
Abstract
The sirtuin family of proteins is categorised as class III histone deacetylases that play complex and important roles in ageing-related pathological conditions such as cancer and the deregulation of metabolism. There are seven members in humans, divided into four classes, and evolutionarily conserved orthologues can be found in most forms of life, including both eukaryotes and prokaryotes. The highly conserved catalytic core domain composed of a large oxidised nicotinamide adenine dinucleotide (NAD+)-binding Rossmann fold subunit suggests that these proteins belong to a family of nutrient-sensing regulators. Along with their function in regulating cellular metabolism in response to stressful conditions, they are implicated in modifying a wide variety of substrates; this increases the complexity of unravelling the interplay of sirtuins and their partners. Over the past few years, all of these new findings have attracted the interest of researchers exploring potential therapeutic implications related to the function of sirtuins. It remains to be elucidated whether, indeed, sirtuins can serve as molecular targets for the treatment of human illnesses.
Similar content being viewed by others

Introduction
Epigenetic modifications of protein, histone and chromatin play an important role in regulating gene expression, cancer formation and life span. Acetylation is a major player in epigenetic modifications, resulting in open chromatin structures and, hence, permissive conditions for transcription-factor recruitment to the promoters, followed by initiation of transcription. By contrast, histone deacetylases (HDACs) oppose the activity of histone acetyltransferases by removing the acetyl groups from lysine residues within specific promoters, leading to gene silencing [1]. In addition, many non-histone proteins have been identified as substrates of HDACs, implicating acetylation as a post-translational modification that affects various aspects of cell physiology [2]. There are two protein families having HDAC activity: the classical HDAC family, which consists of two different phylogenetic classes (class I and class II); and the sirtuin family of proteins, which requires the co-factor nicotinamide adenine dinucleotide (NAD) for its deacetylase activity [3, 4].
The sirtuin family
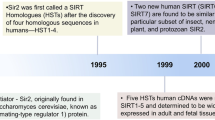
The sirtuin family of proteins is highly conserved, both functionally and structurally. Its members are integrated into most forms of life, including eubacteria, archaea and eukaryotes, and therefore predate both histone and chromatin formation [5]. Sirtuins have been involved in metabolic and chromatin regulation throughout evolution, dating back to the first examples of chromatin-like organisation of DNA in archaea [6, 7]. The silent information regulator 2 gene (Sir2) was first discovered in Saccharomyces cerevisiae and was named after its ability to relieve gene silencing [8]. Once discovered, sirtuins were rapidly characterised in yeast, bacteria, plants and mammals.
Sirtuins belong to the deoxyhypusine synthase (DHS)-like NAD/flavin adenine dinucleotide (FAD)-binding domain clan and all members contain the Rossmann fold structural motif, which can be found in proteins that bind nucleotides. The other members of this clan sharing evolutionary relatedness are the CO dehydrogenase β-subunit/acetyl-coenzyme A (CoA) synthase ε-subunit, DHS, electron transfer flavoprotein FAD-binding domain, NAD(P) transhydrogenase β-subunit and thiamine pyrophosphate enzyme. The mammalian family of sirtuins is categorised as class III histone deacetylases and consists of seven members which are present in nearly all subcellular compartments. SIRT1, SIRT6 and SIRT7 are predominantly localised in the nucleus; SIRT3, SIRT4 and SIRT5 reside within the mitochondria; and SIRT2 is limited to the cytoplasm (Table 1). The highly conserved structure of sirtuins, as well as their NAD+ dependence, suggests that these enzymes are a family of nutrient-sensing regulators cooperating in semi-redundancy to direct cellular metabolism in response to altered nutrition or stress [9]. Whereas this enzyme family is associated with HDACs because of their related nuclear function, the majority of their substrates are non-core histone proteins. Chemically, deacetylation primarily occurs on N- ε-lysine residues and typically regulates protein function. Each isoform maintains a highly conserved catalytic core comprising a large NAD+-binding Rossmann Fold subunit and a small zinc-binding domain.9 The majority of sirtuins function as deacetylases (SIRT1, -2, -3, -5, -6 and -7), where enzymatic activity results in the removal of an acetyl group from N- ε-lysine residues and generates O-acetyl-ADP-ribose and nicotinamide. SIRT4 and -6, however, have also been reported to display mono-adenosine diphosphate (ADP)-ribosyl transferase activity [10, 11]. An increased awareness concerning the regulatory function of sirtuins has resulted in an explosion of research on this family of proteins. The majority of research characterising the functional targets of sirtuins has focused on nuclear SIRT1; however, the other family members are becoming more widely studied.
Phylogenetic analysis of sirtuins from a variety of prokaryotes and eukaryotes has divided the family into five different classes (Table 2) [5, 9, 12]. SIRT1, together with Sir2, homologue of Sir Two (Hst) 1, SIR-2.1 and Drosophila melanogaster Sir2 (D.mel1) orthologues belong to subclass Ia. SIRT2 and SIRT3 share subclass Ib with Hst2, D.mel2 and other fungi and protozoa sirtuins. SIRT4 is in class II and SIRT5 in class III, and both classes include sirtuins from bacteria, archea, nematodes and protozoans. Class IV contains SIRT6 and SIRT7, and can be distinguished from the two previously mentioned classes because only sirtuins from eukaryotes can be found in this category. Finally, class U consists of bacterial Sir2 homologues with undifferentiated motifs. According to this classification of sirtuins, it is believed that sirtuins from classes II, III and U appeared earliest in evolution; the early eukaryotes possessed all four types of sirtuins, whereas later in evolution some eukaryotes lost a few classes, which explains the variable distribution of sirtuins in different organisms [5]. The phylogenesis of all isoforms of human sirtuins is summarised as a dendrogram (Figure 1), demonstrating the splitting of the proteins into distinctly separate branches. This analysis confirms the categorisation of the members in different classes and provides a better assessment of evolutionary divergence.

Phylogenetic analysis of human sirtuin family members. Known peptide sequences for all isoforms of SIRT1-7 from the National Center for Biotechnology Information (NCBI)'s GenBank Database were aligned using ClustalW alignment software, and a dendogram with branch lengths was constructed.
SIRT1
The SIRT1 human gene is located at chromosome (Chr) 10q21.3 [13]. The role of SIRT1 has been the most extensively studied among the sirtuin family members. The first substrate to be identified for SIRT1 was the TATA binding protein-associated factor 168 (TAFI68), a transcription factor necessary for regulating the RNA polymerase I transcriptional complex,[14] where it was shown that deacetylation inhibits transcriptional initiation in vitro. The list of SIRT1 substrates and targets is continuously growing and includes several transcription factors and proteins, implicating it in a variety of cellular functions. For example, p53 was the first non-histone substrate identified for the histone acetyltransferase. It was demonstrated that acetylation activates the DNA-binding activity and target gene expression, as well as increasing its stability [15]. SIRT1-mediated reversal of this post-transcriptional modification can modulate p53 function, weakening the biological effects of acetylation described earlier by repressing p53-dependent transcription [16, 17]. Moreover, it was found that SIRT1-dependent deacetylation regulates localisation of p53, controlling cell fate decisions. For example, in SIRT1-deficient mouse embryonic stem (mES) cells, translocation of the protein to the nucleus is blocked, resulting in cytoplasmic accumulation, passage to mitochondria and induction of the transcription-independent apoptotic pathway following oxidative stress [18]. Consistent with SIRT1 inhibition of p53 function, Sirt1(-/-) knockout mice exhibit p53 hyperacetylation and increased radiation-induced apoptosis, raising the possibility that SIRT1 can facilitate tumour growth by antagonism of p53 [19]. This, together with evidence that sirtinol, an inhibitor of SIRT1, induces a senescence-like cell growth arrest,[20] led to the suggestion that SIRT1 may act as an oncogene. By contrast, other studies [21] demonstrated that SIRT1 seems to act as a tumour suppressor, as Sirt1(+/-) -Trp53(+/-) mice exhibit a higher incidence of tumours, compared with Trp53(+/-) mice.
In white adipose tissue, it was reported that the nuclear peroxisome proliferator-activated receptor gamma (PPARγ) belongs to the category of SIRT1-regulated proteins, as activation of SIRT1 represses it, leading to reduced fat accumulation, whereas its inhibition results in triglyceride accumulation [22, 23]. In the liver, SIRT1 is also involved in nutrient control of glucose homeostasis by modulating the activity of the metabolic co-regulator PPARγ coactivator-1a (PGC-1a), which regulates gluconeogenesis, glycolysis and fatty-acid β-oxidation. Specifically, SIRT1-mediated deacetylation increases hepatic glucose output and use of fat for energy under fasting conditions,[24, 25] whereas SIRT1 was also found to activate PGC-1a, resulting in enhanced mitochondrial function and protection against metabolic disease [26].
Another important group of substrates for SIRT1 are members of the forkhead box factors (FOXO) family of proteins, which are regulated by insulin/AKT. After the finding that longevity in Caenorhabditis elegans depends on the coordinated function of sir-2.1 and the decay accelerating factor 16 gene (daf-16) (the orthologues of FOXO proteins),[27] Brunet et al. were the first to demonstrate that SIRT1 can deacetylate FOXO3 when they are in a complex in response to oxidative stress [28]. In addition, they found that SIRT1 deacetylates FoxO3, tipping FOXO-dependent responses away from apoptosis and towards cell cycle arrest and cellular stress resistance. The beneficial effect of SIRT1-dependent deacetylation of FOXOs was further confirmed by the oxidative stress-resistant phenotype in the heart [29] and the starvation-induced autophagy in cardiac myocytes [30]. Besides the above-mentioned well-characterised targets, several studies have identified more striking partners of SIRT1. Tau protein is deacetylated by SIRT1, affecting the stability of the protein and implying that SIRT1 activators may have therapeutic implications by reducing taumediated neurodegeneration [31]. Recently, Guarani et al. demonstrated that SIRT1 associates with the Notch1 intracellular domain (NICD) and controls stabilisation of the protein,[32] increasing the number and variety of target proteins for SIRT1. Finally, in another recent study, Kim et al. demonstrated that SIRT1 binds to the promoter of SIRT6 and positively regulates its expression, suggesting that different members of the sirtuin family can function collaboratively in maintaining cellular homeostasis [33].
Several studies over the past few years have used gene-targeted mutagenesis experiments in mice to examine the consequences of expressing a mutant SIRT1 protein lacking part of the catalytic domain or deleting the Sirt1 gene completely [19, 21, 34]. McBurney et al. showed that animals homozygous for a null allele of Sirt1 are born at only half the expected frequency, suggesting prenatal lethality [34]. Moreover, they showed that homozygous embryos and pups are smaller than their wild-type and heterozygous littermates and have developmental defects of the eyes, lungs and pancreas. Even under conditions in which Sirt1-null animals survive to adulthood, both sexes are sterile because of a failure of females to ovulate and inefficient spermatogenesis in males. Similar results were obtained in a second study,[19] in which mice were generated either lacking Sirt1 or expressing a mutant Sirt1 gene. Both types of mutant mice were smaller than their wild-type and heterozygous littermates and had developmental defects in the retina and heart, and most died postnatally. Even more severe embryonic lethality was also observed in another Sirt1 mutant mouse model, in which exons 5 and 6 were deleted, suggesting that Sirt1 is essential for normal embryogenesis [21].
Since SIRT1 is involved in many different cellular processes, as already described, several studies have focused primarily on genetic variation of the SIRT1 gene. Studies have attempted to elucidate whether there are any associations between SIRT1 genetic variation and pathological conditions in humans. For example, evidence to date suggests that there might be an association between some aspects of SIRT1 genetic variation and risk for obesity, as well as the response to lifestyle interventions for obesity [35–38]. In addition, mortality in type 2 diabetes mellitus (T2DM) in the presence of factors such as smoking, niacin intake [39] and cardiovascular disease [40] has been linked to SIRT1 genetic variations. Moreover, a recent study reports that four single nucleotide polymorphisms (SNPs) in SIRT1 were associated with diabetic nephropathy in Japanese subjects with T2DM [41]. No association between exceptional longevity and SIRT1 genetic variation was found in this study,[42] however, although life expansion was one of the first phenotypes linked with the role of sirtuins.
SIRT2
Human SIRT2 is located at Chr 19q13.3 [13]. The SIRT2 protein is similar in sequence to yeast Hst2p and both proteins are located in the cytoplasm,[43] making it the first cytoplasmic sirtuin found. SIRT2 co-localises with the microtubule network and deacetylates Lys40 of tubulin, which was the first identified substrate for this protein [44]. SIRT2 also associates with another deacetylase, HDAC6, along microtubules; these two proteins together regulate the level of tubulin acetylation. SIRT2 activity is increased before mitosis [45] and localises to chromosomes, serving as a histone deacetylase during mitosis,[46, 47] suggesting a role for this protein in chromosome condensation. Recent findings showed that, during the cell cycle, SIRT2 is associated with the centrosome, the mitotic spindle and the midbody, further emphasising its role in completion of mitosis [48].
SIRT2 also has been found to interact with FOXO1, as is true for SIRT1. In particular, Jing et al. showed that SIRT2 affects adipocyte differentiation through modulation of both acetylation and phosphorylation of FOXO1 [49]. Recently, SIRT2 was also involved in cellular processes such as neuroprotection and the inflammatory response. It was found that either genetic or pharmacological inhibition of SIRT2 plays a neuroprotective role by reducing sterol levels, which are increased in Huntington's disease [50]. By contrast SIRT2 inhibitors rescue α-synuclein-mediated cytotoxicity in models of Parkinson's disease [51]. Regarding the immune system, hyperacetylated p65 in Sirt2(-/-) cells results in increased expression of a subset of nuclear factor (NF)-κB-dependent target genes [52].
Unfortunately, SIRT2 is the only sirtuin so far that has not been ablated in mice by gene targeting, which is a limitation in the elucidation of its role and its relation to human diseases. Hiratsuka et al. recently reported a role for SIRT2 in cancer pathogenesis by using a proteomic approach [53]. As previously mentioned, the SIRT2 gene is located at Chr 19q13.3 and lies within a region that is frequently deleted in human gliomas; levels of SIRT2 mRNA and protein expression are severely reduced in a large fraction of human glioma cell lines [53]. Moreover, ectopic SIRT2 expression in these cell lines suppresses colony formation and modifies the microtubule network [53]. There have been no studies on genetic variation of the SIRT2 gene so far, due probably to the limited evidence for any association between SIRT2 and human diseases,.
SIRT3
Human SIRT3 is located at Chr 11p15.5 [13] and is located primarily in the mitochondrial matrix,[54, 55] where it is proteolytically processed after entry into the mitochondria. Interestingly, the unprocessed protein is enzymatically inactive in vitro and becomes enzymatically active only after proteolytic processing by 1-methyl-4-phenylpyridine (MPP) [54]. In accordance with its localisation, human acetyl-CoA synthetase-2 was the first substrate to be shown to be deacetylated by SIRT3, thereby activating the acetyl-CoA synthetase activity, linking acetylation with metabolic regulation [56]. Later, it was confirmed that SIRT3 regulates global mitochondrial lysine acetylation, although no metabolic change was observed in Sirt3(-/-) knockout mice [57]. Scher et al., however, demonstrated that in humans SIRT3 can be found not only in mitochondria, but also in the nucleus under normal conditions, where it can deacetylate H4-K16 when recruited to a gene [58]. In support of the nuclear localisation of SIRT3, it was further demonstrated that co-expression of SIRT5 shifts SIRT3 to the nucleus,[59] challenging the concept that mitochondria are the only point of localisation for SIRT3. Focusing again on the mitochondrial role of SIRT3, it was found that SIRT3 mediates cell survival during genotoxic stress by increasing mitochondrial NAD+;[60] SIRT3 is also a major regulator of energy homeostasis, controlling ATP production by direct interaction with complex I [61].
Like SIRT1, SIRT3 has been found to interact with the FOXO family, and has been implicated in the genetic regulation of longevity. SIRT3 interacts with FOXO3a and deacetylation induces FOXO3a-dependent gene expression via an unknown mechanism,[62] as well as blocking hypertrophy in the heart by suppressing cellular levels of reactive oxygen species (ROS) [63]. The role of SIRT3 in oxidative stress has been further highlighted after finding that it regulates enzymes that play a key role in the antioxidant defence mechanism. Two recent studies demonstrated that SIRT3 regulates both manganese superoxide dismutase (MnSOD) activity (by mediating the deacetylation of lysine residues [64, 65]) and isocitrate dehydrogenase 2 (IDH2) activity, thereby increasing the ratio of reduced-to-oxidized glutathione [66]. SIRT3 recently stepped out of the shadow of SIRT1 when several studies demonstrated that SIRT3 regulates cellular processes related to metabolism under both normal and stressful conditions. More specifically, the long-chain acyl-CoA dehydrogenase (LCAD) is hyperacetylated in the absence of SIRT3, resulting in reduced enzymatic activity and decreased levels of fatty acid oxidation during fasting [67]. By contrast, SIRT3 seems to regulate both ketone body production, by affecting mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 (HMGCS2),[68] and the urea cycle through ornithine transcarbamolase (OTC)[69] during caloric restriction. Interestingly, Kim et al. showed for the first time that SIRT3 maintains mitochondrial integrity and oxidative metabolism, which is known to function as a barrier against tumorigenesis as loss of SIRT3 results in a cellular environment that favours carcinogenesis [70]. In accordance with these data, other studies found that SIRT3-mediated regulation of hypoxia-inducible factor (HIF) further induces reprogramming of cancer cell metabolism,[71, 72] emphasising the interplay between ROS and metabolism specifically in cancer cells.
Gene-targeting mutagenesis has contributed to the elucidation of various effects of SIRT3 in pathological conditions. Lombard et al. first inactivated the Sirt3 gene by homologous recombination, deleting exons 2 and 3, which encode the translational start site plus a portion of the catalytic domain. In contrast to Sirt1, Sirt3-deficient mice were born with a normal Mendelian ratio and without any morphological or functional defects [57]. No embryolethality was observed in another Sirt3 mutant mouse model,[61] suggesting that the SIRT3 protein is not essential for normal embryogenesis. This finding has allowed the use of these mouse models further to unravel the role of SIRT3 in the various cellular processes described earlier.
Despite growing evidence that SIRT3 is directly involved in many pathological conditions, only few studies have focused on genetic variation of the SIRT3 gene. One study reported an association between a genetic variant and survival in elderly subjects. Specifically, the TT and GT genotypes relevant to the G477T marker of SIRT3 were associated with increased versus decreased survival in the elderly, respectively [73]. It was also found that the SIRT3 gene shares an intronic enhancer with the PSMD13 gene, which encodes a regulator subunit of the 26S proteasome that has been linked to ageing [74]. Intriguingly, it was reported recently that at least one copy of the SIRT3 gene is deleted in 20 per cent of all human cancers and 40 per cent of breast and ovarian cancers; this is similar to the deletion frequency of the well-known breast cancer tumour suppressor genes, breast cancer susceptibility gene1 (BRCA1) and 2 (BRCA2) [72].
SIRT4
The human SIRT4 gene is located at Chr 12q,[13] and the encoded SIRT4 protein is localised within the mitochondria and has no detectable deacetylase activity. Therefore, it is solely an NAD+-dependent protein ADP-ribosyl transferase [11, 75]. SIRT4 has been detected in a number of tissues -- including brain, kidney, pancreas, liver, thyroid, vascular smooth muscle and striated muscle -- which suggests a role in global metabolic function. One of the first reported interactions of SIRT4 identified its co-localisation with insulin-degrading enzyme and the ADP/ATP carrier proteins, adenine nucleotide translocase (ANT) 2 and ANT3 [75]. Recently, SIRT4 has been implicated in the regulation of fatty acid oxidation and mitochondrial gene expression in liver and muscle cells [76]. Interestingly, SIRT4 knockdown results in an increase in SIRT1 mRNA and protein levels, suggesting a redundant interplay of metabolic regulation among sirtuins. SIRT4 was previously thought to act secondarily to SIRT3 as a minor regulator of mitochondrial function; however, this study indicates that SIRT4 may have a more important role in mitochondrial metabolism, as SIRT4 inhibition increases fat oxidative capacity in liver and mitochondrial function in muscle [76]. These findings may prove beneficial in developing therapeutics for numerous diseases, such as T2DM, in which SIRT1 and SIRT4 have recently been reported to play a role in T2DM pathogenesis [77].
Another known target of SIRT4 activity includes mitochondrial glutamate dehydrogenase (GLUD1), as ADP ribosylation inhibits GLUD1 enzyme activity and opposes the effects of caloric restriction in pancreatic β-cells [11]. SIRT4 has also been linked to non-alcoholic fatty liver disease, in which its location in the cytoplasm was suggested to play a role in the development of insulin resistance [78]. Whereas SIRT4 remains one of the less-studied sirtuins, an increasing body of work supports its involvement in critical metabolic pathways and disease pathologies.
SIRT5
The human SIRT5 gene is located at Chr 6p23,[13] and the gene product functions as a mitochondrial NAD+-dependent deacetylase with well-defined substrates. Supporting a model of conserved evolution, the crystal structures of the core domains of SIRT3 and SIRT5 reveal remarkable structural conservation with other sirtuins, especially human SIRT2 and the ancestral yeast protein [79–81]. Initial characterisation of Sirt5(-/-) knockout mice found no defects in basal glucose, insulin or lipid homeostasis [57, 82]. As fasting increases SIRT5 mRNA levels, however, transgenic mice overexpressing SIRT5 exhibited a novel target of SIRT5 deacetylase activity -- carbamoyl phosphate synthetase-1 (CPS1) [82, 83]. CPS1 is a key enzyme within the urea cycle that catalyses the condensation of ammonia and bicarbonate, yielding carbamoyl phosphate. These findings support a protective role for SIRT5 because the conversion of ammonia to non-toxic urea appears to be regulated tightly by this enzyme through CPS1 deacetylation and activation. SIRT5 was also found to deacetylate and regulate the activity of cytochrome C, a protein regulating oxidative metabolism and apoptosis [80]. It was determined that SIRT5 translocates into the mitochondrial inter-membrane space from the matrix, indicating that varying localisation by means of altered cellular signalling may contribute to SIRT5 regulation and substrate selection.
Two isoforms of SIRT5 have been identified (SIRT5iso1 and SIRT5iso2); the mechanism of subcellular localisation for these two has been examined [84]. The C-termini of the two isoforms differ slightly, whereas each contains a cleavable mitochondrial-targeting sequence at its N-terminus. The cleaved SIRT5iso2 was found localised within the mitochondria, while cleaved SIRT5iso1 was localised in both mitochondria and cytoplasm. Another form, SIRT5DC, contains only the common domain and was found within the mitochondria, similarly to SIRT5iso2. This report suggests that the cytoplasmic localisation of cleaved SIRT5iso1 is regulated by altering the C-terminal sequence. Additionally, it was found that the C-terminus of SIRT5iso2 is rich in hydrophobic amino acid residues and, consequently, functions as a mitochondrial membrane-insertion signal. In sum, these results demonstrate that SIRT5 plays a role in controlling various cellular functions via two isoforms with different intracellular localisations or stabilities [84].
SIRT6
The human SIRT6 gene is located at Chr 19p13.3 [85]. Initial investigation into SIRT6 activity supported a lack of NAD+-dependent protein deacetylase activity and showed that it is a broadly expressed nuclear ADP-ribosyltransferase [10]. These studies were later contradicted, however, revealing that SIRT6 does in fact contain deacetylase activity [86]. The complete genetic characterisation of the human SIRT6 gene showed eight exons, ranging in size from 60 base pairs (bp) (exon 4) to 838 bp (exon 8), encoding a 355-amino acid protein (39.1 kDa) [85]. Another report identified novel functions for the N- and C-terminal domains of SIRT6; the authors were able to demonstrate that the C-terminal extension of SIRT6 directs nuclear localisation, but that it is not required for activity. By contrast, the N-terminal extension of SIRT6 is required for chromatin regulation and catalytic activity [87].
SIRT6 is predominantly a nuclear chromatin-associated protein which aids in the protection of DNA damage and suppresses genomic instability through association with base-excision repair and DNA-end resection [88, 89]. Phenotypically, Sirt6-deficient mice are under-sized and develop a number of abnormalities by two to three weeks of age -- including lymphopenia, fat depletion and severe metabolic defects, resulting in death by four weeks. These findings clearly demonstrate a major role for SIRT6 in genome stability and longevity-related processes; however, specific mechanisms of interaction remain elusive.
A recent study provided exceptional insight into the mechanism of SIRT6-related chromatin regulation [86]. This study outlined a role for SIRT6 in the attenuation of NF-κB-signalling via its interaction with the NF-κB RelA subunit. SIRT6 directly deacetylates histone H3 lysine 9 (H3K9) at NF-κB target promoters. Interestingly, RelA insufficiency rescues the Sirt6-deficient phenotype in the knockout mouse. Additional data indicate that SIRT6 also deacetylates H3 K56 to modulate genomic stability [90]. Given the implied importance of SIRT6 in genomic stability and overall longevity, it is no surprise that Sirt6 deficiency causes the most extreme phenotype among all sirtuin-knockout animal models, generating severely retarded growth rates and lethality.
Sirt6 gene overexpression in adipose tissue resulted in a highly positive outcome, in which Sirt6 expression was related to peroxisome proliferator-activated receptor-responsive genes, and genes associated with the regulation of lipid storage and transport [91]. Additional findings further present a case for metabolic regulation through genetic regulation of the expression of multiple glycolytic genes [92]. More specifically, SIRT6 functions as a co-repressor of HIF1α, regulating nutrient stress-response pathways. Sirt6-deficient cells displayed an increase in HIF1α activity, increased glucose uptake, upregulation of glycolysis and reduced mitochondrial respiration. These results imply that SIRT6 may serve as a 'master regulator' of glucose homeostasis and provide a compelling target for therapeutic approaches to metabolic diseases.
A recent report provided critical data in support of tight regulation and interaction of the sirtuin protein family. The authors determined that SIRT1 forms a complex with FOXO3a and nuclear respiratory factor 1 (NRF1) on the SIRT6 promoter and positively regulates SIRT6 expression. Consequently, this action negatively regulates glycolysis, triglyceride synthesis and fat metabolism through the known deacetylation of H3 K9 [33]. As a result of a liver-specific Sirt6 deletion in mice, alterations in gene expression occur, leading to increases in a host of metabolic pathways, including glycolysis and β-oxidation. Taken together, it seems clear that SIRT6 plays a critical role in glucose and lipid metabolism and is a likely therapeutic target for treating a number of metabolic-related fatty liver diseases [93].
SIRT7
The human SIRT7 gene is located at Chr 17q25 [94]. The exact mechanism of SIRT7 deacetylase activity remains undefined; however, localised within the nucleolus, it is known to play a significant role in cell growth and proliferation. The SIRT7 genomic sequence spans a region of 6.2 kilobases (kb)[94] and the SIRT7 gene comprises ten exons and encodes a 400-amino acid protein (44.9 kDa).
SIRT7 is associated with a number of active rRNA genes (rDNA), including RNA polymerase I (Pol I) and histones [95]. SIRT7 overexpression increases Pol I-mediated transcription, whereas SIRT7 knockdown results in decreased association of Pol I with rDNA and reduced Pol I transcription. Interestingly, silencing of SIRT7 expression stops cell proliferation and triggers apoptosis. SIRT7 also directly interacts with the rDNA transcription factor, upstream binding factor (UBF). Moreover, SIRT7 is phosphorylated via the cyclin-dependent kinase 1 (CDK1)-cyclin B pathway during mitosis, and these phosphorylation events are known to induce conformational modifications to the C-terminal region of SIRT7. This conformational change is likely to alter SIRT7 activity and results in the regulation of rDNA transcription [96]. Recently, it was found that Sirt7-deficient mice experience a reduction in mean and maximum lifespan and develop heart complications, such as fibrosis and inflammatory cardiomyopathy [97]. A noteworthy observation with Sirt7(-/-) knockout mice was an association between hyperacetylated p53 and increased rates of apoptosis in the myocardium [97]. Additional knockout data in primary cardiomyocytes demonstrated a twofold increase in apoptosis and increased susceptibility to oxidative and genotoxic stress. These data suggest a critical role for SIRT7 in stress responses and cell survival in the heart. Furthermore, recent evidence demonstrates that SIRT7 expression inversely correlates with the tumorigenesis potential of numerous mouse cell lines; SIRT7 probably enables cells to maintain a host of critical metabolic pathways through the inhibition of cell growth under severely stressful situations [98].
Future directions/conclusions
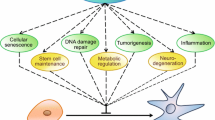
The sirtuin genes encode an important and complex family of proteins that participate in a wide spectrum of physiological processes. In several species, caloric restriction has been shown to increase lifespan and decrease spontaneous rates of illness, such as insulin resistance, neurodegenerative disease and cancer. Because caloric restriction activates specific cellular signalling networks, including sirtuin protein deacetylase activity, it has long been thought that chemical agents that induce sirtuin activity would have similar beneficial therapeutic effects to caloric restriction. This idea is well justified by results demonstrating that several of the sirtuin knockout mouse models develop illnesses similar to those observed in humans. Thus, it seems reasonable to propose that the sirtuin protein family will prove to represent novel molecular targets for the treatment of human diseases having a strong association with increasing age. As such, these novel findings for the potential role of sirtuins in human illnesses should allow for the identification of potential molecular targets and biomarkers to determine risk and the development of agents that may be chemopreventive.
References
Marks P, Rifkind RA, Richon VM, Breslow R, et al: Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer. 2001, 1: 194-202. 10.1038/35106079.
Yang XJ, Seto E: Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol Cell. 2008, 31: 449-461. 10.1016/j.molcel.2008.07.002.
Imai S, Armstrong CM, Kaeberlein M, Guarente L: Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000, 403: 795-800. 10.1038/35001622.
Glozak MA, Seto E: Histone deacetylases and cancer. Oncogene. 2007, 26: 5420-5432. 10.1038/sj.onc.1210610.
Frye RA: Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000, 273: 793-798. 10.1006/bbrc.2000.3000.
Bell SD, Botting CH, Wardleworth BN, Jackson SP, et al: The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science. 2002, 296: 148-151. 10.1126/science.1070506.
Starai VJ, Celic I, Cole RN, Boeke JD, et al: Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002, 298: 2390-2392. 10.1126/science.1077650.
Klar AJ, Fogel S, Macleod K: MAR1: A regulator of the HMa and HMalpha loci in Saccharomyces cerevisiae. Genetics. 1979, 93: 37-50.
Greiss S, Gartner A: Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol Cells. 2009, 28: 407-415. 10.1007/s10059-009-0169-x.
Liszt G, Ford E, Kurtev M, Guarente L: Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005, 280: 21313-21320. 10.1074/jbc.M413296200.
Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, et al: SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006, 126: 941-954. 10.1016/j.cell.2006.06.057.
Michan S, Sinclair D: Sirtuins in mammals: Insights into their biological function. Biochem J. 2007, 404: 1-13. 10.1042/BJ20070140.
Frye RA: Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999, 260: 273-279. 10.1006/bbrc.1999.0897.
Muth V, Nadaud S, Grummt I, Voit R: Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J. 2001, 20: 1353-1362. 10.1093/emboj/20.6.1353.
Brooks CL, Gu W: Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr Opin Cell Biol. 2003, 15: 164-171. 10.1016/S0955-0674(03)00003-6.
Luo J, Nikolaev AY, Imai S, Chen D, et al: Negative control of pp53 by Sir2alpha promotes cell survival under stress. Cell. 2001, 107: 137-148. 10.1016/S0092-8674(01)00524-4.
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, et al: hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001, 107: 149-159. 10.1016/S0092-8674(01)00527-X.
Han MK, Song EK, Guo Y, Ou X, et al: SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell. 2008, 2: 241-251. 10.1016/j.stem.2008.01.002.
Cheng HL, Mostoslavsky R, Saito S, Manis JP, et al: Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci USA. 2003, 100: 10794-10799. 10.1073/pnas.1934713100.
Ota H, Tokunaga E, Chang K, Hikasa M, et al: Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006, 25: 176-185.
Wang RH, Sengupta K, Li C, Kim HS, et al: Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008, 14: 312-323. 10.1016/j.ccr.2008.09.001.
Picard F, Auwerx J: PPAR(gamma) and glucose homeostasis. Annu Rev Nutr. 2002, 22: 167-197. 10.1146/annurev.nutr.22.010402.102808.
Picard F, Kurtev M, Chung N, Topark-Ngarm A, et al: Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004, 429: 771-776. 10.1038/nature02583.
Rodgers JT, Lerin C, Haas W, Gygi SP, et al: Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005, 434: 113-118. 10.1038/nature03354.
Purushotham A, Schug TT, Xu Q, Surapureddi S, et al: Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9: 327-338. 10.1016/j.cmet.2009.02.006.
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, et al: Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006, 127: 1109-1122. 10.1016/j.cell.2006.11.013.
Tissenbaum HA, Guarente L: Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001, 410: 227-230. 10.1038/35065638.
Brunet A, Sweeney LB, Sturgill JF, Chua KF, et al: Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004, 303: 2011-2015. 10.1126/science.1094637.
Alcendor RR, Gao S, Zhai P, Zablocki D, et al: Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007, 100: 1512-1521. 10.1161/01.RES.0000267723.65696.4a.
Hariharan N, Maejima Y, Nakae J, Paik J, et al: Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010, 107: 1470-1482. 10.1161/CIRCRESAHA.110.227371.
Min SW, Cho SH, Zhou Y, Schroeder S, et al: Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010, 67: 953-966. 10.1016/j.neuron.2010.08.044.
Guarani V, Deflorian G, Franco CA, Kruger M, et al: Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011, 473: 234-238. 10.1038/nature09917.
Kim HS, Xiao C, Wang RH, Lahusen T, et al: Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12: 224-236. 10.1016/j.cmet.2010.06.009.
McBurney MW, Yang X, Jardine K, Hixon M, et al: The mammalian SIR2alpha protein has a role in embryogenesis and gameto-genesis. Mol Cell Biol. 2003, 23: 38-54. 10.1128/MCB.23.1.38-54.2003.
Weyrich P, Machicao F, Reinhardt J, Machann J, et al: SIRT1 genetic variants associate with the metabolic response of Caucasians to a controlled lifestyle intervention: The TULIP Study. BMC Med Genet. 2008, 9: 100-10.1186/1471-2350-9-100.
Peeters AV, Beckers S, Verrijken A, Mertens I, et al: Association of SIRT1 gene variation with visceral obesity. Hum Genet. 2008, 124: 431-436. 10.1007/s00439-008-0567-8.
Zillikens MC, van Meurs JB, Rivadeneira F, Amin N, et al: SIRT1 genetic variation is related to BMI and risk of obesity. Diabetes. 2009, 58: 2828-2834. 10.2337/db09-0536.
Shimoyama Y, Suzuki K, Hamajima N, Niwa T: Sirtuin 1 gene polymorphisms are associated with body fat and blood pressure in Japanese. Transl Res. 2011, 157: 339-347. 10.1016/j.trsl.2011.02.004.
Zillikens MC, van Meurs JB, Sijbrands EJ, Rivadeneira F, et al: SIRT1 genetic variation and mortality in type 2 diabetes: Interaction with smoking and dietary niacin. Free Radic Biol Med. 2009, 46: 836-841. 10.1016/j.freeradbiomed.2008.12.022.
Kuningas M, Putters M, Westendorp RG, Slagboom PE, et al: SIRT1 gene, age-related diseases, and mortality: The Leiden 85-plus study. J Gerontol A Biol Sci Med Sci. 2007, 62: 960-965. 10.1093/gerona/62.9.960.
Maeda S, Koya D, Araki SI, Babazono T, et al: Association between single nucleotide polymorphisms within genes encoding sirtuin families and diabetic nephropathy in Japanese subjects with type 2 diabetes. Clin Exp Nephrol. 2011, 15: 381-390. 10.1007/s10157-011-0418-0.
Flachsbart F, Croucher PJ, Nikolaus S, Hampe J, et al: Sirtuin 1 (SIRT1) sequence variation is not associated with exceptional human longevity. Exp Gerontol. 2006, 41: 98-102. 10.1016/j.exger.2005.09.008.
Afshar G, Murnane JP: Characterization of a human gene with sequence homology to Saccharomyces cerevisiae SIR2. Gene. 1999, 234: 161-168. 10.1016/S0378-1119(99)00162-6.
North BJ, Marshall BL, Borra MT, Denu JM, et al: The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003, 11: 437-444. 10.1016/S1097-2765(03)00038-8.
Dryden SC, Nahhas FA, Nowak JE, Goustin AS, et al: Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 2003, 23: 3173-3185. 10.1128/MCB.23.9.3173-3185.2003.
Inoue T, Hiratsuka M, Osaki M, Yamada H, et al: SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007, 26: 945-957. 10.1038/sj.onc.1209857.
Vaquero A, Scher MB, Lee DH, Sutton A, et al: SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20: 1256-1261. 10.1101/gad.1412706.
North BJ, Verdin E: Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One. 2007, 2: e784-10.1371/journal.pone.0000784.
Jing E, Gesta S, Kahn CR: SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6: 105-114. 10.1016/j.cmet.2007.07.003.
Luthi-Carter R, Taylor DM, Pallos J, Lambert E, et al: SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci USA. 2010, 107: 7927-7932. 10.1073/pnas.1002924107.
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, et al: Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007, 317: 516-519. 10.1126/science.1143780.
Rothgiesser KM, Erener S, Waibel S, Luscher B, et al: SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 2010, 123: 4251-4258. 10.1242/jcs.073783.
Hiratsuka M, Inoue T, Toda T, Kimura N, et al: Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem Biophys Res Commun. 2003, 309: 558-566. 10.1016/j.bbrc.2003.08.029.
Schwer B, North BJ, Frye RA, Ott M, et al: The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002, 158: 647-657. 10.1083/jcb.200205057.
Onyango P, Celic I, McCaffery JM, Boeke JD, et al: SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA. 2002, 99: 13653-13658. 10.1073/pnas.222538099.
Schwer B, Bunkenborg J, Verdin RO, Andersen JS, et al: Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006, 103: 10224-10229. 10.1073/pnas.0603968103.
Lombard DB, Alt FW, Cheng HL, Bunkenborg J, et al: Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007, 27: 8807-8814. 10.1128/MCB.01636-07.
Scher MB, Vaquero A, Reinberg D: SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21: 920-928. 10.1101/gad.1527307.
Nakamura Y, Ogura M, Tanaka D, Inagaki N: Localization of mouse mitochondrial SIRT proteins: Shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem Biophys Res Commun. 2008, 366: 174-179. 10.1016/j.bbrc.2007.11.122.
Yang H, Yang T, Baur JA, Perez E, et al: Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007, 130: 1095-1107. 10.1016/j.cell.2007.07.035.
Ahn BH, Kim HS, Song S, Lee IH, et al: A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008, 105: 14447-14452. 10.1073/pnas.0803790105.
Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, et al: SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008, 4: 291-299.
Sundaresan NR, Gupta M, Kim G, Rajamohan SB, et al: Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009, 119: 2758-2771.
Tao R, Coleman MC, Pennington JD, Ozden O, et al: Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010, 40: 893-904. 10.1016/j.molcel.2010.12.013.
Qiu X, Brown K, Hirschey MD, Verdin E, et al: Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12: 662-667. 10.1016/j.cmet.2010.11.015.
Someya S, Yu W, Hallows WC, Xu J, et al: Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010, 143: 802-812. 10.1016/j.cell.2010.10.002.
Hirschey MD, Shimazu T, Goetzman E, Jing E, et al: SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010, 464: 121-125. 10.1038/nature08778.
Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, et al: SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010, 12: 654-661. 10.1016/j.cmet.2010.11.003.
Hallows WC, Yu W, Smith BC, Devries MK, et al: Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell. 2011, 41: 139-149. 10.1016/j.molcel.2011.01.002.
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, et al: SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010, 17: 41-52. 10.1016/j.ccr.2009.11.023.
Bell EL, Emerling BM, Ricoult SJ, Guarente L: SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011.
Finley LW, Carracedo A, Lee J, Souza A, et al: SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011, 19: 416-428. 10.1016/j.ccr.2011.02.014.
Rose G, Dato S, Altomare K, Bellizzi D, et al: Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Exp Gerontol. 2003, 38: 1065-1070. 10.1016/S0531-5565(03)00209-2.
Bellizzi D, Dato S, Cavalcante P, Covello G, et al: Characterization of a bidirectional promoter shared between two human genes related to aging: SIRT3 and PSMD13. Genomics. 2007, 89: 143-150. 10.1016/j.ygeno.2006.09.004.
Ahuja N, Schwer B, Carobbio S, Waltregny D, et al: Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007, 282: 33583-33592. 10.1074/jbc.M705488200.
Nasrin N, Wu X, Fortier E, Feng Y, et al: SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem. 2010, 285: 31995-32002. 10.1074/jbc.M110.124164.
Song R, Xu W, Chen Y, Li Z, et al: The expression of Sirtuins 1 and 4 in peripheral blood leukocytes from patients with type 2 diabetes. Eur J Histochem. 2011, 55: e10-
Chen YR, Fang SR, Fu YC, Zhou XH, et al: Calorie restriction on insulin resistance and expression of SIRT1 and SIRT4 in rats. Biochem Cell Biol. 2010, 88: 715-722. 10.1139/O10-010.
Schuetz A, Min J, Antoshenko T, Wang CL, et al: Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 2007, 15: 377-389. 10.1016/j.str.2007.02.002.
Schlicker C, Gertz M, Papatheodorou P, Kachholz B, et al: Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008, 382: 790-801. 10.1016/j.jmb.2008.07.048.
Jin L, Wei W, Jiang Y, Peng H, et al: Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem. 2009, 284: 24394-24405. 10.1074/jbc.M109.014928.
Nakagawa T, Lomb DJ, Haigis MC, Guarente L: SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009, 137: 560-570. 10.1016/j.cell.2009.02.026.
Nakagawa T, Guarente L: Urea cycle regulation by mitochondrial sirtuin, SIRT5. Aging (Albany NY). 2009, 1: 578-581.
Matsushita N, Yonashiro R, Ogata Y, Sugiura A, et al: Distinct regulation of mitochondrial localization and stability of two human Sirt5 isoforms. Genes Cells. 2011, 16: 190-202. 10.1111/j.1365-2443.2010.01475.x.
Mahlknecht U, Ho AD, Voelter-Mahlknecht S: Chromosomal organization and fluorescence in situ hybridization of the human Sirtuin 6 gene. Int J Oncol. 2006, 28: 447-456.
Kawahara TL, Michishita E, Adler AS, Damian M, et al: SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009, 136: 62-74. 10.1016/j.cell.2008.10.052.
Tennen RI, Berber E, Chua KF: Functional dissection of SIRT6: Identification of domains that regulate histone deacetylase activity and chromatin localization. Mech Ageing Dev. 2010, 131: 185-192. 10.1016/j.mad.2010.01.006.
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, et al: Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006, 124: 315-329. 10.1016/j.cell.2005.11.044.
Kaidi A, Weinert BT, Choudhary C, Jackson SP: Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010, 329: 1348-1353. 10.1126/science.1192049.
Yang B, Zwaans BM, Eckersdorff M, Lombard DB: The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009, 8: 2662-2663. 10.4161/cc.8.16.9329.
Kanfi Y, Peshti V, Gil R, Naiman S, et al: SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. 2010, 9: 162-173. 10.1111/j.1474-9726.2009.00544.x.
Zhong L, D'Urso A, Toiber D, Sebastian C, et al: The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010, 140: 280-293. 10.1016/j.cell.2009.12.041.
Yang SJ, Choi JM, Chae SW, Kim WJ, et al: Activation of peroxisome proliferator-activated receptor gamma by rosiglitazone increases sirt6 expression and ameliorates hepatic steatosis in rats. PLoS One. 2011, 6: 17057-10.1371/journal.pone.0017057.
Voelter-Mahlknecht S, Letzel S, Mahlknecht U: Fluorescence in situ hybridization and chromosomal organization of the human Sirtuin 7 gene. Int J Oncol. 2006, 28: 899-908.
Ford E, Voit R, Liszt G, Magin C, et al: Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20: 1075-1080. 10.1101/gad.1399706.
Grob A, Roussel P, Wright JE, McStay B, et al: Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J Cell Sci. 2009, 122: 489-498. 10.1242/jcs.042382.
Vakhrusheva O, Smolka C, Gajawada P, Kostin S, et al: Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008, 102: 703-710. 10.1161/CIRCRESAHA.107.164558.
Vakhrusheva O, Braeuer D, Liu Z, Braun T, et al: Sirt7-dependent inhibition of cell growth and proliferation might be instrumental to mediate tissue integrity during aging. J Physiol Pharmacol. 2008, 59 (Suppl. 9): 201-212.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vassilopoulos, A., Fritz, K.S., Petersen, D.R. et al. The human sirtuin family: Evolutionary divergences and functions. Hum Genomics 5, 485 (2011). https://doi.org/10.1186/1479-7364-5-5-485
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-7364-5-5-485