
Abstract
Background
Ectodermal dysplasias (ED) are a group of diseases that affects the development or function of the teeth, hair, nails and exocrine and sebaceous glands. One type of ED, ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC or Hay-Wells syndrome), is an autosomal dominant disease characterized by the presence of skin erosions affecting the palms, soles and scalp. Other clinical manifestations include ankyloblepharon filiforme adnatum, cleft lip, cleft palate, craniofacial abnormalities and ectodermal defects such as sparse wiry hair, nail changes, dental changes, and subjective hypohydrosis.
Case presentation
We describe a patient presenting clinical features reminiscent of AEC syndrome in addition to recurrent infections suggestive of immune deficiency. Genetic testing for TP63, IRF6 and RIPK4 was negative. Microarray analysis revealed a 2 MB deletion on chromosome 1 (1q21.1q21.2). Clinical exome sequencing uncovered compound heterozygous variants in CHUK; a maternally-inherited frameshift variant (c.1365del, p.Arg457Aspfs*6) and a de novo missense variant (c.1388C > A, p.Thr463Lys) on the paternal allele.
Conclusions
To our knowledge, this is the fourth family reported with CHUK-deficiency and the second patient with immune abnormalities. This is the first case of CHUK-deficiency with compound heterozygous pathogenic variants, including one variant that arose de novo. In comparison to cases found in the literature, this patient demonstrates a less severe phenotype than previously described.
Similar content being viewed by others


Background
Ectodermal dysplasias (ED, OMIM:604292) are a group of diseases affecting the teeth, hair, nails and exocrine and sebaceous glands. In some cases, part of the skin, eyes, inner ears, fingers, toes and central nervous system can also be affected. There are approximately 150 different types of ED, the most commonly recognized syndromes being the ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC, OMIM: 129900), Rapp-Hodgkin syndrome (OMIM: 129400) and ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC, OMIM:106260) [1]. AEC syndrome, also known as Hay-Wells syndrome, is caused by heterozygous pathogenic variants in TP63 [2, 3]. A classical feature of AEC syndrome is the presence of skin erosions affecting the palms, soles and scalp. Other clinical manifestations include ankyloblepharon filiforme adnatum, cleft lip, cleft palate, craniofacial abnormalities, and ectodermal defects such as sparse wiry hair, nail changes, dental changes, and subjective hypohydrosis [4,5,6].
IRF6-related disorders are a group of inherited disorders associated with heterozygous pathogenic variants in IRF6, including Van der Woude syndrome (VWS, OMIM:119300) and popliteal pterygium syndrome (PPS, OMIM:119500). VWS is characterized by orofacial clefting and lip pits whereas PPS is characterized by similar lip/palate abnormalities in combination with ankyloblepharon in some cases, as well as digital and genital abnormalities [7, 8]. Bartsocas-Papas syndrome (BPS, OMIM: 263650), a severe form of PPS, is associated with homozygous pathogenic variants in RIPK4 and CHUK [9,10,11]. Recently, a de novo missense variant in CHUK was reported in one patient with ectodermal dysplasia, orofacial clefting, limb anomalies and hypogammaglobulinemia [12].
Copy number variants affecting the 1q21.1 region have been associated with genomic disorders. Phenotypic features of 1q21.1 deletion syndrome include microcephaly (50%), mild intellectual disability (30%), mildly dysmorphic facial features, and eye abnormalities (26%). Other findings can include cardiac defects, genitourinary anomalies, skeletal malformations, and seizures (~ 15%). Psychiatric and behavioral abnormalities can include autism spectrum disorders, attention deficit hyperactivity disorder, and sleep disturbances (OMIM: 612474). The majority of microdeletions are inherited, and incomplete penetrance and variable expressivity have been noted [13,14,15]. In this report, we describe, for the first time, a patient with compound heterozygous variants in CHUK. Interestingly, one variant arose de novo. To our knowledge, this is the second patient with CHUK-deficiency and immune abnormalities associated with de novo variant in CHUK. However, based on our data, it is unclear if, in some cases, de novo heterozygous CHUK variants are sufficient to cause disease. Clinical features of the patient are consistent, although less severe, with previously reported cases. This patient is also carrier of a 2 MB deletion on chromosome 1 which might contribute to some of his features.
Case presentation
Our patient is a male born to healthy non consanguineous parents weighing 2.375 kg, measuring 48 inches at birth. Maternal and paternal age were 27 and 25 years old, respectively. During the pregnancy there were no exposures to drugs, alcohol, tobacco or medications. The fetal movements were described as normal up until approximately 32 weeks gestation, when they were noted to be decreased. He was delivered by induced vaginal delivery at 37 + 4 weeks gestation due to intrauterine growth retardation and reduced fetal movements. He was transferred to the Children’s Mercy Hospital Neonatal Intensive Care Unit (NICU) on day 1 of life due to cleft lip and palate and ankyloblepharon filiforme adnatum. Physical examination revealed sparse eyelashes and eyebrows, hypoplasia of the teeth, abnormal palmar creases, 5th finger clinodactyly, mild 2nd, 3rd toe syndactyly and hypohidrosis (Fig. 1). The patient had recurrent bacterial and viral infections. His infections included recurrent otitis media despite bilateral myringotomy and tube placement, Staphylococcus aureus impetigo, coxsackie hand foot mouth disease, recurrent upper and lower respiratory infections including respiratory syncytial virus (RSV) bronchiolitis and multiple episodes of non-RSV viral bronchiolitis. His immune work up showed mild abnormalities including low immunoglobulin (Ig) M (31 mg/dL) and low normal IgG levels (355 mg/dL). His IgA was normal (17 mg/dL). His lymphocyte subsets showed normal T cells (CD3; 1860 mm3) but mildly low CD4 (1333 mm3) and CD8 (372 mm3) subsets. The patient’s developmental history was appropriate. He had a head ultrasound, abdominal ultrasound, echocardiogram and bone survey which were unremarkable. This clinical presentation led to the suspicion of an ectodermal dysplasia syndrome such as AEC syndrome, Bartsocas-Papas syndrome or Van der Woude syndrome. Gene testing for TP63, RIPK4 and IRF6 was negative. Microarray analysis revealed a 2 MB deletion on chromosome 1 encompassing 18 genes (arr [hg19] 1q21.1q21.2 (145,885,645–147,929,115)). Parental studies were requested but not performed.

Clinical photographs of the index patient. a-b Mild 2nd, 3rd toe syndactyly, eczema, recurrent onychomadesis, recurrent skin infections causing desquamation; c the patient at 2 years of age - sparse hair, eyelashes and eyebrows, depressed flat nasal bridge, hypoplastic alae nasi, thin vermillion border, mild epicanthus, ankyloblepharon and unilateral left cleft lip and palate s/p repair
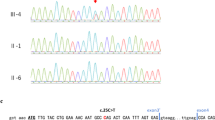
Clinical exome sequencing was performed on the affected individual with methods as previously published [16,17,18]. Variants were filtered to 1% minor allele frequency, then prioritized by the American College of Medical Genetics (ACMG) categorization [19], OMIM identity and phenotypic assessment. This individual was found to be compound heterozygous for a frameshift variant c.1365del (p.Arg457Aspfs*6) and a missense variant c.1388C > A (p.Thr463Lys) in CHUK (NM_001278.3) (Fig. 2a). Both variants are located in exon 13 and occurred in trans, as visualized by the Integrative Genomics Viewer (IGV) tool (Additional file 1: Figure S1) [20, 21]. Sanger sequencing confirmed that the p.Arg457Aspfs*6 variant was maternally-inherited and the p.Thr463Lys was not detected in either parental sample (Fig. 2b). Paternity was confirmed using short-tandem repeat analysis. This indicates that the p.Thr463Lys variant arose de novo, but germline mosaicism in the father can’t be excluded. These variants were absent from population databases.

CHUK pathogenic variants and segregation studies. a Family pedigree showing segregation studies; b Sanger sequencing indicates that the c.1365del variant was maternally-inherited. The c.1388C > A (p.Thr463Lys) was not detected in either parental sample. Paternity was confirmed, indicating that the c.1388C > A variant arose de novo
Discussion & conclusion
Pathogenic variants in CHUK have been reported in 3 families to date (Table 1; Additional file 2: Figure S2): In 2010, Lahtela et al., described a Finnish family in which a homozygous loss of function variant in CHUK (c.1264C > T; p.Gln422*) was associated with Cocoon syndrome, an autosomal recessive lethal condition characterized by severe fetal malformations. Prenatal ultrasound of 2 fetuses revealed an abnormal cyst in the cranial region, a large defect in the craniofacial area, an omphalocele and immotile and hypoplastic limbs. Abnormalities of the heart, lungs, skin, bones and skeletal muscles were also observed. Both parents were heterozygous for this variant and genealogical analysis revealed a common ancestor [22]. In 2015, Leslie et al., reported a homozygous variant in the splice acceptor site of exon 10 (c.934-2A > G) in a female patient with Bartsocas-Papas syndrome born to healthy first degree cousins. Clinical manifestations included alopecia totalis (with absent eyebrows and eyelashes), wide cranial suture and fontanelle, nose and ear dysmorphisms, bilateral microophthalmia, ankyloblepharon, bilateral cleft lip and palate, genital hypoplasia, popliteal webs and skeletal abnormalities [11]. In 2017, Khandelwal et al., reported a 10-year-old female born to non-consanguineous Caucasian parents with a de novo missense variant in CHUK (c.425A > G, p.His142Arg). Clinical features of the patient included sparse hair, absent eyebrows and eyelashes, ankyloblepharon and dysplastic nails. X-rays of the hands and feet showed complex anomalies consisting of, among others, hypoplastic thumbs and 3rd–5th toe syndactyly. Other features included posterior cleft palate, retrognathia, buccal synechia, hypoplastic external genitalia, conical and fragile primary teeth and short stature (height -3.5SD and weight -3SD). Her development was marked by growth retardation, gastrointestinal reflux with swallowing problems and lower respiratory tract infections. She also had hypogammaglobulinemia. To our knowledge, a second pathogenic variant was not detected in this patient, but additional screening methods such as deletion/duplication analysis were not performed [12].
The CHUK gene encodes for Ikka (Inhibitor of nuclear factor kappa-B kinase subunit alpha), a catalytic subunit of the multiprotein complex IbK kinase. Studies in mice show that the Ikka protein is ubiquitously expressed with the highest levels in the developing spine, limb buds and head. It plays an important role in limb development, apoptosis of interdigital tissue and proliferation and differentiation of epidermal keratinocytes. Embryos from the Ikka-deficient mice developed to term, but died shortly after birth. The fetuses displayed several skeletal abnormalities affecting the size and the morphology of the spine, skull, forepaws and the hindpaws. Limb bones were relatively smaller and of normal shape. Microscopic evaluation of the skin revealed hyperplasia of the suprabasal layer (stratum spinosum) and absent stratum granulosum and stratum corneum. Mice heterozygous for the CHUK gene deletion are normal, viable and fertile [23].
In this report, we describe a male patient presenting with an AEC syndrome-like phenotype and recurrent infections suggestive of immune deficiency. Targeted sequencing of TP63, RIPK4 and IRF6 was negative. Microarray analysis identified a 2 MB deletion on chromosome 1 covering the distal part of the 1q21.1 region deletion. Although this pathogenic deletion is unlikely to account for all the clinical features of the patient, it could contribute to his dysmorphic facial features, small size and failure to thrive (Fig. 1, Additional file 3: Figure S3). Additionally, exome sequencing revealed that he was compound heterozygous for two novel variants in CHUK. The c.1365del (p.Arg457Aspfs*6) frameshift variant, was inherited from his unaffected mother and the c.1388C > A (p.Thr463Lys) missense variant arose de novo. This genotype is compatible with autosomal recessive inheritance and consistent with previously reported families [11, 22]. To our knowledge, only one patient has been reported so far with a de novo missense variant in CHUK [12]. Since deletion/duplication testing was not performed, the presence of a second undetected variant cannot be ruled out. Interestingly, our patient shares several clinical features with this individual. However, skeletal defects appeared less severe in our patient and we cannot rule out progressive hypogammaglobulinemia needing Ig replacement at follow up. Therefore, based on our findings, it is unclear if, in some cases, the inheritance pattern could be dominant and that de novo heterozygous CHUK variants are sufficient to cause the disease. Even if the majority of pathogenic de novo variants are involved in dominant genetic disorders, there are growing examples of recessive disorders that can be caused by the combination of an inherited variant on one allele and a de novo variant on the other.
Abbreviations
- ACMG:
-
American College of Medical Genetics
- AEC:
-
Ankyloblepharon-ectodermal defects-cleft lip/palate syndrome
- BPS:
-
Bartsocas-Papas syndrome
- ED:
-
Ectodermal dysplasia
- EEC:
-
Ectrodactyly-ectodermal dysplasia-clefting syndrome
- Ig:
-
Immunoglobulin
- IGV:
-
Integrative Genomics Viewer
- NICU:
-
Neonatal Intensive Care Unit
- OMIM:
-
Online Mendelian Inheritance in Man
- PPS:
-
Popliteal pterygium syndrome
- RSV:
-
Respiratory syncytial virus
- VWS:
-
Van der Woude syndrome
References
Deshmukh S, Prashanth S. Ectodermal dysplasia: a genetic review. Int J Clin Pediatr Dent. 2012;5(3):197–202.
van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, et al. p63 gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001;69(3):481–92.
Rinne T, Bolat E, Meijer R, Scheffer H, van Bokhoven H. Spectrum of p63 mutations in a selected patient cohort affected with ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (AEC). Am J Med Genet A. 2009;149A(9):1948–51.
Sutton VR, van Bokhoven H. TP63-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, LJH B, Bird TD, Ledbetter N, Mefford HC, RJH S, et al., editors. GeneReviews(R). Seattle; 1993.
Koch PJ, Dinella J, Fete M, Siegfried EC, Koster MI. Modeling AEC-new approaches to study rare genetic disorders. Am J Med Genet A. 2014;164A(10):2443–54.
Sutton VR, Plunkett K, Dang DX, Lewis RA, Bree AF, Bacino CA. Craniofacial and anthropometric phenotype in ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (hay-wells syndrome) in a cohort of 17 patients. Am J Med Genet A. 2009;149A(9):1916–21.
Schutte BC, Saal HM, Goudy S, Leslie E. IRF6-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, LJH B, Bird TD, Ledbetter N, Mefford HC, RJH S, et al., editors. GeneReviews(R). Seattle; 1993.
Hall JG, Reed SD, Rosenbaum KN, Gershanik J, Chen H, Wilson KM. Limb pterygium syndromes: a review and report of eleven patients. Am J Med Genet. 1982;12(4):377–409.
Kalay E, Sezgin O, Chellappa V, Mutlu M, Morsy H, Kayserili H, Kreiger E, Cansu A, Toraman B, Abdalla EM, et al. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genet. 2012;90(1):76–85.
Mitchell K, O'Sullivan J, Missero C, Blair E, Richardson R, Anderson B, Antonini D, Murray JC, Shanske AL, Schutte BC, et al. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet. 2012;90(1):69–75.
Leslie EJ, O'Sullivan J, Cunningham ML, Singh A, Goudy SL, Ababneh F, Alsubaie L, Ch’ng GS, van der Laar IM, Hoogeboom AJ, et al. Expanding the genetic and phenotypic spectrum of popliteal pterygium disorders. Am J Med Genet A. 2015;167A(3):545–52.
Khandelwal KD, Ockeloen CW, Venselaar H, Boulanger C, Brichard B, Sokal E, Pfundt R, Rinne T, van Beusekom E, Bloemen M, et al. Identification of a de novo variant in CHUK in a patient with an EEC/AEC syndrome-like phenotype and hypogammaglobulinemia. Am J Med Genet A. 2017;
Digilio MC, Bernardini L, Consoli F, Lepri FR, Giuffrida MG, Baban A, Surace C, Ferese R, Angioni A, Novelli A, et al. Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur J Med Genet. 2013;56(3):144–9.
Haldeman-Englert CR, Jewett T. 1q21.1 recurrent microdeletion. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, LJH B, Bird TD, Ledbetter N, Mefford HC, RJH S, et al., editors. GeneReviews(R). Seattle; 1993.
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40(12):1466–71.
Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168.
Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, Andraws N, Patterson ML, Krivohlavek LA, Fellis J, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra135.
Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra64.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92.
Lahtela J, Nousiainen HO, Stefanovic V, Tallila J, Viskari H, Karikoski R, Gentile M, Saloranta C, Varilo T, Salonen R, et al. Mutant CHUK and severe fetal encasement malformation. N Engl J Med. 2010;363(17):1631–7.
Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284(5412):316–20.
Acknowledgements
Authors would like to thank the patient and the family involved in this study. We thank our colleagues in the Center for Pediatric Genomic Medicine, Children’s Mercy Kansas City.
Funding
This work was supported by the Marion Merrell Dow Foundation, Children’s Mercy - Kansas City, Patton Trust, W.T. Kemper Foundation, Pat & Gil Clements Foundation, Claire Giannini Foundation, and Black & Veatch.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: IT, MCD, CS. Performed the experiments: MCD, IT, CS. Contributed reagents/materials/analysis tools: NM, EF. Wrote the paper: MCD, IT. Contributed to the recruitment and clinical investigations of the patient for the study: NS, KE, NR, KC, LZ, ER. All authors reviewed and agreed to the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The project was approved by the research ethics committee of the Children’s Mercy Hospitals.
Consent for publication
Written informed consent for publication of this case, including age, relevant medical history, symptoms and full facial photographs, was obtained from the patient’s father.
Competing interests
Authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. DNA alignment of NGS data using Integrative Genomics Viewer (IGV). IGV snapshot of exon 13 of CHUK gene (NM_001278.3), located on chromosome 10, showing that the two variants (c.1365del, p.Arg457Aspfs*6; c.1388C > A, p.Thr463Lys) are present on different reads, indicating that they occurred on different chromosome (in trans). (TIFF 307 kb)
Additional file 2:
Figure S2. The protein is composed of a protein kinase domain (orange), a leucine zipper region (green) and the NEMO binding-region (blue). Missense (gray) and loss of function (red) pathogenic variants found in the CHUK gene. Reported in this study (Bold). (TIFF 128 kb)
Additional file 3:
Figure S3. Growth chart. A) Weight for age (kg). B) Length for age (cm). (TIFF 602 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cadieux-Dion, M., Safina, N.P., Engleman, K. et al. Novel heterozygous pathogenic variants in CHUK in a patient with AEC-like phenotype, immune deficiencies and 1q21.1 microdeletion syndrome: a case report. BMC Med Genet 19, 41 (2018). https://doi.org/10.1186/s12881-018-0556-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0556-2