
Abstract
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disorder characterized by the selective degeneration of defined subgroups of motoneuron in the brainstem, spinal cord and motor cortex with signature hallmarks of mitochondrial Ca2+ overload, free radical damage, excitotoxicity and impaired axonal transport. Although intracellular disruptions of cytosolic and mitochondrial calcium, and in particular low cytosolic calcium ([Ca2+]c) buffering and a strong interaction between metabolic mechanisms and [Ca2+]i have been identified predominantly in motoneuron impairment, the causes of these disruptions are unknown. The existing evidence suggests that the mutant superoxide dismutase1 (mtSOD1)-mediated toxicity in ALS acts through mitochondria, and that alteration in cytosolic and mitochondria-ER microdomain calcium accumulation are critical to the neurodegenerative process. Furthermore, chronic excitotoxcity mediated by Ca2+-permeable AMPA and NMDA receptors seems to initiate vicious cycle of intracellular calcium dysregulation which leads to toxic Ca2+ overload and thereby selective neurodegeneration. Recent advancement in the experimental analysis of calcium signals with high spatiotemporal precision has allowed investigations of calcium regulation in-vivo and in-vitro in different cell types, in particular selectively vulnerable/resistant cell types in different animal models of this motoneuron disease. This review provides an overview of latest advances in this field, and focuses on details of what has been learned about disrupted Ca2+ homeostasis and mitochondrial degeneration. It further emphasizes the critical role of mitochondria in preventing apoptosis by acting as a Ca2+ buffers, especially in motoneurons, in pathophysiological conditions such as ALS.
Similar content being viewed by others


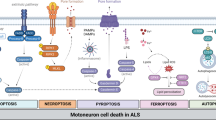
Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable, adult-onset, deadly neurodegenerative disorder distinguished by the progressive degeneration of a defined motoneuron (MN) population in the brain stem, spinal cord and motor cortex. ALS leads to paralysis, atrophy and death within 5 years [1]. Currently, there are no effective drug formulations for cures and riluzole is the only FDA approved treatment available for this devastating disease. The majority of ALS cases are sporadic, but ~10% of ALS cases are familial ALS (fALS). About 20% of these familial cases are caused by dominantly inherited mutations in the gene encoding the enzyme Cu/Zn-superoxide dismutase (SOD1) and are the most common [2]. Mutations in a number of other genes also cause familial ALS; including mutations in autosomal dominant familial MN disease include fALS types 3 [3], 5 [4], 6 or FUS gene [5, 6], 7 [7], 8 [8], 9 or ANG gene [9], 10 or TDP-43 (TARDBP gene) [10–12], 11 or Figure four gene [13], NF-H gene [14], DAO gene [15], X-linked [16], C90RF72 [17–19], alsin [20] and MND with FTD [21].
Unfortunately, the discovery of these mutant genes has not yet advanced into useful ALS model organisms, allowing most work described in literature have been conducted on mutant SOD1 cells, rodents or on patients (both familial and sporadic). Although the process of MN degeneration both in sporadic and familial forms is still little understood, it is generally agreed that there are cell-specific features, particularly impaired uptake of Ca2+ into mitochondria and low content of Ca2+ binding proteins which modulate physiological as well as pathophysiological processes and may render MNs selectively vulnerable to degeneration [22–25]. In both SOD1-mediated and sporadic sALS, reports demonstrate that mitochondrial alterations, including morphological changes, enhanced activity of complexes I, III, and IV, and increased reactive oxygen species (ROS) generation, are key factors in its pathogenesis [8, 26–30]. These factors are further exacerbated by observations indicating that the impaired spinal cord and vulnerable spinal cord neurons trigger the functional decline of MNs in neighboring regions, leading to the onset of pathology in ALS [31–34]. The breakdown of mitochondrial membrane potential (ΔΨm), excitotoxic stimulation of AMPA/kainite receptors and age associated MN damage reported by several groups may also be a factor to ALS pathogenesis [35–38].
Impaired intracellular Ca2+ homeostasis, rather than direct mitochondrial disruption, is supported by observations that neurons lost early in the disease progression have intrinsically poor cytoplasmic Ca2+ buffering capabilities, due to the absence of Ca2+ binding proteins such as calbindin D28K (CB-D28K) and parvalbumin (PV) [22, 36]. These findings are in agreement with observations that low cytosolic Ca2+ buffering ability acts as a primary risk factor for MN deterioration, while increases in cytosolic Ca2+ buffering capacity protects vulnerable MNs from degeneration, both in-vitro and in-vivo[25, 36, 39, 40]. In addition to the mechanisms of Ca2+ toxicity present in most cell types, there are several other characteristics of neurons that make them especially vulnerable. Disturbances of glutamate-mediated neurotransmission and the subsequent glutamate release triggered by Ca2+ entry [41] increase extracellular glutamate levels leading to excitotoxicity. This problem is further compounded by reduced glial glutamate uptake caused by oxidative damage to excitatory amino acid transporter 2 (EAAT2) [42]. Studies of fALS in cell lines and in various mouse models that induce Ca2+ disturbance via the inhibition of glial glutamate transport by mtSOD1 yield phenotypes similar to effects proposed in sALS [43]. In addition, in cell culture experiments, partial protection was also obtained by treatment with the Ca2+channel-blocker nifedipine, implicating Ca2+ entry through voltage-gated Ca2+ channels in mediating the toxicity of mtSOD1G93A in MNs [44]. In addition to damaging the cell of origin, ROS generated in MNs can cross the plasma membrane and damage glutamate transporters in neighboring astrocytes [41, 45]. Further evidence also indicates that the patients with minor SOD1 mutations had the shortest disease durations. In other words, an accelerated disease course is found for the mutants that are more functionally impaired. Since hydrogen peroxide plays an essential role in the formation of SOD1 monomers, dissociation of the dimeric SOD1 molecule monomers should be as essential important process for APS pathogenesis [46].
However, in ALS pathology, the cause of dysfunction has become highly contentious after latest discoveries where ALS astrocytes were shown to secrete substances that are selectively toxic to MNs [47, 48]. This means that multiple cell types are involved in the disease process. By adding or deleting mtSOD1 in specific cell types e.g. astrocytes [49, 50], microglia [51], schwann cells [52], motor neurons [53] and fibroblast [54] it is possible to influence the disease. Nonetheless, MNs are the cells that directly cause the loss of muscle and limb movement. It is shown that mtSOD1 expressed solely in MNs is sufficient to kick off the disease, although disease progression is slow compared to ubiquitous expression of mtSOD1 [53]. In patients, MNs in the motor cortex, HMNs and FMNs of brain stem and spinal cord undergo cell death selectively. There are a number of hypothesis that explain cell type selectivity, including the extraordinary long axons, the large soma and the poor intracellular calcium buffering capacity and excitotoxicity. It is the latter hypothesis that shall be elaborated on in this review, the only mechanism proved to play a role in patients. Calcium, which causes neuronal death in excitotoxicity, can originate from either the extracellular space or from intracellular stores. In general resting calcium in the extracellular space is ~ 3–4 orders of magnitude higher than in the intracellular stores [55]. As reported previously, MNs are excited by glutamate from the pre-synaptic neuron that binds to glutamate receptors on the postsynaptic MN, which are, among others, the AMPA receptors (AMPAR). The AMPAR found on MNs are mainly calcium permeable in vitro and in-vivo [56–59], which may explain the selective vulnerability of MNs to excitotoxic cell death. In addition, extracellular calcium entry via these calcium permeable AMPAR is responsible for selective MN death, as MN death is inhibited by selective blockers of calcium permeable AMPAR[57, 58]. Also, electrophysiological experiments showed that the AMPAR receptors of MNs had a lower rectification index and a higher relative calcium permeability ratio than other neurons [59]. In conclusion, MNs express calcium permeable AMPAR receptors, which could partially explain their pronounced and selective vulnerability to excitotoxic insults [59]. Further explanation for selective MN vulnerability in this cell type has to deal with the increased amount of intracellular calcium. Studies into the calcium homeostasis in MNs have shown a diminished calcium buffering capacity distinguishing ALS-vulnerable from resistant MN types. To begin with, ALS-vulnerable spinal and brain stem MNs in mice display a low endogenous calcium buffering capacity as demonstrated by patch clamp and microfluorometirc calcium measurements [60, 61]. In addition, ALS-resistant oculomotor neurons contain a larger calcium buffering capacity than ALS-vulnerable MNs, as measured by similar microfluorometirc calcium measurements [62].
The kinetics of calcium signals in MNs are shaped by multiple mechanisms including Ca2+ influx, Ca2+ uptake and release phenomenon, MN specific Ca2+ buffering and extrusion across cellular membrane and microdomains in vicinity [63, 64]. Over the past decade, there has been an increased understanding of local communications between Ca2+ microdomains and its upstream and downstream target signaling pathways. In particular, an impaired interaction between calcium signaling and mitochondrial processes has been identified as one cellular factor contributing to neurodegenerative processes like those found in motoneuron diseases. In addition, previous studies also indicate the importance of calcium and mitochondria for normal physiological function of MNs [62, 65]. Under pathophysiological conditions, low [Ca2+]i buffering and a robust interaction between metabolic processess and [Ca2+]i have been related with selective and severe MN damage resulting from excitotoxic stress and disruptions of cellular and mitochondrial Ca2+ homeostasis [66, 67]. Considering the prime participation of mitochondria not only in calcium homeostasis directly but also in the energy transduction (to operate other Ca2+ clearing mechanisms) and in enacting apoptosis, this aspect of cellular function is of immense importance.
The influence of mitochondrial dysfunction on intracellular Ca2+ homeostasis and its role in MN death are fascinating issues that warrant in depth debate. Despite rigorous research, since description of ALS by Charcot ~ 130 years ago, the cellular and molecular abnormalities, which lead to loss of specific MNs, are still dodging the scientific community. Obviously, unraveling the connecting association between mitochondrial dysfunction, calcium dysregulation, and neuronal demise is vital for the understanding of ALS pathogenesis. The aim of this review is to discuss and determine the role of mtSOD1 toxicity in cellular Ca2+ homeostasis and mitochondrial dysfunction in MNs of ALS. This review will focus on what has been learned about motoneuron specific calcium dysregulation and perturbed cellular calcium homestasis in ALS from genetically modified animals and cell culture models. Taken together, this review proposes an intregative view, describing mechanisms and critical elements of the pathology of mtSOD1-mediated motoneuron degeneration in ALS.
Review
Disease mechanism in amyotrophic lateral sclerosis
Many pathophysiological mechanisms have been suggested to play a role in the etiology of ALS. Corresponding to clinical features, ALS is characterized by a progressive loss of spinal, brains stem and cortical MNs. MN damage as a result of oxidative stress and excitotoxicity is one of the key hypothesis in ALS etiology. The present evidence also supports the hypothesis of mitochondrial dysfunction acting with oxidative stress to cause neurodegeneration via apoptotic mechanisms. Oxidative stress is also linked with other suggested disease mechanisms such as excitotoxicity causing an increase in [Ca2+]i, which subsequentially leads to increased nitric oxide formation. Peroxynitrite, produced by the reaction of superoxide anions and nitric oxide, can consequently lead to oxidative disruption [42]. Glutamate excitotoxicity is another mechanism implicated in ALS pathogenesis through disruption of [Ca2+]i homeostasis and free radical production. For example, in human ALS (hALS), overexpression of glutamatergic synapses leading to excessive Ca2+ influx has been linked with MN degeneration [68]. The oxidative stress manifested in ALS might also prop up increased excitotoxicity, as glutamate transporters are predominantly susceptible to disruption by oxidants, and oxidative modifications to the transporters have been reported in ALS and the mtSOD1 mouse model [69]. The roles of individual cellular domains at the organelle level suggest that high calcium buffering enhances MNs vulnerability [70, 71]. In summary, ALS comprises the interplay of numerous mechanisms from initiation and spread of MN cell death by mitochondrial dysfunction and/or by enhanced MN excitability by intracellular calcium overload. Therefore the etiology of the disease is most likely to be multifactorial [72, 73]. This article will focus on and further discuss the hypothesis and key mechanism that have been most influential in the present and past decade of ALS research.
Oxidative stress sensitivity and mitochondrial dysfunction
Oxidative stress occurs from an imbalance between the production of ROS and the ability of the system to remove ROS or repair the damage caused because of it, and to reinstate the existing reducing ambiance [68]. Oxidative stress and mitochondrial dysfunction are implicated in the pathogenesis of both normal aging and neurodegenerative diseases. There is ongoing debate as to whether oxidative stress is a primary cause of degeneration or it is merely an end result of some other toxic insult. Although oxidative stress is implausible to be the solitary cause of disease initiation, in several cases it may be enhance vulnerability of homeostatic control mechanisms from handling with a toxic insult into a vicious cycle of cellular insults that results in MN degeneration. MN damage as a consequence of oxidative stress is supposed to be the key premise in ALS. A number of studies have established the existence of elevated oxidative metabolism in ALS, for example finding of increased biochemical markers of oxidative injury in post-mortem samples from ALS patients [38]. Free radical scavenging proteins like SOD1, mitochondrial manganese SOD (SOD2), catalase, and cytochrome c can neutralize free radicals but can not prevent cellular damage by ROS [38, 72]. Increased .OH generation may occur as an end result of either enhanced peroxidase activity or decreased Cu-binding affinity of mtSOD1. mtSOD1 transgenic (Tg) mice show elevated levels of protein and lipid oxidation at both pre- and post-symptomatic stages of MN dysfunction [74, 75]. Oxidative stress is also associated with many other proposed disease mechanisms such as excitotoxicity and axonal transport defects [76–80]. Studies suggest that ROS generated in MNs can cross the plasma membrane to produce oxidative damage to glutamate transporters in neighboring astrocytes via the excitotoxic stimulation of AMPA/kainate receptors, followed by locally restricted excitotoxicity. This initiates a vicious cycle of MN overactivation causing damage, which in turn further activates MNs [37, 46, 62].
Morphological and ultrastructural abnormalities observed in mitochondria of both, sporadic and familial forms of ALS point towards a crucial involvement of mitochondria in ALS [81–84]. Localization and aggregates of SOD1 in mitochondria of transgenic mouse models were shown previously [85–87]. There are now several observations suggest that mitochondria play a crucial role in disturbing energy metabolism by predisposing calcium-mediated excitotoxicity, leading to ROS generation and initiation of the apoptotic pathway, thereby jeopardizing cell function and normal cellular metabolism [88, 89]. Defects in mitochondrial function have been found in fALS and some sALS by histopathological observations of mitochondrial swelling and vacuolization in ALS transgenic mouse models and in ALS patients [28, 30, 31, 39, 62, 90]; Figure 1A. Morphological abnormalities were not only confined to CNS, but were also found in skeletal muscles, intramuscular nerve fibers and proximal horns of the spinal cord [91–93]. Recently, in a SOD1-transfected cell culture model of MN disease, our laboratory has shown impairement of mitochondrial calcium handling and impaired cross-talk between mitochondria-endoplasmic reticulum (ER) microdomains [94]; Figure 1B-D. Many other studies have recently focused on mitochondrial dysfunction, specifically on the increased activity of the mitochondrial respiratory chain necessary for ATP synthesis, resulting in an increased ROS production. Furthermore, the excessive electron transport chain activity can deplete energy stores, resulting in the loss of integrity of neuronal cell membranes and leaving them permeable to ions and water which can cause damage. Deficits in the activities of complex I and complex IV, as a result of mutations in mitochondrial DNA, have been identified in the skeletal muscles and spinal cord of sALS patients [33, 95, 96].

Mitochondrial structure of motor neurons in mutant SOD1 transgenic mice and calcium load in microdomains in a cell culture model of motoneuron disease. (A) a, Shows abnormalities like dilated cristae (asterisk) and leaking outer membrane (indicated with arrow) in mitochondrion. (A) b, Swollen dendritic mitochondria with dilated and disorganized cristae (adapted from ref. 31). (B-D) The simultaneous measurement of cytosolic calcium (Fura-2) and mitochondrial calcium (Rhod-2) concentrations in WT and G93A transfected SH-SY5Y cells during FCCP-evoked mitochondrial Ca2+ release. (B) The kinetic profile of the FCCP-evoked Ca2+ release in the WT transfected SH-SY5Y neuroblastoma cells; the cytosolic (Error bar green, black square trace) and mitochondrial (Error bar red, black circle trace) compartment were measured simultaneously. The trace represents the mean of 5 cells in focus stimulated with 2 μM FCCP. (C) The corresponding kinetic profile of the FCCP-evoked Ca2+ release in the G93A transfected SH-SY5Y neuroblastoma cells; the cytosolic (Error bar green, black square trace) and mitochondrial (Error bar red, black circle trace) compartment were measured simultaneously. The trace represents the mean of 5 cells in focus stimulated with 2 μM FCCP. FCCP-evoked [Ca2+]mito signals were smaller in amplitude and exhibited slower kinetics in G93A transfected SH-SY5Y cells compared to WT transfected cells and were altered from [Ca2+]i efflux. (D) A bar diagram of the cytosolic (green bar) and mitochondrial (red bar) fluorescence signals (F/F0) from WT (F/F0 = 0.1569 ± 0.0235 for [Ca2+]i and F/F0 = −0.1069 ± 0.0181 for [Ca2+] mito; hollow; N = 5, n = 17) and G93A (F/F0 = 0.1008 ± 0.0248 for [Ca2+]i and F/F0 = −0.0486 ± 0.0043 for[Ca2+]mito; striped pattern, N = 4; n = 17) transfected SH-SY5Y neuroblastoma cells. Values represent means ± SD, **p < 0.001. N = Number of experiments; n = Number of cells (adapted from ref. 94).
The query of whether changes in the mitochondrial genome can cause variations in mitochondrial function has been addressed by transferring mitochondrial DNA from ALS subjects to mitochondrial DNA-depleted human neuroblastoma cells. This manipulation resulted in anomalous electron transport chain function, increases in the activity of free radical scavenging enzymes, disturbed Ca2+ homeostasis and altered mitochondrial structure, suggesting a pathophysiological role for mitochondrial DNA mutations in different forms of ALS [94, 97]. A striking recent set of publications provides evidence that mtSOD1 might disrupt the association of respiratory complex IV (cytochrome c) with the inner mitochondrial membrane, thus obstructing the mitochondrial respiratory system. Cultured MNs expressing mtSOD1 and MNs in ex-vivo brain slices where respiratory chain complex IV was blocked by cyanide or azide also shows mitochondrial involvement [98–100].
The major question of whether mitochondrial anomalies are involved in the disease progression or simply a derivative of neuronal degeneration is still far from over. Pathological features like the occurrence of membrane-bound vacuoles in MNs in Tg mice expressing G93A or G37R suggest that mitochondrial alterations are an early consequence eliciting the beginning of the disease, instead of merely a derivative of neuronal degeneration [101, 102]. Mitochondrial vacuolization occurs by detachment of the outer membrane from the inner membrane and increase of the intermembrane space, confirmed by biomarkers studies for mitochondrial compartments. After membrane expansion, mature vacuoles form which leads to the inner membrane disintegrations [103, 104]. A recent publication demonstrates the localization of a significant fraction of SOD1 in intermitochondrial space thereby causing toxicity. Inhibition of mitochondrial respiratory metabolism is reported in Tg ALS mice models [105, 106]. Certainly, MNs are highly susceptible to mitochondrial damage. Studies using mitochondrial respiratory chain inhibition by cyanide and azide result in selective MN death, which can be counteracted by ROS scavengers and AMPAR blockers [62, 107]. Furthermore, ALS-like symptoms can be induced by deletion of vascular endothelial-cell growth factor (VEGF) that eliminates the ability to respond to tissue mild and chronic hypoxia [108–110]. Cross-breeding these mice with the mtSOD1 severely enhanced MN degeneration, while treatment of SOD1-Tg mice with VEGF hindered progression of disease symptoms and extend mice survival [62, 111–117].
Characteristically low Ca2+ buffering capacity of motoneurons and its impact on selective motoneuron vulnerability in amyotrophic lateral sclerosis
Several groups have reported that, the disruption of intracellular Ca2+ homeostasis plays a prominent role in the etiology of ALS. The involvement of Ca2+ as a risk factor was suggested by the observation that Ca2+-binding proteins such as CB-D28k and PV were absent in MN populations lost early in ALS. In contrast, MNs less prone to damage expressed markedly higher levels of calcium-binding proteins CB-D28k and/or PV [22, 62], and were relatively insensitive to mitochondrial calcium buffering. In dorsal vagal neurons, which contain an abundance of Ca2+ sequestering proteins [118], the delay in the decay time constant (τ) of Ca2+ transients (FCCP influx) is not caused by mitochondrial permeability. This observation identified a low cytosolic Ca2+ buffering capacity as an important risk factor for MN degeneration. Data from different groups shows that the vulnerable populations of MNs display low endogenous calcium buffering capacity [119], due to low expression levels of Ca2+-buffering proteins. Although potentially essential under physiological conditions, as it allows for rapid Ca2+ transients relaxation times during high frequency rhythmic activity, these characteristics make MNs more susceptible to an excessive influx of Ca2+ ions. This susceptibility increases the risk of activation of excitotoxic second messenger cascades and related cellular damages [62, 119]. Another argument in favor of this hypothesis is that high concentrations of mobile buffers accelerate the distribution of local Ca2+ gradients by a mechanism known as buffering diffusion (Figure 2A, B). According to this concept, under pathophysiological conditions, differential buffering reflects a basic diversity in the spatio-temporal organization of Ca2+ signaling rather than a singular difference in single cellular parameter [120–122]. Likewise, an increase in [Ca2+]i buffering capacity could defend vulnerable MNs and protect from degeneration both in vitro and in-vivo[25, 123].

Ca2+homeostasis and its correlation with weakly and strongly buffered motoneurons under physiological and pathophysiological conditions. (A) The Ca2+ buffering capacity (KS) of a cell, reflecting relative fraction of bound versus free Ca2+, can be calculated by using the ‘added buffer’ approach by linear one-compartment model. The recovery time of [Ca2+]i elevations (τ) depends on the amount of endogenous buffer (S; denotes Ca2+-binding proteins), the amount of exogenous buffer (B; i.e. Fura-2) and the transport rate (γ) of Ca2+ across cellular membranes. KB indicates the buffer capacity of the exogenous buffer (i.e. Fura-2). (B) Ca2+ homeostasis in weakly and strongly buffered MNs. The amplitude of Ca2+ transients is several times larger in weakly buffered cells (e.g. HMNS and SMNs) than in strongly buffered cells (e.g. oculomotor neurons), and the recovery time is significantly accelerated (τ). (C) Low Ca2+ buffering in ALS-vulnerable HMNs exposes mitochondria to higher Ca2+ loads compared to high-buffered cells. Under normal physiological conditions the neurotransmitter opens glutamate, NMDA and AMPA receptor channels along with VDCC with high glutamate release and reuptake by EAAT1 and EAAT2. This results in a small rise in intracellular calcium that can be buffered by the cell. In ALS disorder, the glutamate receptor channels possess high calcium conductivity and thereby high Ca2+ loads; increase the risk for mitochondrial damage. This triggers mitochondrial production of reactive oxygen species (ROS), which then inhibit glial EAAT2 function. This leads to further increase in glutamate concentrations in the synapse and further rises in postsynaptic calcium levels which contributes to the selective vulnerability of MNs in ALS. Low cytosolic Ca2+ buffering capacity promotes Ca2+ accumulation and formation of subcellular domains around influx sites (red), and thus facilitates the interaction of elevated calcium levels with intracellular organelles such as mitochondria (modified from refs. [62, 63, 73, 94, 118]).
In order to check whether cytosolic [Ca2+]i buffering can protect cells from dysfunction and degeneration, our group performed Ca2+ imaging studies following a depolarization stimulus (60 mM K+) in primary neuronal cells obtained from mice cortex at E18 expressing low and high CB-D28k. Results show that indeed CB-D28k buffer [Ca2+]i where low CB-D28k transfected neuronal cells display a significant (~2 times) reduction in the peak amplitude of the sustained [Ca2+]i increase compare to high CB-D28k transfected neuronal cells (data not shown). τ in CB-D28k transfected cells is also slower (~60s) compared to non-transfected cells where baseline recovery time is ~30-35 s while showing little differences in the area under the time - concentration curve (AUC). This observation is in good agreement with the ∁low buffering hypothesis” which states that low buffer capacity allows for rapid Ca2+ dynamics during physiological activity, but represents a significant risk factor during ALS-related MN disease [124]. The observation of high cytosolic buffering capacity in selectively resistant MNs is consistent with earlier immunocytochemical studies of endogenous calcium buffering proteins. Moreover, in-vitro cell culture models have shown that elevated [Ca2+]i buffer concentration reduces ALS specific MN damage providing further support in favor of the notion that increased buffer concentrations create beneficial protection [125]. The low Ca2+ buffering properties synergize with a high AMPA/kainate current density to clarify the susceptibility of MNs to increased stimulation by glutamate and associated Ca2+ influx [59, 126]. Earlier observation also indicates interactions of mitochondrial proteins e.g. VAPBP56S and PTPIP51 implicated in regulating calcium homestasis [127, 128].
In the absence of adequate cytoplasmic buffering, or in cases where the existing cytoplasmic buffering system is overwhelmed, there is a shift in mitochondrial Ca2+ uptake from a beneficial physiological regulatory mechanism to a possibly detrimental process leading towards MNs death. In this process MNs death is clearly Ca2+ dependent. The downstream mechanism that links the rise in [Ca2+]i to MNs death is not clear though these processes have been mostly credited to mitochondrial Ca2+ accumulation or glutamate excitotoxicity. Whether the change in ΔΨm is simply an inevitable consequence of an unusually enormous Ca2+ load or there is other downstream pathway contributing to glutamate response is not known yet, however depolarisation of ΔΨm is one consequences of it. Data also suggested the role for other factor in addition to Ca2+ such as nitric oxide synthase (NOS) inhibitors. Results also suggest that mitochondrial damage arbitrated at least in part by Ca2+ induced MPTP opening and may contribute to surge of neighbouring cell activation. In conclusion, we believe that two of the most important features for MNs in ALS are: (i) low buffer capacity generates exceptionally large Ca2+ domains, but not in case of serious end stage ALS like symptoms and (ii) in ALS vulnerable MNs buffering capacity critically depends upon the domain size of mitochondria and ER. Therefore, we proposed a model where a portion of MN mitochondria interacts with areas of high [Ca2+] around influx sites due to low buffering induced excitotoxicity shown in Figure 2C.
Another proposed factor for calcium disregulation could be an ALS-related immune response targeted at voltage-dependent calcium channels (VDCC), where a disruption of Ca2+ homeostasis results from compromised voltage-dependent calcium influx [129]. Furthermore, synaptic glutamate transport is also believed to be involved in other forms of ALS and related MN neurodegeneration. In cell-culture, limited protection was achieved by treatment with nifedipine, implicating Ca2+ entry through voltage-gated Ca2+ channels, in addition to glutamate receptors, in mediating the toxicity of mtSOD1 in MNs. The crucial role of Ca2+-permeable AMPAR was further emphasized by cross-breeding of Tg SOD1 mice with mice that exhibited markedly reduced Ca2+ permeability of AMPA/kainate receptors, due to GluR2 overexpression [45, 126]. Finally, impaired mitochondrial calcium transport capacity in mtSOD1 mice may play an important role. Firstly, it links mitochondrial dysfunction to glutamate excitotoxicity and secondly, elevation of [Ca2+]c concentrations in neurons compromises mitochondrial integrity and function by inducing enhanced production of ROS from mitochondria [130]; Figure 2C.
Glutamate transmission and excitotoxicity
Glutamate is known as the predominant excitatory neurotransmitter in the CNS acting at both ionotropic and metabotropic receptors. It is synthesized and stowed in synaptic nerve components and released in response to depolarization of the neuron. Excessive glutamate exposure is toxic to neurons via glutamate-triggered Ca2+ influx [73, 131]. Several lines of proof implicate increases in glutamate neurotransmission and glutamate-triggered Ca2+ entry as significant ALS risk-factors. Increased extracellular glutamate levels result from reduced glial glutamate uptake due to oxidative damage to excitatory amino acid transporter 2 (EAAT2) or by aberrations in its production. Increased glutamate levels in the cerebrospinal fluid (CSF) of a subset of ALS patients have been shown by many groups. The elevation of this glutamate level may be attributed to deficient glutamate transporter capacity (loss of EAAT2 function), as low levels of the transport protein have been found in some post mortem ALS brains [66, 73, 132]. Furthermore, glutamate uptake inhibitors causes selective MN damage in organotypic slices [66] and in dissociated spinal cord culture [133], suggesting that reduction of glutamate transport could contribute to the MN damage seen in the ALS disease. The leading argument for a role of glutamate excitotoxicity in ALS is the efficacy of FDA approved drug riluzole, the lone drug which proved effective against disease progression in patients and has anti-excitotoxic properties. It was shown that riluzole inhibits the release of glutamate through the inactivation of voltage-dependent Na+ channels on glutamatergic nerve terminals as well as to activate a G-protein-dependent signal transduction cascades.
In-vivo evidence for a possible role of GluR1-4 (AMPA receptor subunits) in ALS comes from several studies. Transgenic mice lacking GluR2 (GluR2 subunit is a component in the AMPA receptor complex, which renders them particularly impermeable to calcium) do not hurt from MN disease. This suggests that a low GluR2 level is a modifier of MN degeneration rather than being sufficient to cause ALS [134]. Furthermore, glutamate excitotoxicity in sALS is caused by a selective loss of astrocytic glutamate transporter-1 (GLT-1) and is reproduced in mice by knockout of GLT-1, a homologue of EAAT-2 [135]. Oral administration of glutamate inhibitors prolonged the life span of SOD1G93A mice [136]. Further studies have pointed to the significance of GluR2 in neuronal survival, in which alterations in RNA editing at the Q/R site lead to the generation of a lethal phenotype involving seizure and acute neurodegeneration [137]. Furthermore, Glu2-N overexpression induces a progressive decline in the function of spinal cord, most likely due to the long-onset degeneration of spinal MNs [138].
Mechanism underlying mitochondria-ER Ca2+ stores coupling
Diverse microdoamin intracellular pools contribute in causing Ca2+ signals in neuronal cells and in shaping their spatio-temporal patterns and cell fate. Kinetic and “hot spot” hypothesis of mitochondria, different channels with distinct properties and highly defined expression patterns on ER are all capable of regulating [Ca2+]i in many systems [139, 140]. In an attempt to understand more about the Ca2+ metabolism of hypoglossal MNs, Jaiswal and colleagues studied the role of the ER in Ca2+ handling where it was shown that the ER in MNs retained a comparatively lower quantity of calcium than mitochondria after [Ca2+]i elevation, indicating a relative inability to sequester Ca2+ in the MNs of SOD1G93A mice as compared to WT lettermates. These results indicate that the conventional Ca2+ storing function of mitochondria is dominating over ER Ca2+ accumulation in these MNs. These results are in good agreement with the “hotspot” hypothesis that suggests that mitochondria preferentially accumulate Ca2+ at microdomains of elevated Ca2+concentration ([Ca2+]i), predominantly near ER Ca2+ release sites and other Ca2+ channels. Accordingly, mitochondria can affect both Ca2+ release from the ER and capacitative Ca2+ entry across the plasma membrane, thereby shaping the size and duration of the intracellular Ca2+ signal in MNs of WT and SOD1G93A mice. These events determined by the Ca2+ sensitivity of the Ca2+ channels and capability of mitochondria to remove Ca2+ from the subcellular microdomain at the opening of the ion channel. This effect has been confirmed in-vitro, but the condition appears markedly different in various cell models [141, 142]. This indicates that several modulatory mechanisms occur, many of which still await reasonable clarification at cellular and molecular level. As discussed above, vast evidence supports the notion that the measured high degree of Ca2+ accumulation of MNs mitochondria in-situ mainly influenced by the vicinity of mitochondria to the ion channels through which Ca2+ enters the cytosol. A fundamental, but still unanswered, question is the precise mechanism by which stochastic versus specific localization of Ca2+ influx occur and the amount to which mitochondrial function differs within different cell types, are critical quest in the field of ALS research.
Mitochondrial dysfunction, Ca2+ homeostasis and ALS: a multifactorial disease mechanism
The Ca2+-dependent signaling mechanisms that result in the enhanced vulnerability of MNs in ALS disease and associated mouse models are also critical for normal cellular function. Earlier studies suggest that unrestrained Ca2+ entry compounded with an inability to sequester this calcium leads to the degeneration of mitochondria in the MNs of the mouse model of ALS [29, 31]. In other MNs types have a low Ca2+-buffering capacity because of small concentrations of Ca2+-buffering proteins and a high quantity of Ca2+-permeable AMPAR[126, 143–146]. These two properties appear to be specific to MNs and are possibly vital for their normal function. However, this increased sensitivity to calcium also means that MNs are more easily over stimulated by glutamate and astounded by Ca2+. It is unknown whether Ca2+ dependent pathways are different in MNs. More recently, focus was shifted to the role of mitochondria as an effective regulator of [Ca2+]i signals [147, 148]. The use of mitochondria-targeted Ca2+ probes reveals a fast, intense surge in free intra-mitochondrial Ca2+ upon cellular stimulation. Increased Ca2+ uptake by mitochondria leads to up-regulation of the enzymes activity in oxidative metabolism, resulting in cell-specific metabolic changes [149–151]. This hypothesis is further strengthened by the appearance of abnormalities in mitochondrial ultrastructure and vacuoles formation resulted from degenerating mitochondria found in post mortem samples of ALS [28–31]. Even though the precise molecular mechanism is still not known, we hypothesize that MN vulnerability in ALS is a outcome of physiological features, mainly highly specialized Ca2+ surroundings that are required for appropriate neuronal function, uninterrupted activity-dependent mitochondria-ER Ca2+ cycling and the leading role of mitochondria in buffering Ca2+ transients.
In vulnerable MNs, low instrinsic cytosolic Ca2+ buffering requires that mitochondria play the dominant role in the regulation of [Ca2+]i transients; even during small cytosolic Ca2+ increases, mitochondria are known to take up more than 50% of intracellular Ca2+. Outsized and enduring Ca2+ microdomains around Ca2+ influx sites increase the danger of toxic Ca2+ buildups and a successive activation of Ca2+ dependent neurodegenerative pathways under excitotoxic conditions. Excitotoxicity associated with these influx domains is primarily suppressed by mitochondrial contributions, rather than ER Ca2+ uptake [36, 94, 99]. The prominent role of mitochondria in regulating adequate Ca2+ loads in MNs has significant implications for pathological conditions such as in ALS. First, the quantity of Ca2+ taken up by the mitochondria is greater in MNs than in many others cell types and therefore MN mitochondria are more susceptible to Ca2+ mediated damage, such as ROS generation [37, 46, 152]. Second, our experiments provide indication that the restriction of cytosolic Ca2+ to non-pathoological levels is determined by intact Ca2+ uptake into mitochondria. Hence, when mitochondrial Ca2+ uptake is disturbed as seen by low Ca2+ uptake in SOD1G93A MNs compare to WT, MNs are directly endangered by elevated Ca2+ levels, especially during high-frequency, repetitive Ca2+ oscillations. The reduced capability to limit Ca2+ transient amplitudes in the cytosol, particularly in local microdomains of extraordinary Ca2+ influx when mitochondria are depolarised, heightens the hazard of initiating Ca2+ dependent neurodegenerative pathways leading to cell demise.
Combining the lessons learned from multiple animal and cell culture models of ALS, the central insight is that mitochondrial dysfunction and Ca2+ homeostasis [152, 153] are strong contributors to the selective vulnerability of MNs. Considering the association and significance of mitochondrial dysfunction and Ca2+ homeostasis, we postulate that MN possess large number of voltage and ligand gated Ca2+ channels that, when activated, cause rapid Ca2+ influx. Since cytoplasmic Ca2+ buffering is relatively weak in these cell types, significant demand is placed on MN mitochondria. If the mitochondria are already damaged or weakened, this results in mitochondrial Ca2+ overload and ROS production and in some cases prone for infection [154]. Furthermore, long-lasting ΔΨm depolarisation due to Ca2+ entry can be a basis for the release of pro-apoptotic proteins and activate enzymes involved in apoptotic pathways [155, 156].
Multidrug therapies in ALS: where should we focus for the treatment?
Even though scientific discoveries are speeding up with an exceptional pace and years of experimentation using cell cultures, mice and rat made Tg for human mtSOD1 has yielded precious data about the mechanisms that underlie ALS as well as suggestions for therapy, to date approaximately ~50 clinical trials have failed and ended with disappointment and frustration. The main cause for the failure to translate experimentation in animals to therapies for patients is that there are too many compounds that increase life span in mice and rats, but fail to improve human patients condition. To increase the chances of success for future clinical trials we might consider the fact that ALS is not simply a multifactorial disease but also a multisystemic disease that is the consequence of a complex neurotoxic mechanism that involves molecular and cellular cross-talk between MNs, glia and astrocytes. Prelinical trials have not yet resulted in favorable outcomes, due to the fact that data are generally collected in animals of the identical age and with the same mutation expressed in a homogeneous genetic background whereas the age of beginning of disease, the progression and the severity of ALS in human patients are heterogeneous, signifying that possible genetic risk factors and modifying causes exist for sALS and certain drugs are effective only if given before onset of the disease [157–159]. In addition, the trial suffers from the deficiency of presumed negative clinical data set, which is different from the null result (i.e. data that do not affect the outcome). Interestingly, in most of the clinical trials, a subset of the patient population showed better condition with the possibility that each clinical trial has been successful within only a select subset of the patient population [158].
It is also important to keep in mind that ALS is a multifactorial disease, and it might be unrealistic to envision that one drug will have a broad spectrum of efficacy on pathologies that are widespread and, at times unrelated. In view of recent findings of a non-cell-autonomous demise of MNs [47, 48], design of multi drug combination therapies should be targeted at the intersection of various aspects of this cascade, rather than a single-drug cure [157, 158]. We suggest that the forthcoming clinical trials should include combinatorial studies, and that patients who display progress in their condition should not be considered as ‘outliers’, because they might indeed represent the target population, especially for the drugs tested [157]. Until now animal studies have shown that multi-drug combination therapies and the method of delivery of a drug is also often have synergistic effects in ALS, for example, riluzole administered with melatonin and vitamine E (inhibits Na+-current activation and the apoptotic cascade), minocycline administered with creatine (inhibits microglia activation and the apoptotic signaling cascade), or treatment with IGF-1 or VEGF retrogradely transported in MNs through viral vectors [111–119, 136, 158–163]. A substitute to pharmacological cures, the latest developments in stem-cell therapy might offer possibilities for neural grafting in patients with ALS [114, 158, 164]. The identification and characterization of early detection markers in ALS, and the establishment of dependable biomarkers for disease progression in a select set of clinical trials studies on blood, plasma and cerebrospinal fluid (CSF), obtained from patients and control subjects bounds to improved improved clinical trials [165]. The lessons learned from a decade of research using the mtSOD1 animal model might help scientists in finding cures for neurodegeneration where single-drug treatments have confirmed insufficient for effective treatment of ALS.
Conclusions
In spite of rigourous research for years there are several important questions still unanswered; these include: (a) does the expression of mtSOD1 at physiological levels causes morphological and structural abnormalities of mitochondrial assembly and its calcium buffering capacity? (b) At pathophysiological levels, does the mitochondria-ER and EMRCC Ca2+ sequestration source specificity and spatiotemporal properties of [Ca2+]i signaling varies at sub cellular level in and around microdomain? (c) In the presence of mtSOD1 gene, what are the consequences of alterations in mitochondrial function on Ca2+ homeostasis and ERMCC? Numerous developments and improvements in visualization of diseased MNs and spatiotemporal resolution of mitochondria-ER calcium signaling cascades have the potential to bring novel insights. MN possess numerous Ca2+ channels that cause rapid Ca2+ influx because of comparatively weak [Ca2+]i buffering, results in mitochondrial Ca2+ overload and strong ROS generation in mtSOD1 Mice. Further studies of ER-mitochondria calcium cycle (ERMCC) with targeted calcium probes show a rapid and dramatic increase in free intra mitochondrial and ER calcium. The defects in mitochondrial assembly and vacuoles derived from defective mitochondria found in post mortem ALS patients further support the suggestion. Design of novel antioxidant strategies to selectively target the oxidative stress and redox imbalance is the other evenue need to be explored. Furthermore, check on selective loss of MNs causing the discharge of pro-apoptotic proteins which activate enzymes leads to activation of apoptotic pathways and apoptotic cells observed in MNs of ALS is beneficial. Dysregulation between mitochondria-ER and ERMCC are well known features and therapeutic drugs aiming to stabilize these cycles reduce ROS and oxidative stress and may be effective in wide range of MN diseases. Another nice direction would be to develop so called “smart drugs” and “combination therapies or multi drug therapies” which have mutifactorial imact on disease mechanism due to mulfactorial nature of ALS disease. Furthermore, it is hypothesized that disrupted Ca2+ homeostasis and oxidative stress induced ROS have a vital role in propagating injury by increasing the excitability of MNs and by targeting neighboring glia. Perhaps as a consequence, excitotoxicty builds up with increased activity-dependent Ca2+ influx and associated mitochondrial Ca2+ cycling. Given the mitochondrial disturbances, Ca2+ buffering becomes inefficient and increase in cytosolic Ca2+ levels. Protective options are to elevate the resistance of MNs to high intracellular Ca2+ concentrations by inducing defense mechanism and/or to inhibit the downstream apoptotic and death cycle pathways activated by increased intracellular Ca2+ concentrations. However, severely impaired MNs are not amendable to taking functional advantage of neuronal protection in ALS.
Therefore perhaps we should focus on new tools such as recently discovered genes that cause ALS and induced pluripotent stem cells taken from ALS patients and derived into MNs to identify potential cytosolic pathways and barriers that could lead to MN degeneration in ALS. Forthcoming studies will hopefully add to the understanding of why these processes preferentially damage MNs and the role non-cell autonomous cell death might play. In conclusion, data indicates that ALS is a multifactorial disease and therefore a combined therapeutic interference (combination therapy) with many facets of target site both at the MNs and glial cells will be most likely essential for survival of ALS patients. While keeping in mind the previous failures in clinical trials for ALS, further studies in this direction to better understand the pathogenesis of cell death in ALS and targeted therapy therefore will be of great interest.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- aCSF:
-
Artificial cerebrospinal fluid
- AMPA:
-
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- [Ca2+]i:
-
AMPAR, AMPA receptors
- [Ca2+]c:
-
Cytosolic calcium
- CB-D28K:
-
Calbidin D28K
- CSF:
-
Cerebrospinal fluid
- EAAT2:
-
Excitatory amino acid transporter 2
- ER:
-
Endoplasmic reticulum
- ERMCC:
-
ER-mitochondria calcium cycle
- fALS:
-
Familial amyotrophic lateral sclerosis
- FCCP:
-
Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- FMNs:
-
Facial motoneurons
- GluR:
-
Glutamate receptors
- GLT:
-
Glutamate transporter
- hALS:
-
Human amyotrophic lateral sclerosis
- HMNs:
-
Hypoglossal motoneurons
- PV:
-
Parvalbumin
- ROS:
-
Reactive oxygen species
- SOD1:
-
Cu/Zn superoxide dismutase 1
- mtSOD1:
-
Mutant superoxide dismutase1
- MNs:
-
Motoneurons
- NMDA:
-
N-Methyl-D-aspartic acid
- PV:
-
Parvalbumin
- Tg:
-
Transgenic
- VDCC:
-
Voltage-dependent calcium channels
- VEGF:
-
Vascular endothelial-cell growth factor
- WT:
-
Wild-type
- Δψm:
-
Mitochondrial membrane potential
- τ:
-
Decay time constant.
References
Rowland LP, Shneider NA: Amyotrophic lateral sclerosis. N Engl J Med. 2001, 344: 1688-1700.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993, 362 (6415): 59-62.
Hand CK, Khoris J, Salachas F, Gros-Louis F, Lopes AAS, Mayeux-Portas V, Brown RH, Meininge V, Camu W, Rouleau GA: A novel locus for familial amyotrophic lateral sclerosis, on chromosome 18q. Am J Hum Genet. 2002, 70 (1): 251-256.
Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, Griffin JW, Cornblath DR: Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet. 1998, 62 (3): 633-640.
Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH: Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009, 323 (5918): 1205-1208.
Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y: Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010, 75 (9): 807-814.
Sapp PC, Hosler BA, McKenna-Yasek D, Chin W, Gann A, Genise H, Gorenstein J, Huang M, Sailer W, Scheffler M, Valesky M, Haines JL, Pericak-Vance M, Siddique T, Horvitz HR, Brown RH: Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am J Hum Genet. 2003, 73 (2): 397-403.
Nishimura AL, Mitne-Neto M, Silva HC, Oliveira JR, Vainzof M, Zatz M: A novel locus for late onset amyotrophic lateral sclerosis/motor neuron disease variant at 20q13. J Med Genet. 2004, 41 (4): 315-320.
Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Hardiman O: ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006, 38 (4): 411-413.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE: TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008, 319 (5870): 1668-1672.
Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L: Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4: e1000193-
Conforti FL, Sproviero W, Simone IL, Mazzei R, Valentino P, Ungaro C, Magariello A, Patitucci A, La Bella V, Sprovieri T, Tedeschi G, Citrigno L, Gabriele AL, Bono F, Monsurrò MR, Muglia M, Gambardella A, Quattrone A: TARDBP gene mutations in south Italian patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011, 82: 587-588.
Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH: Deleterious variants of figure four, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009, 84 (1): 85-88.
Al-Chalabi A, Andersen PM, Nilsson P, Chioza B, Andersson JL, Russ C, Shaw CE, Powell JF, Leigh PN: Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999, 8 (2): 157-164.
Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, Habgood J, Panoutsou S, Winkler S, Tisato V, Hajitou A, Smith B, Vance C, Shaw C, Mazarakis ND, de Belleroche J: Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A. 2010, 107 (16): 7556-7561.
Figlewicz DA, Orrell RW: The genetics of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003, 4 (4): 225-231.
Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung WY, McKenna-Yasek D, Haines JL, Pericak-Vance MA, Horvitz HR, Brown RH: Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA. 2000, 284 (13): 1664-1669.
Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis-Dusenbery BN, Eggan K: The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013, 16 (12): 1725-1727.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson P, Blair IP, Soo KY, King AE, Atkin JD: C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014, Feb 18. [Epub ahead of print]
Eymard-Pierre E, Lesca G, Dollet S, Santorelli FM, di Capua M, Bertini E, Boespflug-Tanguy O: Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am J Hum Genet. 2002, 71 (3): 518-527.
Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmad A, Yang Y, Rimmler J, Hung W, Schlotter B, Ahmed A, Ben Hamida M, Hentati F, Siddique T: Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics. 1998, 2 (1): 55-60.
Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH: The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994, 36: 846-858.
Jaiswal MK, Keller BU: Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol. 2009, 75: 478-489.
Lips MB, Keller BU: Endogenous calcium buffering in motoneurones of the nucleus hypoglossus from mouse. J Physiol. 1998, 511: 105-117.
Van Den Bosch L, Schwaller B, Vleminckx V, Meijers B, Stork S, Ruehlicke T, Van Houtte E, Klaassen H, Celio MR, Missiaen L: Protective effect of parvalbumin on excitotoxic motor neuron death. Exp Neurol. 2002, 174: 150-161.
Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD: Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000, 23: 222-229.
Radi R, Rubbo H, Bush K, Freeman BA: Xanthine oxidase binding to glycosaminoglycans: kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch Biochem Biophys. 1997, 339: 125-135.
Sasaki S, Iwata M: Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1996, 204 (1–2): 53-56.
Sasaki S, Warita H, Murakami T, Abe K, Iwata M: Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004, 107: 461-474.
Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL: An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995, 14: 1105-1116.
Kong J, Xu Z: Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998, 18: 3241-3250.
Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G: Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002, 277: 29626-29633.
Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA: Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002, 80: 616-625.
Fujita K, Yamauchi M, Shibayama K, Ando M, Honda M, Nagata Y: Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J Neurosci Res. 1996, 45: 276-281.
Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM: Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999, 46: 787-790.
Jaiswal MK: Calcium, mitochondria and the pathogenesis of ALS: the good, the Bad and the ugly. Front Cell Neurosci. 2013, 7: 199-
Carriedo SG, Sensi SL, Yin HZ, Weiss JH: AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000, 20: 240-250.
Beal MF: Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002, 32: 797-803.
Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ: Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002, 125: 1522-1533.
Beers DR, Ho BK, Siklos L, Alexianu ME, Mosier DR, Habib Mohamed A, Otsuka Y, Kozovska ME, Smith RE, McAlhany RG, Appel SH: Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem. 2001, 79: 499-509.
Heath PR, Shaw PJ: Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve. 2002, 26 (4): 438-458.
Rao SD, Weiss JH: Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004, 27: 17-23.
Maragakis NJ, Rothstein JD: Glutamate transporters in neurologic disease. Arch Neurol. 2001, 58 (3): 365-370.
Trotti D, Rolfs A, Danbolt NC, Brown RH, Hediger MA: SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999, 2: 427-433.
Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H, Takahashi R: Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum Mol Genet. 2004, 13: 2183-2196.
Rao SD, Yin HZ, Weiss JH: Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J Neurosci. 2003, 23: 2627-2633.
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K: Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007, 10 (5): 608-614.
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S: Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007, 10: 615-622.
Boillée S, Vande Velde C, Cleveland DW: ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006, 52 (1): 39-59.
Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW: Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008, 11 (3): 251-253.
Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW: Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006, 312 (5778): 1389-1392.
Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW: Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci U S A. 2009, 106 (11): 4465-4470.
Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC: Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008, 28 (9): 2075-2088.
Meyer K, Ferraiuolo L, Miranda CJ, Likhite S, McElroy S, Renusch S, Ditsworth D, Lagier-Tourenne C, Smith RA, Ravits J, Burghes AH, Shaw PJ, Cleveland DW, Kolb SJ, Kaspar BK: Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014, 111: 829-832.
Foskett JK, White C, Cheung KH, Mak DO: Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007, 87 (2): 593-658.
Lu YM, Yin HZ, Weiss JH: Ca2+ permeable AMPA/kainate channels permit rapid injurious Ca2+ entry. Neuroreport. 1995, 6 (8): 1089-1092.
Van Den Bosch L, Vandenberghe W, Klaassen H, Van Houtte E, Robberecht W: Ca2+-permeable AMPA receptors and selective vulnerability of motor neurons. J Neurol Sci. 2000, 180 (1–2): 29-34.
Van Den Bosch L, Van Damme P, Vleminckx V, Van Houtte E, Lemmens G, Missiaen L, Callewaert G, Robberecht W: An alpha-mercaptoacrylic acid derivative (PD150606) inhibits selective motor neuron death via inhibition of kainate-induced Ca2+ influx and not via calpain inhibition. Neuropharmacology. 2002, 42 (5): 706-713.
Van Damme P, Van Den Bosch L, Van Houtte E, Callewaert G, Robberecht W: GluR2-dependent properties of AMPA receptors determine the selective vulnerability of motor neurons to excitotoxicity. J Neurophysiol. 2002, 88 (3): 1279-1287.
Palecek J, Lips MB, Keller BU: Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J Physiol. 1999, 520 (Pt 2): 485-502.
DePaul R, Abbs JH, Caligiuri M, Gracco VL, Brooks BR: Hypoglossal, trigeminal, and facial motoneuron involvement in amyotrophic lateral sclerosis. Neurology. 1988, 38 (2): 281-
von Lewinski F, Keller BU: Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 2005, 28 (9): 494-500.
Neher E: The use of fura-2 for estimating Ca2+ buffers and Ca2+ fluxes. Neuropharmacology. 1995, 34: 1423-1442.
Palecek J, Keller BU: Differential calcium buffering in motoneuron populations that are selectively vulnerable and resistant in motoneuron disease. Pflugers Arch. 2000, Supplement, Vol. 439, pR333
Ladewig T, Keller BU: Simultaneous patch-clamp recording and calcium imaging in a rhythmically active neuronal network in the brainstem slice preparation from mouse. Pflugers Arch. 2000, 440: 322-332.
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW: Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995, 38 (1): 73-84.
Carriedo SG, Yin HZ, Sensi SL, Weiss JH: Rapid Ca2+ entry through Ca2+ - permeable AMPA/Kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998, 18: 7727-7738.
Shaw PJ, Ince PG: Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J Neurol. 1997, 244 (Suppl. 2): S3-S14.
Cleveland DW: From charcot to SOD1: Mechanisms of selective motor neuron death in ALS. Neuron. 1999, 24: 515-520.
Nägerl UV, Mody I: Calcium-dependent inactivation of high-threshold calcium currents in human dentate gyrus granule cells. J Physiol. 1998, 509: 39-45.
Kruman II, Pedersen WA, Springer JE, Mattson MP: ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999, 160 (1): 28-39.
Simpson EP, Yen AA, Appel SH: Oxidative stress: a common denominator in the pathogenesis of amyotrophic lateral sclerosis. Curr Opin Rheumatol. 2003, 15 (6): 730-736.
Goodall EF, Morrison KE: Amyotrophic lateral sclerosis (motor neuron disease): proposed mechanisms and pathways to treatment. Expert Rev Mol Med. 2006, 8 (11): 1-22.
Strong MJ: The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol Ther. 2003, 98: 379-414.
Liu D, Wen J, Liu J, Li L: The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999, 13: 2318-
Sau D, Rusmini P, Crippa V, Onesto E, Bolzoni E, Ratti A, Poletti A: Dysregulation of axonal transport and motorneuron diseases. Biol Cell. 2011, 103: 87-107.
Gunawardena S, Goldstein LS: Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J Neurobiol. 2004, 58: 258-271.
Perlson E, Jeong GB, Ross JL, Dixit R, Wallace KE, Kalb RG, Holzbaur EL: A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009, 29: 9903-9917.
Beal MF: Mitochondria and the pathogenesis of ALS. Brain. 2000, 123: 1291-1292.
Kawamata H, Manfredi G: Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010, 131: 517-526.
Magrané J, Cortez C, Gan WB, Manfredi G: Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2013, Nov. 25 [Epub ahead of print]
Magrané J, Sahawneh MA, Przedborski S, Estévez ÁG, Manfredi G: Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci. 2012, 32: 229-242.
Cozzolino M, Ferri A, Valle C, Carri MT: Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci. 2013, 55: 44-49.
Cozzolino M, Carrì MT: Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012, 97 (2): 54-66.
Vijayvergiya C, Beal MF, Buck J, Manfredi G: Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005, 25 (10): 2463-2470.
Vande Velde C, Miller TM, Cashman NR, Cleveland DW: Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008, 105 (10): 4022-4027.
Vande Velde C, McDonald KK, Boukhedimi Y, McAlonis-Downes M, Lobsiger CS, Bel Hadj S, Zandona A, Julien JP, Shah SB, Cleveland DW: Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One. 2011, 6 (7): e22031-doi:10.1371/journal.pone.0022031
Bowling AC, Schulz JB, Brown RH, Beal MF: Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993, 61 (6): 2322-2325.
Browne SE, Bowling AC, Baik MJ, Gurney M, Brown RH, Beal MF: Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J Neurochem. 1998, 71: 281-287.
Jaarsma D, Rognoni F, Duijn WV, Verspaget HW, Haasdijk ED, Holstege JC: Cu- Zn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropath. 2001, 102: 293-305.
Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G: Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984, 43: 471-480.
Hirano A: Cytopathology of amyotrophic lateral sclerosis. Adv Neurol. 1991, 56: 91-101.
Siklos L, Engelhardt J, Harati Y, Smith RG, Joo F, Appel SH: Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann Neurol. 1996, 39: 203-216.
Jaiswal MK, Zech W, Goos M, Leutbecher C, Ferri A, Zippelius A, Carrì MT, Nau R, Keller BU: Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci. 2009, 10: 64-
Wiedemann FR, Winkler K, Kuznetsov AV, Bartels C, Vielhaber S, Feistner H, Kunz WS: Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998, 156 (1): 65-72.
Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, Heinze HJ, Elger CE, Schubert W, Kunz WS: Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000, 123 (Pt 7): 1339-1348.
Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, Sheehan JP, Maguire RS, Pattee G, Juel VC: Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol. 1998, 153: 135-142.
Kruman II, Mattson MP: Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem. 1999, 72 (2): 529-540.
Bergmann F, Keller BU: Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurons from mouse. J Physiol. 2004, 555: 45-59.
Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT: Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005, 5 (25): 164-172.
Dal Canto MC, Gurney ME: Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (fALS). Brain Res. 1995, 676: 25-40.
Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S: Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001, 191: 25-33.
Higgins CMJ, Jung C, Xu Z: ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4: 16-
Xu Z, Jung C, Higgins C, Levine J, Kong J: Mitochondrial degeneration in amyotrophic lateral sclerosis. J Bioenerg Biomembr. 2004, 36: 395-399.
Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW: Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004, 43: 5-17.
Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW: Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013, 33: 4657-4671.
Kaal EC, Vlug AS, Versleijen MW, Kuilman M, Joosten EA, Bar PR: Chronic mitochondrial inhibition induces selective motoneuron death in vitro: a new model for amyotrophic lateral sclerosis. J Neurochem. 2000, 74: 1158-1165.
Tovar-y-Romo LB: Tapia, R: Delayed administration of VEGF rescues spinal motor neurons from death with a short effective time frame in excitotoxic experimental models in vivo. ASN Neuro. 2012, 4: e00081-
Lunn JS, Sakowski SA, Kim B, Rosenberg AA, Feldman EL: Vascular endothelial growth factor prevents G93A-SOD1-induced motor neuron degeneration. Dev Neurobiol. 2009, 69: 871-884.
Bogaert E, Van Damme P, Poesen K, Dhondt J, Hersmus N, Kiraly D, Scheveneels W, Robberecht W, Van Den Bosch L: VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiol Aging. 2010, 31: 2185-2191.
Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM: Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001, 28: 131-138.
Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W: VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003, 34: 383-394.
Zheng C, Nennesmo I, Fadeel B, Henter JI: Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004, 56: 564-567.
Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND: VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004, 429: 413-417.
Wang Y, Ou Mao X, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K: Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007, 27: 304-307.
Lambrechts D, Poesen K, Fernández-Santiago R, Al-Chalabi A, Del Bo R, Van Vught PW, Khan S, Marklund SL, Brockington A, van Marion I, Anneser J, Shaw C, Ludolph AC, Leigh NP, Comi GP, Gasser T, Shaw PJ, Morrison KE, Andersen PM, Van den Berg LH, Thijs V, Siddique T, Robberecht W, Carmeliet P: Meta-analysis of vascular endothelial growth factor variations in amyotrophic lateral sclerosis: increased susceptibility in male carriers of the -2578AA genotype. J Med Genet. 2009, 46: 840-846.
Dodge JC, Haidet AM, Yang W, Passini MA, Hester M, Clarke J, Roskelley EM, Treleaven CM, Rizo L, Martin H, Kim SH, Kaspar R, Taksir TV, Griffiths DA, Cheng SH, Shihabuddin LS, Kaspar BK: Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol Ther. 2008, 16: 1056-1064.
Vanselow BK, Keller BU: Calcium dynamics and buffering in oculomotor neurones from mouse those are particularly resistant during amyotrophic lateral sclerosis (ALS)-related motoneuron disease. J Physiol. 2000, 525: 433-445.
Elliott JL, Snider WD: Parvalbumin is a marker of ALS-resistant motor neurons. Neuroreport. 1995, 6 (3): 449-452.
Zhou Z, Neher E: Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol. 1993, 469: 245-273.
Klingauf J, Neher E: Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997, 72: 674-690.
Lips MB, Keller BU: Activity-related calcium dynamics in motoneurons of the nucleus hypoglossus from mouse. J Neurophysiol. 1999, 82: 2936-2946.
Dekkers J, Bayley P, Dick JR, Schwaller B, Berchtold MW, Greensmith L: Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death. Neuroscience. 2004, 123 (2): 459-466.
Rizzuto R, Brini M, Pozzan T: Intracellular targeting of the photoprotein aequorin: a new approach for measuring, in living cells, Ca2+ concentrations in defined cellular compartments. Cytotechnology. 1993, 11: 44-46.
Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD: Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998, 18: 9673-9684.
Maragakis NJ, Rothstein JD: Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004, 15 (3): 461-473.
Morotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CC: Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet. 2012, 21: 1979-1988.
De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC: VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2012, 21: 1299-1311.
Appel SH, Smith RG, Alexianu M, Siklos L, Engelhardt J, Colom LV, Stefani E: Increased intracellular calcium triggered by immune mechanisms in amyotrophic lateral sclerosis. Clin Neurosci. 1995, 6: 368-374.
Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ: Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998, 1 (5): 366-373.
Choi D, Koh J, Peters S: Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988, 8: 185-196.
Shaw PJ, Forest V, Ince PG, Richardson JP, Wastell HJ: CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration. 1995, 4 (2): 209-216.
Carriedo SG, Yin HZ, Weiss JH: Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996, 16: 4069-4079.
Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J: Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996, 17: 945-956.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF: Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996, 16: 675-686.
Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED: Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996, 39 (2): 147-157.
Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH: Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000, 406: 78-81.
Kuner R, Groom AJ, Bresink I, Kornau HC, Stefovska V, Müller G, Hartmann B, Tschauner K, Waibel S, Ludolph AC, Ikonomidou C, Seeburg PH, Turski L: Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc Natl Acad Sci U S A. 2005, 102: 5826-5831.
Schinder AF, Olson EC, Spitzer NC, Montal M: Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996, 16: 6125-6133.
Arnaudeau S, Kelley WL, Walsh JV, Demaurex N: Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001, 276: 29430-29439.
Szabadkai G, Simoni AM, Rizzuto R: Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J Biol Chem. 2003, 278: 15153-15161.
Rothstein JD, Martin LJ, Kuncl RW: Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992, 326 (22): 1464-1468.
Baron KT, Wang GJ, Padua RA, Campbell C, Thayer SA: NMDA-evoked consumption and recovery of mitochondrially targeted aequorin suggests increased Ca2+ uptake by a subset of mitochondria in hippocampal neurons. Brain Res. 2003, 993: 124-132.
Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, Graier WF: Sustained Ca2+ transfer across mitochondria is essential for mitochondrial Ca2+ buffering, store-operated Ca2+entry, and Ca2+ store refilling. J Biol Chem. 2003, 45: 44769-44779.
Rizzuto R, Bernardi P, Pozzan T: Mitochondria as all-round players of the calcium game. J Physiol. 2000, 529: 37-47.
Corona JC, Tapia R: Ca2+-permeable AMPA receptors and intracellular Ca2+ determine motoneuron vulnerability in rat spinal cord in vivo. Neuropharmacology. 2007, 52: 1219-1228.
Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD: Calcium and mitochondria. FEBS Lett. 2004, 567: 6-102.
Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T: Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994, 126: 1183-1194.
Rizzuto R: Calcium mobilization from mitochondria in synaptic transmitter release. J Cell Biol. 2003, 163: 441-443.
Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavaré JM, Denton RM: Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: Significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci U S A. 1996, 93: 5489-5494.
Ladewig T, Kloppenburg P, Lalley PM, Zipfel WR, Webb WW, Keller BU: Spatial profiles of store-dependent calcium release in motoneurones of the nucleus hypoglossus from newborn mouse. J Physiol. 2003, 547: 775-787.
Duchen MR: Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000, 28: 339-348.
Rizzuto R, Duchen MR, Pozzan T: Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004, 13 (215): re1-
Goos M, Zech WD, Jaiswal MK, Balakrishnan S, Ebert S, Mitchell T, Carrì MT, Keller BU, Nau R: Expression of a Cu,Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of neuroblastoma cells to infectious injury. BMC Infect Dis. 2007, 12 (7): 131-
Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB: Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004, 24: 5611-5622.
Shaw PJ: Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005, 76: 1046-1057.
Genç B, Ozdinler PH: Moving forward in clinical trials for ALS: motor neurons lead the way please. Drug Discov Today. 2013, doi:10.1016/j.drudis.2013.10.014. [Epub ahead of print]
Carri MT, Grignaschi G, Bendotti C: Targets in ALS: designing multidrug therapies. Trends Pharmacol Sci. 2006, 27 (5): 267-273.
Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH: Retrograde viral delivery of IGF-1 prolongs sur vival in a mouse ALS model. Science. 2003, 301: 839-842.
Tolosa L, Mir M, Asensio VJ, Olmos G, Llado J: Vascular endothelial growth factor protects spinal cord motoneurons against glutamate-induced excitotoxicity via phosphatidylinositol 3-kinase. J Neurochem. 2008, 105: 1080-1090.
Tovar-y-Romo LB, Tapia R: VEGF protects spinal motor neurons against chronic excitotoxic degeneration in vivo by activation of PI3-K pathway and inhibition of p38MAPK. J Neurochem. 2010, 115: 1090-1101.
Zhang W, Narayanan M, Friedlander RM: Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann Neurol. 2003, 53: 267-270.
Garbuzova-Davis S, Willing AE, Zigova T, Saporta S, Justen EB, Lane JC, Hudson JE, Chen N, Davis CD, Sanberg PR: Intravenous administration of human umbilical cord blood cells in a mouse model of amyotrophic lateral sclerosis: distribution, migration, and differentiation. J Hematotherapy Stem Cell Res. 2003, 12: 255-270.
Lepore AC, Maragakis NJ: Targeted stem cell transplantation strategies in ALS. Neurochem Internat. 2007, 50: 966-975.
Bova MP, Kinney GG: Emerging drug targets in amyotrophic lateral sclerosis. Expert Opin Orphan Drugs. 2013, 1 (1): 5-20.
Acknowledgments
I would like to thank Drs Bernhard U. Keller, Zygmunt Galdzicki and Fritz Lischka for their valuable discussions. Thanks to Derek Holman for his insightful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Jaiswal, M.K. Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol and Cell Ther 2, 26 (2014). https://doi.org/10.1186/2052-8426-2-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2052-8426-2-26