G}}\) by keeping genes in \(\mathop G\limits^{\prime }\) (See Additional file 1, Additional file 2).
In the fourth step, the algorithm uses WGCNA v. 1.70.3 package [25] to construct the control co-expression network as the reference network for \({\mathbb{E}}_{21 \times 4704}^{{S^{C} {,}\mathop G\limits^{\prime } }}\) matrix. In order to do this, the “adjacency” function is applied for building an adjacency matrix from the gene expression matrix according to the following parameters: type = “signed hybrid”, power = 8, corFnc = “bicor”. Then, the adjacency matrix is converted to the Topological Overlap Matrix (TOM) for achieving the dissimilarity matrix (1-TOM) by the “TOMsimilarity” function. At the end of this step, we use the “flashClust” and “cutreeDynamic” functions from the flashClust [26] package in R to extract modules from the reference network based on the dissimilarity matrix. Also, the “cutreeDynamic” function is performed according to “minModuleSize” = 30 and “deepSplit” = 2 parameters to cluster the tightly connected genes into groups called modules. We find 12 modules named using colors. Genes with no module are gathered in a group named “Gray”.
The fifth step seeks the modules with \(Z_{Summery}\) value less than 2 as non-preserved modules, according to [27]. The \(Z_{Summery}\) statistic is defined based on two other statistics, \(Z_{density}\) and \(Z_{connectivity}\). The \(Z_{density}\) statistic shows whether nodes in a module in the reference network are connected as high as in the test network. The \(Z_{connectivity}\) statistic determines the similarity of the connectivity pattern between nodes in each module of the reference and test networks. So, to find modules that are not reproducible (are non-preserved) from control to autism, we consider the control network as a reference and the autistic network as a test network. We calculate \(Z_{Summery}\) statistic using “modulePreservation” function with parameters: type = “signed hybrid”, corFnc = “bicor”, nPermutations = 200. At the end of this step, the “Turquoise” module is found with 1173 genes as the non-preserved module.
In the sixth step, a PPI network named \(G_{PPI}\) is constructed based on the genes in the “Turquoise” module by “STRINGdb” [28] package in R to find genes with the most critical roles in the module. This package finds 1021 proteins. So, a network is achieved with 1021 nodes and 18,286 edges. (For \(G_{PPI}\), see Additional file 3).
In the last step, 20 genes whose corresponding proteins have the highest degree in the protein network \(G_{PPI}\) are collected in the set \(G^{A}\) as abnormal genes in autism.
DMN_miRNA algorithm: detecting the minimum number of miRNAs in autism
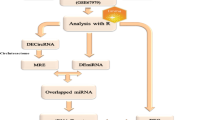
We propose the DMN_miRNA algorithm (see Fig. 2) for detecting the set of miRNAs \(\mathop R\limits^{\prime } \subseteq R\) to target abnormal genes in the autism for regulation.

DMN_miRNA algorithm to detect the minimum number of miRNAs in autism
In the first step, the set \(G^{A}\) as abnormal genes and three mRNA–miRNA databases, Tarbase v8.0 [29], mirTarbase v8.0 [30] and miRecords [31], are given as inputs. These databases contain mRNA and their miRNA regulators, whose regulatory relationships were validated experimentally or computationally. In the second step, we submit set \(G^{A}\) to an online tool called miRNet [32] for retrieving mRNA–miRNA pairs from these databases. In this regard, the set \(R = \left\{ {r_{1} , \ldots r_{l} } \right\}\), \(l = 35\), is obtained for genes in the set \(G^{A}\).
In the third step, we use the miRNet website to construct a bipartite network called \(G_{miR}\). This network includes two node sets based on miRNA set \(R\) and the abnormal gene set \(G^{A}\). Each edge from the miRNA \(r \in R\) to the gene \(g \in G^{A}\) shows that \(r\) targets \(g\) to regulate. \(G_{miR}\) is available in Additional file 4.
In the last step, we would like to obtain a minimum set of effective biomarkers (miRNAs), \(\Re \subseteq R\), which regulate genes in the \(G_{miR}\). In other words, we are looking for the smallest set of miRNAs whose gene targets cover \(G^{A}\). So, we consider \(G^{A}\) as the set that is desired to be covered, target genes of each miRNA as a subset and perform an extended greedy set cover algorithm on the network \(G_{miR}\). According to this definition, every subset assigns to a miRNA. Therefore, we are selecting the smallest set of miRNAs that covers \(G^{A}\) by finding the minimum set cover.
Results
In this section, we show the selected miRNAs as regulators for abnormal genes in autism by performing the proposed framework.
Finding abnormal genes involved in autism
We perform the FA_gene algorithm (see Method section) to find a small set of genes involved in autism. Based on the second step of the algorithm, we reduce 18,801 genes to 4707 \(\left( {\mathop G\limits^{\prime } } \right)\) due to the variation in gene expression of control and autistic samples.
According to the fourth step of the algorithm, we use set \(\mathop G\limits^{\prime }\) to construct the co-expression control network as a reference network to extract modules. Figure 3A shows the 12 detected modules from the gene co-expression network. Figure 3B displays the inter-modular relationships between greenyellow, black and red modules, blue, cyan, and magenta modules, salmon, brown, pink, and turquoise modules, green and gray modules.

Hierarchical clustering dendrogram of genes in the control network (x-axis) based on dissimilarity between them (y-axis); each main branch forms a module, and the corresponding module is named by a color (A). Clustering of modules eigengenes (B)
Based on the fifth step of the FA_gene algorithm, we evaluate the preservation of modules between ASD and control samples by \(Z_{Summery}\). Figure 4 shows the preservation \(Z_{Summery}\) plot which includes the correlation between module size (genes) and \(Z_{Summery}\). According to [27], modules with \(Z_{Summery}\) less than 2 can be considered as non-preserved modules between reference (control) and test (autism) co-expression networks. As it can be seen in the figure, all modules are preserved except for the “Turquoise” module, with \(Z_{Summery} = - 0.94\). This module is valuable because it is distinct in autism and control co-expression networks. This module in the control co-expression network includes \({\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 4}}\right.\kern-0pt} \!\lower0.7ex\hbox{$4$}}\) number of genes in the control co-expression network (1173 out of 4704 nodes) and is also dense with 651,335 edges. While it has 1127 nodes and 64,566 edges in the autistic network. In comparison with the autistic network, 49 nodes and 586,769 connections are lost.

The module preservation based on \({\text{Z}}_{{{\text{Summery}}}}\) statistic (y-axis) and module size (x-axis)
According to the sixth step of the algorithm, we make the PPI network by the corresponding genes in the “Turquoise” module to find important genes whose abnormal expression may lead to the loss of the nodes and connections in the non-preserved co-expression module. A PPI network, \(G_{PPI}\), with 1021 nodes (proteins) and 18,286 edges achieved for 1173 genes. Figure 5A shows the degree distribution of nodes in the PPI network. Most nodes (more than 600 nodes) have degrees less than 50. The maximum degree is 333, and the minimum degree is 1. Figure 5B displays the sub-network of \(G_{PPI}\) on the subset of top 20 nodes with the highest degrees which is plotted using Cytoscape 3.9.1 [33]. The nodes in the extracted sub-network are colored based on the degree of centrality values. The set \(G^{A}\) is obtained based on the top 20 corresponding proteins from \(G_{PPI}\). Also, these top 20 nodes with the highest degree in \(G_{PPI}\) with their corresponding degree are listed in Table 2.

Degree histogram for nodes in \({\text{G}}_{{{\text{PPI}}}}\) (A), the extracted PPI sub-network \({\text{G}}_{{{\text{PPI}}}}\) based on the degree centralities (B)
Finding the minimum set of miRNAs to regulate abnormal genes in autism
We perform DMN_miRNA (see Method section) to find the minimum number of miRNAs targeting abnormal genes in autism for regulation. Based on the second step of the DMN_miRNA algorithm, set \(R\) is constructed by 35 miRNAs, \(\left| R \right| = 35\), which are retrieved from the miRNet website [32] as the regulator of genes in the set \(G^{A}\). There is no miRNA regulator for LCK, CD19, and ZAP70 genes in the set \(G^{A}\). So, the analysis is followed on 17 out of 20 genes in the set \(G^{A}\).
According to the third step of the algorithm, the bipartite network \(G_{miR}\) is constructed with 17 genes, 35 miRNAs, and 119 edges (see Fig. 6A). The List of miRNAs in set \(R\) and corresponding degrees in the network, \(G_{miR}\), is available in Fig. 6B. Then, critical nodes based on the set cover algorithm (step fourth of the algorithm) are predicted as the effective biomarkers for autism and named \(\Re = \{\)hsa-mir-155-5p, hsa-mir-17-5p, hsa-mir-181a-5p, hsa-mir-18a-5p, hsa-mir-92a-1-5p\(\}\). Table 3 shows the miRNAs in the set \(\Re\), their degrees, and their target genes in \(G_{miR}\).

mRNA–miRNA network for genes in \({\text{G}}^{{\text{A}}}\); red circles represent genes, and blue ones represent miRNAs in \({\text{R}}\) (A). miRNAs in \({\text{R}}\) and the corresponding degree in \({\text{G}}_{{{\text{miR}}}}\) (B)
We also submit miRNA set \(\Re\) to miRNet to extract all target genes (7498 genes), which are regulated by set \(\Re\). These target genes are used to evaluate the regulatory impact of identified miRNAs and for further analysis.
To investigate the influence of miRNA set \(\Re\) in autism, a list of autism-related genes is collected based on the extracted genes from [34] and autism-related genes in the SFARI (Simons Foundation Autism Research Initiative) human genes database [35]. This list contains 1079 genes and is named autistic genes. By comparing the genes in this list with extracted targets from miRNet, we found that the miRNAs in the set \(\Re\) regulate 822 from the autistic genes. Therefore, considering that a large number of autistic genes (about 76%) are affected by these miRNAs, they can play a significant role in autism.
Enrichment analysis of target genes for predicted miRNAs
In this section, to ensure the effectiveness of the miRNAs (set \(\Re\)) in Biological Processes (BP) and pathways related to autism, we perform Gene Ontologies (GOs) and pathway analysis for all target genes (7498 genes), which are obtained using miRNet.
The online tool DAVID (Database for Annotation Visualization and Integrated Discovery) [36, 37] was used to extract biological processes and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways [38] for all gene targets of the set \(\Re\). We select the top 10 BPs and pathways according to the repeated target genes to compare them with the BPs and pathways of autistic genes.
The corresponding occurrences of target genes and autistic genes in each BP and pathway are shown in Fig. 7A and B, respectively. Focusing on the event of autistic genes indicates that there are five common BPs with gene targets of \(\Re ,\) which are positive and negative regulation of transcription, DNA-templated, transcription from RNA polymerase II promoter, and negative regulation of the apoptotic process. Although three BPs are not observed in autistic genes, they are all about the regulation of transcription from RNA polymerase II promoters. Also, six of the top 10 pathways are common between autistic genes and gene targets: pathways in cancer, MAPK signaling, focal adhesion, regulation acting cytoskeleton, viral carcinogenesis, and proteoglycans in cancer.

Enrichment analysis for miRNAs in R: Biological process (A), Pathway analysis (B)
Discussion
In this study, we proposed a two-step framework including two algorithms (FA_gene and DMN_miRNA) to find critical genes and the minimum number of miRNAs involved in autism. The low number of samples and the high number of genes are always challenging in studying gene expression datasets. There are different ways to reduce the number of genes, mainly by using machine learning and statistical methods. However, there is a risk of overfitting for reducing the number of genes by machine learning methods. Meanwhile, most statistical tests do not consider the biological relationships between genes. Therefore, in the FA_gene algorithm, we made a co-expression network of control samples by considering positive correlation values between gene expressions to cluster genes into modules with the same expression pattern to find the relations between genes. Then, the non-preserved module, compared with the autistic network, was detected, which was the biggest module with 1173 genes. The PPI network of these genes was constructed to find critical genes in this module that encoded proteins with crucial roles inside the cells. A small number of highly connected proteins in the PPI network can maintain the global network structure and lead us to essential proteins [39] in the non-preserved module. The expression of the top 20 nodes (\(G^{A}\)) in this PPI network were compared between autism and control samples. Although, these genes were not in the list of DEGs but eleven of them (TP53, TNF, MAPK3, ACTB, RAC2, ZAP70, CD19, CDKN1A, CDK4, CTSD, CD74) were downregulated in the autism vs control samples with a logFC value between -0.36 to -0.8. On the other hand, we searched the literature to find the relationships between genes in \(G^{A}\) and autism or any other neurological disorder. TP53 as the highest degree node was identified as a critical driver in ASD [3]. Also, developmental abnormalities were detected in altered TP53 and TP53-associated pathways in autism [40]. A high level of TP53 was detected in some brain areas in autistic samples vs controls [41]. TNF was related to autism [42], and TNF high blood concentration was observed in autistic children associated with symptom severity [43]. MAPK3 was associated with autism in several genome-wide association studies, copy number variants studies [44, 45], and also one of the biomarkers proposed to detect the early stage of Alzheimer [46]. ACTB is another gene related to autism [35]. Studies displayed that mutations in ACTB can cause Baraitser-Winter syndrome [47]. TLR7 was related to cognition and dendrite growth [48]. Studies showed that LCK is downregulated in the whole blood of ASD patients compared to controls [49]. RAC2 is a member of the Rho family GTPases. This family has an important role in brain development and is implicated in some neuropsychiatric and neurodegenerative diseases [50]. Also, its downregulation in Schizophrenia was reported [51]. Variations and differentially expression of EEF2 cause neurodevelopmental disorders like Alzheimer [52, 53]. CAT encodes catalase enzyme, which is decreased in autistic patients [54]. ZAP70 is another gene that is downregulated in the whole blood of ASD patients compared to controls [49]. CD19 is a biomarker for normal and neoplastic B cells, as well as follicular dendritic cells [55]. A statistically decrease in the number of B cells (CD19) in persons with Alzheimer's disease was depicted [56]. CDKN1A was identified as a critical driver in ASD [3, 57]. The higher levels of CCL2 and CCL5 were detected in ASD patients while lower plasma levels of these genes were detected in ASDs with fragile X syndrome [58,59,60]. CDK4 expression is decreased in the acute state of schizophrenia [61]. CTSD is one of 55 gene signatures for autism according to [9] and was reported related to autism [42]. CD4 protein is an important mediator of neuronal damages in central nervous system [62]. CD74 is another gene that inhibits β-Amyloid production [63], and β-Amyloid is amplified in autistic patients [64]. A decrease in RACK1 was reported in the cortex of down syndrome patients [65]. There is evidence that a decrease in the PKC activity in the aging brain or Alzheimer is related to the loss of RACK1 [66]. All the above reports demonstrate that genes in \(G^{A}\) are related to autism, other neurological issues, or the nervous system. Although the association of these genes with autism is not entirely determined yet, they could be a potential signature for autism since all of them except for RPLP0 are reported to be involved in neurological issues.
In the second step of the proposed framework, we used the DMN_miRNA algorithm to make an mRNA–miRNA network constructed based on the selected genes and corresponding regulators. miRNAs are key post-transcriptional regulators in different cellular biological processes, which led us to evaluate common miRNAs as critical regulators in ASD. Instead of finding miRNAs as regulators for all genes in the non-preserved module, we focus on the genes in \(G^{A}\). Since genes of \(G^{A}\) are connected (positively correlated) to almost all nodes (1170 out of 1173 genes) in the module, it can be assumed that there are common miRNA regulators for genes in \(G^{A}\) and the rest of the genes in the module. Thus, by investigating an mRNA–miRNA network reduced to genes in \(G^{A}\) and their regulators, a minimum set of miRNAs can be obtained that target almost all genes in the non-preserved module. To find a small set (as much as possible) of miRNAs that covers all selected genes, we used a set cover algorithm, with a bit of change in the definition of the original set cover. Our algorithm found five miRNAs that were reported to be related to autism by previous studies. The upregulation of “mir-155-5p” was reported in the cerebellar cortex [67], amygdala [68], and frontal cortex [69]. The “miR-17” and “miR-181a” were up-regulated in peripheral blood [70] and lymphoblastoid cell lines [71], respectively. Also, the upregulation of “miR-181a” was reported in lymphoblastoid cell lines [71]. These reports were in agreement with our results, which showed that most genes in \(G^{A}\) which are targeted by these miRNAs were downregulated in autism according to control samples. These miRNAs enrich pathways that are reported related to autism by previous studies. “Pathways in cancer” was introduced as one of the common pathways between autism and cancer [72]. The relation of “endocytosis” to autism was reported in [73]. The involvement of “focal adhesion” and “actin cytoskeleton” in the pathogenesis of autism was announced by [74]. Although relation of “Regulation acting cytoskeleton”, “Viral carcinogenesis,” and “Proteoglycans in cancer” were not found in the litrature but these pathways were common with autistic-gene pathways. Therefore, 9 out of 10 top pathways for the miRNA set \(\Re\) were specially reported or potentially related to autism. It shows that miRNAs in \(\Re\) should have a regulatory role in autism.
Conclusions
In this study, using both co-expression and PPI networks led us to critical genes in ASD, which were reported as genes related to the nervous system or neurological disorders but couldn’t be detected with DEG analysis. This set of critical genes (TP53, TNF, MAPK3, ACTB, TLR7, LCK, RAC2, EEF2, CAT, ZAP70, CD19, RPLP0, CDKN1A, CCL2, CDK4, CCL5, CTSD, CD4, RACK1, CD74) were co-expressed with almost all genes in the non-preserved module of co-expression network that guided us to assume common miRNA regulators for most genes in this module. The DMN_miRNA algorithm detected a minimum number of (five) miRNA regulators for these critical genes; hsa-mir-155-5p, hsa-mir-17-5p, hsa-mir-181a-5p, hsa-mir-18a-5p, hsa-mir-92a-1-5p. These miRNAs regulate the most involved genes in autism which were approved by the previous studies and enrichment analyses. So, we suggest using FA_gene and DMN_miRNA algorithms to detect critical genes and a minimum number of miRNAs from expression analysis. The critical genes and regulators for ASDs indicated in this study can be examined in experimental studies and then suggested for diagnostic tests.
Availability of data and materials
The gene expression dataset analyzed in this study was extracted from Gene Expression Omnibus (GEO) and is available under Accession Number GSE18123.
Abbreviations
- ASD:
-
Autism spectrum disorder
- FA_gene:
-
Finding abnormal genes in autism
- DMN_miRNA:
-
Detecting the minimum number of miRNAs in autism
- GBPSO-SVM:
-
Geometric binary particle swarm optimization-support vector machine
- mRNA:
-
Messenger RNAs
- DEG:
-
Differentially expressed gene
- PPI:
-
Protein–protein interaction
- GEO:
-
Gene expression omnibus
- PDD-NOS:
-
Pervasive developmental disorder-not otherwise specified
- TOM:
-
Topological overlap matrix
- SFARI:
-
Simons foundation autism research initiative
- BP:
-
Biological processes
- GO:
-
Gene ontologies
- DAVID:
-
Database for annotation visualization and integrated discovery
- KEGG:
-
Kyoto encyclopedia of genes and genomes
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
Remington B, Hastings RP, Kovshoff H, Degli Espinosa F, Jahr E, Brown T, et al. Early intensive behavioral intervention: outcomes for children with autism and their parents after two years. Am J Ment Retard. 2007;112:418–38.
Rahman MR, Petralia MC, Ciurleo R, Bramanti A, Fagone P, Shahjaman M, et al. Comprehensive analysis of RNA-Seq gene expression profiling of brain transcriptomes reveals novel genes, regulators, and pathways in autism spectrum disorder. Brain Sci. 2020;10:1–16.
Chaste P, Leboyer M. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin Neurosci. 2008;14:198–207.
Lord C, Brugha TS, Charman T, Cusack J, Dumas G, Frazier T, et al. Autism spectrum disorder. Nat Rev Dis Prim. 2020;6:1–23.
Saffari A, Arno M, Nasser E, Ronald A, Wong CCY, Schalkwyk LC, et al. RNA sequencing of identical twins discordant for autism reveals blood-based signatures implicating immune and transcriptional dysregulation. Mol Autism. 2019;10:1–15.
Latkowski T, Osowski S. Computerized system for recognition of autism on the basis of gene expression microarray data. Comput Biol Med. 2015;56:82–8.
Hameed SS, Hassan R, Muhammad FF. Selection and classification of gene expression in autism disorder: use of a combination of statistical filters and a GBPSO-SVM algorithm. PLoS ONE. 2017;12:e0187371.
Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee IH, et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS ONE. 2012;7:e49475.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem. 2011;67:129–39.
Vasu MM, Anitha A, Thanseem I, Suzuki K, Yamada K, Takahashi T, et al. Serum microRNA profiles in children with autism. Mol Autism. 2014;5:1–9.
Sarachana T, Zhou R, Chen G, Manji HK, Hu VW. Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med. 2010;2(2):1–18.
Vaccaro TDS, Sorrentino JM, Salvador S, Veit T, Souza DO, De Almeida RF. Alterations in the MicroRNA of the blood of autism spectrum disorder patients: effects on epigenetic regulation and potential biomarkers. Behav Sci (Basel). 2018;8:75.
Shen L, Lin Y, Sun Z, Yuan X, Chen L, Shen B. Knowledge-guided bioinformatics model for identifying autism spectrum disorder diagnostic microRNA biomarkers. Sci Rep. 2016;6:1–9.
Gao H, Zhong J, Huang Q, Wu X, Mo X, Lu L, et al. Integrated systems analysis explores dysfunctional molecular modules and regulatory factors in children with autism spectrum disorder. J Mol Neurosci. 2021;71:358–68.
Xiong C, Sun S, Jiang W, Ma L, Zhang J. ASDmiR: a step-wise method to uncover miRNA regulation related to autism spectrum disorder. Front Genet. 2020;11:1139.
Ansel A, Rosenzweig JP, Zisman PD, Melamed M, Gesundheit B. Variation in gene expression in autism spectrum disorders: an extensive review of transcriptomic studies. Front Neurosci. 2017;10:601.
Segura M, Pedreño C, Obiols J, Taurines R, Pàmias M, Grünblatt E, et al. Neurotrophin blood-based gene expression and social cognition analysis in patients with autism spectrum disorder. Neurogenetics. 2015;16:123–31.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10.
Gautier L, Cope L, Bolstad BM, Irizarry RA. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–15.
Gentleman R. annotate: Annotation for microarrays. R package version 1.72.0. 2021.
MacDonald JW. hugene10sttranscriptcluster. db: Affymetrix hugene10 annotation data (chip hugene10sttranscriptcluster). R Packag version. 2017;8.
Ritchie ME, Phipson B, Wu DI, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:47.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:1–13.
Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012;46:1–17.
Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–13.
Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018;46:D239–45.
Huang HY, Lin YCD, Li J, Huang KY, Shrestha S, Hong HC, et al. miRTarBase 2020: updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020;48:D148–54.
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–10.
Chang L, Zhou G, Soufan O, Xia J. miRNet 2.0: network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020;48:W244–51.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Vaishnavi V, Manikandan M, Munirajan AK. Mining the 3′ UTR of autism-implicated genes for SNPs perturbing microRNA regulation. Genomics Proteomics Bioinforma. 2014;12:92–104.
Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. 2013;4:36.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022. https://doi.org/10.1093/nar/gkac194.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27.
He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2:0826–34.
Wong S, Napoli E, Krakowiak P, Tassone F, Hertz-Picciotto I, Giulivi C. Role of p53, mitochondrial DNA deletions, and paternal age in autism: a case-control study. Pediatrics. 2016;137:e20151888.
Araghi-Niknam M, Fatemi SH. Levels of Bcl-2 and P53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell Mol Neurobiol. 2003;23:945–52.
Carter CJ. Autism genes and the leukocyte transcriptome in autistic toddlers relate to pathogen interactomes, infection and the immune system. A role for excess neurotrophic sAPPα and reduced antimicrobial Aβ. Neurochem Int. 2019;126:36–58.
Xie J, Huang L, Li X, Li H, Zhou Y, Zhu H, et al. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget. 2017;8:82390–8.
Park SM, Park HR, Lee JH. MAPK3 at the autism-linked human 16p11.2 locus influences precise synaptic target selection at drosophila larval neuromuscular junctions. Mol Cells. 2017;40:151–61.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Upadhyaya Y, Xie L, Salama P, Cao S, Nho K, Saykin AJ, et al. Differential co-expression analysis reveals early stage transcriptomic decoupling in alzheimer’s disease. BMC Med Genom. 2020;13:1–10.
Rivière JB, Van Bon BWM, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser–Winter syndrome. Nat Genet. 2012;44:440–4.
Liu HY, Hong YF, Huang CM, Chen CY, Huang TN, Hsueh YP. TLR7 negatively regulates dendrite outgrowth through the Myd88—c-Fos–IL-6 pathway. J Neurosci. 2013;33:11479–93.
Cirnigliaro M, Barbagallo C, Gulisano M, Domini CN, Barone R, Barbagallo D, et al. Expression and regulatory network analysis of miR-140-3p, a new potential serum biomarker for autism spectrum disorder. Front Mol Neurosci. 2017;10:250.
Bai Y, Xiang X, Liang C, Shi L. Regulating Rac in the nervous system: molecular function and disease implication of Rac GEFs and GAPs. Biomed Res Int. 2015. https://doi.org/10.1155/2015/632450.
Hoseth EZ, Krull F, Dieset I, Mørch RH, Hope S, Gardsjord ES, et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl Psychiatry. 2018;8:1–10.
Garcia-Esparcia P, Sideris-Lampretsas G, Hernandez-Ortega K, Grau-Rivera O, Sklaviadis T, Gelpi E, et al. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am J Neurodegener Dis. 2017;6:15.
Nabais Sá MJ, Olson AN, Yoon G, Nimmo GAM, Gomez CM, Willemsen MA, et al. De Novo variants in EEF2 cause a neurodevelopmental disorder with benign external hydrocephalus. Hum Mol Genet. 2021;29:3892–9.
Zoroglu SS, Armutcu F, Ozen S, Gurel A, Sivasli E, Yetkin O, et al. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur Arch Psychiatry Clin Neurosci. 2004;254:143–7.
Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1:1–17.
Sharma R, Chiao JW, Turo K, Ammon R, Escher JE, Gambert SR. Distribution of lymphocytes in persons with Alzheimer’s disease (AD) as compared to age matched nondemented (ND) control subjects. Clin Gerontol. 1997;18:13–9.
Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L, et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012;8:e1002592.
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81.
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah IN, Van de Water J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J Neuroimmunol. 2011;232:196–9.
Ashwood P, Nguyen DV, Hessl D, Hagerman RJ, Tassone F. Plasma cytokine profiles in Fragile X subjects: is there a role for cytokines in the pathogenesis? Brain Behav Immun. 2010;24:898–902.
Okazaki S, Boku S, Otsuka I, Mouri K, Aoyama S, Shiroiwa K, et al. The cell cycle-related genes as biomarkers for schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry. 2016;70:85–91.
Buttini M, Westland CE, Masliah E, Yafeh AM, Wyss-Coray T, Mucke L. Novel role of human CD4 molecule identified in neurodegeneration. Nat Med. 1998;4:441–6.
Matsuda S, Matsuda Y, D’Adamio L. CD74 interacts with APP and suppresses the production of Aβ. Mol Neurodegener. 2009;4:1–10.
Omura Y, Lu D, Jones MK, Nihrane A, Duvvi H, Shimotsuura Y, et al. Early Detection of Autism (ASD) by a non-invasive quick measurement of markedly reduced acetylcholine & DHEA and increased β-amyloid (1–42), asbestos (chrysotile), titanium dioxide, Al, Hg & often coexisting virus infections (CMV, HPV 16 and 18), bacteria. Acupunct Electrother Res. 2015;40:157–87.
Liu W, Dou F, Feng J, Yan Z. RACK1 is involved in β-amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiol Aging. 2011;32:1818–26.
Peyrl A, Weitzdoerfer R, Gulesserian T, Fountoulakis M, Lubec G. Aberrant expression of signaling-related proteins 14-3-3 gamma and RACK1 in fetal down syndrome brain (trisomy 21). Electrophoresis. 2002;23:152–7.
Wu YE, Parikshak NN, Belgard TG, Geschwind DH. Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nat Neurosci. 2016;19:1463–76.
Almehmadi KA, Tsilioni I, Theoharides TC. Increased expression of miR-155p5 in amygdala of children with autism spectrum disorder. Autism Res. 2020;13:18–23.
Mor M, Nardone S, Sams DS, Elliott E. Hypomethylation of miR-142 promoter and upregulation of microRNAs that target the oxytocin receptor gene in the autism prefrontal cortex. Mol Autism. 2015;6:1–11.
Jyonouchi H, Geng L, Streck DL, Dermody JJ, Toruner GA. MicroRNA expression changes in association with changes in interleukin-1ss/interleukin10 ratios produced by monocytes in autism spectrum disorders: their association with neuropsychiatric symptoms and comorbid conditions (observational study). J Neuroinflamm. 2017;14:229.
Seno MMG, Hu P, Gwadry FG, Pinto D, Marshall CR, Casallo G, et al. Gene and miRNA expression profiles in autism spectrum disorders. Brain Res. 2011;1380:85–97.
Gabrielli AP, Manzardo AM, Butler MG. GeneAnalytics pathways and profiling of shared autism and cancer genes. Int J Mol Sci. 2019;20:1166.
Torshizi AD, Duan J, Wang K. Transcriptional network analysis on brains reveals a potential regulatory role of PPP1R3F in autism spectrum disorders. BMC Res Notes. 2018;11:1–7.
Shen L, Zhang K, Feng C, Chen Y, Li S, Iqbal J, et al. iTRAQ-based proteomic analysis reveals protein profile in plasma from children with autism. PROTEOMICS Clin Appl. 2018;12:1700085.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the concept and design of the study. Material preparation, data collection, and analysis were performed by MR, NS and FZ. The first draft of the manuscript was written by MR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The autistic gene expression matrix (\({\mathbb{E}}_{{n_{A} \times \mathop m\limits^{\prime } }}^{{S^{A} ,\mathop G\limits^{\prime } }}\)).
Additional file 2.
The control gene expression matrix (\({\mathbb{E}}_{{n_{C} \times \mathop m\limits^{\prime } }}^{{S^{C} ,\mathop G\limits^{\prime } }}\)).
Additional file 3.
The PPI network (\(G_{PPI}\)) for genes in the non-preserved module.
Additional file 4.
The mRNA–miRNA network (\(G_{miR}\)) for genes in \(G^{A}\).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rastegari, M., Salehi, N. & Zare-Mirakabad, F. Biomarker prediction in autism spectrum disorder using a network-based approach. BMC Med Genomics 16, 12 (2023). https://doi.org/10.1186/s12920-023-01439-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01439-5