
Abstract
Background
Based on the glutamatergic dysfunction hypothesis for schizophrenia pathogenesis, we have been performing systematic association studies of schizophrenia with the genes involved in glutametergic transmission. We report here association studies of schizophrenia with SLC1A4, SLC1A5 encoding neutral amino acid transporters ASCT1, ASCT2, and SLC6A5, SLC6A9 encoding glycine transporters GLYT2, GLYT1, respectively.
Methods
We initially tested the association of 21 single nucleotide polymorphisms (SNPs) distributed in the four gene regions with schizophrenia using 100 Japanese cases-control pairs and examined allele, genotype and haplotype association with schizophrenia. The observed nominal significance were examined in the full-size samples (400 cases and 420 controls).
Results
We observed nominally significant single-marker associations with schizophrenia in SNP2 (P = 0.021) and SNP3 (P = 0.029) of SLC1A4, SNP1 (P = 0.009) and SNP2 (P = 0.022) of SLC6A5. We also observed nominally significant haplotype associations with schizophrenia in the combinations of SNP2-SNP7 (P = 0.037) of SLC1A4 and SNP1-SNP4 (P = 0.043) of SLC6A5. We examined all of the nominal significance in the Full-size Sample Set, except one haplotype with insufficient LD. The significant association of SNP1 of SLC6A5 with schizophrenia was confirmed in the Full-size Sample Set (P = 0.018).
Conclusion
We concluded that at least one susceptibility locus for schizophrenia may be located within or nearby SLC6A5, whereas SLC1A4, SLC1A5 and SLC6A9 are unlikely to be major susceptibility genes for schizophrenia in the Japanese population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Schizophrenia is a devastating mental disorder that affects about 1% of worldwide populations [1], and genetic factors are known to play a crucial role in its pathogenesis [2]. The successful treatment with dopamine antagonists on the positive symptomatology of the disease suggests a crucial role of dopamine in the pathophysiology of schizophrenia. However, due to the poor effects of dopamine antagonists against the negative and cognitive symptoms of schizophrenia, other neurotransmitter systems than dopamine, such as glutamate are suggested to be involved in the pathogenesis of schizophrenia. Based on the fact that phencyclidine (PCP), the antagonist of N-methyl-D-aspartate (NMDA) glutamate receptor, induces schizophreniform psychosis, a glutamatergic dysfunction hypothesis has been proposed for the pathogenesis of schizophrenia [3–5]. This hypothesis has been supported by recent multiple reports of significant association of schizophrenia with glutamate receptor genes and with the genes related to glutamatergic transmission [Review, [6, 7]]. The dopamine and glutamate hypothesis of schizophrenia are not independent, and in fact, glutamate-dopamine interaction has been supported by many preclinical and clinical findings [Review, [8]].
Other synaptic elements related to glutamate, such as transporters, also potentially affect glutamatergic neurotransmission. Excitatory amino acid transporters (EAATs) maintain extracellular glutamate concentrations within physiological levels by reuptaking synaptically released glutamate. Abnormalities of mRNA expression of EAATs were reported in the thalamus, prefrontal cortex, parahippocampal gyrus and striatum in schizophrenia [9–12]. Recently, we have reported the positive association of SLC1A2 and SLC1A6, the genes encoding EAAT2 and EAAT4, respectively with schizophrenia [13, 14], providing support for the potential important roles of EAATs in schizophrenia.
Neutral amino acid transporters (ASCTs), which transport neutral amino acid (alanine, serine, cysteine and threonin) were identified based on nucleotide sequence homology to the EAATs [15, 16]. The amino acid identity between EAATs and ASCTs is 40–44%. The functions of ASCTs in glutamate transmission have also been reported. ASCT1 not only mediates the efflux of glutamate from the neuron into the synaptic junction via Calcium-independent release, but also mediates the efflux of L-serine from glial cells and its uptake by neurons [17–19]. L-serine is used for syntheses of various biomolecules, including the co-agonists at NMDA glutamate receptor, D-serine and glycine. ASCT2 appears to play an important role in the glutamine-glutamate cycle between neurons and glia by facilitation the efflux of glutamine from glial cells [20]. Recently, Weis et al. reported significant decrease in ASCT1 immunoreactivity in the cingulate cortex, white matter, and striking loss of ASCT1 immunoreactivity in the hippocampus in schizophrenia. [21].
Glycine acts as an obligatory co-agonist at NMDA glutamate receptor to promote NMDA receptor function. In the central system, the actions of glycine are terminated by its rapid uptake into the nerve terminal and adjacent glial cells via high-affinity glycine transporters (GLYTs) [22]. Therefore, increasing synaptic level of glycine by inhibiton of its uptake could lead to enhance the activation of NMDA receptor. Both preclinical and clinical evidence have provided support for the utility of this modulatory approach, as well as the potential therapeutic value of GLYT1 inhibitors in the treatment of schizophrenia [Review, [23]]. Therefore the ASCTs and GLYTs genes are strong candidates for schizophrenia, as well as glutamate receptor and glutamate transporter genes.
In this study we report association studies of schizophrenia with total 21 SNPs distributed in genes SLC1A4, SLC1A5, SLC6A5 and SLC6A9 that encoding the neutral amino acid transporters ASCT1, ASCT2 and the glycine transporters GLYT2, GLYT1, respectively. SNPs were selected to cover the entire gene regions by linkage disequilibria (LD). To enhance the detection power of the study, we also examined the haplotype associations with the disease.
Methods
Human subjects
Blood samples were obtained from unrelated Japanese individuals who had provided written informed consent. We used 400 cases (mean age 47.2; 44.8% female) recruited from hospitals in Kyushu and Aichi areas and 420 unrelated controls (mean age 43.6; 44.0% female) recruited from the Kyushu and Aichi areas. We initially tested the association of the genes with schizophrenia using the Screening Sample Set: 100 out of 400 cases (mean age 49.5; 44.0% female) and 100 out of 420 controls (mean age 51.2; 44.0% female) recruited from the Kyushu area. All patients were diagnosed by the Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria [24]. The patients are all consecutive inpatients. The schizophrenia diagnoses were confirmed by several psychiatrists. We used another 16 healthy Japanese samples to test the frequencies of the candidate SNPs selected from the database. This study was approved by the Ethics Committee of Kyushu University, Faculty of Medicine. DNA samples were extracted from leukocytes by standard methods [25].
SNP selection
We retrieved the primary SNP information from the dbSNP database [26]. Assuming the same size of the half length of LD (60 kb) as reported in Caucasians [27], we initially intended to select common SNPs every 30 kb in the three genes including all of the exonic SNPs. We tested the frequencies of the candidate SNPs, in the 16 healthy Japanese samples by the direct sequencing method. Out of them, common SNPs with minor allele frequencies over 10% were selected for further analyses. The SNPs in which significant deviation from Hardy-Weinberg equilibrium (HWE) observed in the 100 control samples were replaced by another SNP nearby. Since LD gaps (D ' < 0.3) were observed in the initial SNP set after the LD analyses described below, we selected additional SNPs to fill the LD gaps.
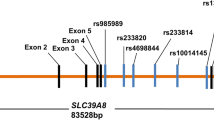
We finally selected the following 21 common SNPs distributed across the gene regions for further analyses: 7 of SLC1A4, SNP1, rs10211524; SNP2, rs7559202; SNP3, rs7592468; SNP4, rs3732062; SNP5, novel SNP in intron 3 (located on 33bp 5' to SNP rs7583682); SNP6, rs759458; SNP7, rs2540969, 5 of SLC1A5, SNP1, rs918486; SNP2, rs3027956; SNP3, rs313853; SNP4, rs2070246; SNP5, rs11673198, 6 of SLC6A5, SNP1, rs894747; SNP2, rs3781742; SNP3, rs3758807; SNP4, rs3819252; SNP5, rs2000959, SNP6, rs1792970 and 3 of SLC6A9, SNP1, rs783307; SNP2, rs2248829; SNP3, rs7555. The locations of the 21 SNPs are shown in Figure 1.

Genomic organizations of each gene and locations of the SNPs. (a) SLC1A4 spans over 34 kb and is composed of 8 exons. (b) SLC1A5 spans over 14 kb and is composed of 8 exons. (c) SLC6A5 spans over 55.6 kb and is composed of 16 exons. (d) SLC6A9 spans over 19.8 kb and is composed of 14 exons. Exons are shown as vertical bars with exon numbers. SNPs we analyzed are indicated by circles. Exonic SNPs are indicated by filled circles.
Genotyping
The 21 SNPs were amplified as individual fragments by PCR as previously described [14]. The nucleotide sequences of each primer, PCR conditions and genotyping methods for each SNP are shown in Additional File 1. Because of the GC rich sequences of SLC1A5 region, we used Fail Safe PCR system (Epicentre Technologies) to optimize the PCR conditions when amplified SNP1, SNP2 and SNP5 of SLC1A5. We genotyped samples for SNP5 of SLC6A5 by polymerase chain reaction/restriction fragment length polymorphism (PCR-RFLP) and the other 20 SNPs by direct sequencing, as previously described [28]. The raw data of direct sequencing were compiled on PolyPhred [29] and/or Mutation Surveyor (SoftGenetics LLC).
Statistical analyses
To control genotyping errors, Hardy-Weinberg equilibrium (HWE) for the genotype frequencies was checked in the control samples by the χ2-test (d.f. = 1). We evaluated the statistical differences in genotype and allele frequencies between cases and controls by Fisher's exact probability test. The magnitude of linkage disequilibrium (LD) was evaluated by caculating D ' using the haplotype frequencies estimated by the EH program, version 1.14 [30], and D' is represented graphically using the software Graphical Overview of Linkage Disequilibrium (GOLD) [31]. Statistical analysis of the haplotype association was carried out as previously described [32]. The significance level for all statistical tests was 0.05. We adjusted the P values of association studies for multiplicity using a false discovery rate (FDR) controlling procedure [33].
Results
Genotyping and SNP association analyses
We selected total 21 SNPs at an average interval of 7.8 kb for SLC1A4, 4.5 kb for SLC1A5, 14.2 kb for SLC6A5 and 18.9 kb for SLC6A9 to cover each entire gene region with LD. Since the average allele frequencies of the SNPs are 0.35, 0.30, 0.37 and 0.30 respectively, the expected detection powers for SLC1A4, SLC1A5, SLC6A5 and SLC6A9 are 0.84, 0.82, 0.84 and 0.82, respectively, under the multiplicative model with genotype relative risk = 1.8 [34]. Considering the high expected detection powers, we initially tested the single-marker association of the 21 SNPs with schizophrenia using the Screening Sample Set (100 cases and 100 controls) by the method described above, and investigated the association in the Full-size Sample Set (400 cases and 420 controls) only for the SNPs that showed significant single-marker or haplotype association with schizophrenia in the Screening Sample Set.
Table 1 shows the results of genotype and allele frequencies of SNPs in 100 case and 100 control samples. No significant deviation from HWE in control samples was observed in these SNPs (data not shown). We observed significant associations with schizophrenia in allele frequencies of SNP2 and SNP3 of SLC1A4 (P = 0.021, P = 0.029, respectively), and in genotype frequencies of SNP1 and SNP2 of SLC6A5 (P = 0.009, P = 0.022, respectively), although none of them survived after controlling the FDR at level 0.05 (n = 7 for SLC1A4 and n = 6 for SLC6A5).
Pairwise linkage disequilibrium and haplotype association analyses
We compared the magnitude of LD for all possible pairs of the SNPs in each gene region in controls and cases by calculating D' (Figure 2). No essential difference was shown in the LD pattern of any genes between cases and controls. Strong or modest LD (D' > 0.3) were observed in all combinations of adjacent SNPs in SLC1A5 and SLC6A9 regions. Whereas in each small subregion of the other two gene regions, LD drops abruptly: SNP4-SNP5 of SLC1A4 and SNP2-SNP3 of SLC6A5 (D' = 0.061 and D' = 0.125, respectively).

Pairwise LD analyses using GOLD for control (upper diagonal) and case (lower diagonal) haplotypes of each gene. The relative location of markers used to construct the haplotypes is represented on the horizontal and vertical axes, which is more clearly depicted in Figure. 1. LD measure, D', is graphically represented adjacent to each GOLD plot (red and dark blue are opposite ends of the scale).
We constructed pairwise haplotypes for all of the possible SNP pairs (Table 2). We observed significant associations with schizophrenia in combinations of SNP2-SNP7 of SLC1A4 (P = 0.037) and SNP1-SNP4 of SLC6A5 (P = 0.043). However, neither of them survived after controlling the FDR at level 0.05.
Association analyses using the Full-size Sample Set
Since nominally significant single-marker and haplotype associations with schizophrenia were observed in the Screening Sample Set, we genotyped the Full-size Sample Set for the SNPs involved in the significance, SNP2, SNP3 of SLC1A4 and SNP1, SNP2, SNP4 of SLC6A5 to examine these significant associations in the Full-size Sample Set. We excluded SNP7 of SLC1A4 from further analyses in the Full-size Sample Set because of the insufficient D' observed in the combination of SNP2-SNP7 in both cases and controls (D' = 0.064 and D' = 0.171, respectively). The genotype and allele frequencies of each SNP in the Full-size Sample Set are shown in the Additional File 2. The significant association of SNP1 of SLC6A5 with schizophrenia was confirmed in both genotype and allele frequencies in the Full-size Sample Set (P = 0.032, P = 0.018, respectively). We failed to detect other single-marker associations (P value range 0.065 – 0.355) and the haplotype association (P = 0.088) observed in the initial screening.
Discussion
SLC1A4, SLC1A5, SLC6A5 and SLC6A9 were located on chromosome 2p13-15, 19q13.3, 11p15.2-p15.1, and 1p33, respectively. Suggestive evidence for linkage of chromosome 2p14-p13, where SLC1A4 is located, with schizophrenia has been reported in schizophrenia families from Palau and Ireland [35, 36]. However, the subsequent mutation screening failed to find any sequence polymorphism segregated with the illness in the SLC1A4 region of the Palauan families [37]. In addition, negative association of SLC1A4 with schizophrenia was reported in the German population [38]. There has been no linkage with schizophrenia reported to the chromosome regions where SLC1A5, SLC6A5 or SLC6A9 are located [36]. Moreover, exclusion of linkage between schizophrenia and SLC1A5 in 23 English and Icelandic schizophrenia families was reported [39]. Recently, negative associations of schizophrenia with polymorphisms in SLC6A9 and SLC6A5 were reported in the Chinese and the German population, respectively [40, 41]. We investigated the association of SLC1A4, SLC1A5, SLC6A5 and SLC6A9 genes with schizophrenia in the Japanese population by analysing total 21 common SNPs.
Since the frequencies of genotyped SNPs are over 0.3, the expected detection powers of the four genes are over 0.80, assuming the genotype relative risk of 1.8. However, assuming lower genotype relative risk of 1.5 or 1.3, the expected detection powers for the four genes dropped to 0.50 – 0.53 or 0.24 – 0.25, respectively. Consequently, the negative finding for genes and SNPs excluded from the analyses using the Full-size Sample Set in this study may be due to type II error at lower relative risks, and they need to be investigated further in an enlarged sample size.
Out of the 21 SNPs analyzed, two within SLC1A4, (SNP4 and SNP6, 330 cases and 319 controls) and one within SLC6A5, (SNP5, 328 cases and 307 controls) have recently been reported to show no association with schizophrenia in the German population [38, 41]. We also observed no association of these SNPs with the disease in our Screening Sample Set. The SNP1 in SLC6A5 of which we observed a significant association with the disease, was not included in the report mentioned above.
In LD analysis of the initial screening of the 21 SNPs distributed in the four genes, modest LD (D' > 0.3) was observed in all combinations of adjacent SNPs in controls except for the combinations of SNP4-SNP5 of SLC1A4 and SNP2-SNP3 of SLC6A5, suggesting recombination hot spots in the two regions (6.6 kb and 7.6 kb, respectively) (Figure 2). We compared the LD structure to the publically open database, HapMap [42]. The LD gap we observed in the SLC6A5 region was not observed in the HapMap LD structure from either Japanese or Chinese population data (D' = 0.817 and D' = 1, respectively). The other LD gap, which was observed in the SLC1A4 region, failed to be compared due to the absence of the novel SNP we found.
We observed significant single-marker associations in SNP2 and SNP3 of SLC1A4 in the Screening Sample Set. However, we failed to confirm these findings in the Full-size Sample Set. We attribute to type I error due to the small sample size used in the Screening Sample Set. On the other hand, the single-marker association of SNP1 (rs894747) in SLC6A5 region, although it does not show the significant association with the disease in the independent 300 case and 320 control samples (0.092), it does show the significant association in the Full-size Sample Set (P = 0.018). We consider that the nonsignificant result observed in the enlarged samples may be due to the small sample size. SNP1 is located in the intergenic region, 2,355-bp upstream from the transcription start site. In the negative association report of SLC6A5 in German population described above, four SNPs and one short-tandem-repeat distributed in intron 1~intron 11, but no SNP located in the upstream region were analysed [39]. In our Full-size Sample Set, the G allele was more frequently observed in schizophrenics (44.4%) than in controls (38.6%). Therefore, the G allele may be in LD with a risk allele for schizophrenia (odds ratio, 1.27; 95% confidence interval, 1.04~1.55). We conclude that at least one susceptibility locus for schizophrenia is located within or nearby SLC6A5, whereas SLC1A4 SLC1A5 and SLC6A9 are unlikely to be major susceptibility genes for schizophrenia in the Japanese population. No potential regulatory elements were previously identified in the region where SNP1 is located [43]. It is necessary to search for functional SNPs in the haplotype block where SNP1 is located. A copy number variation (CNV) has been reported in the European population on the chromosome 11p15.1, containing exon 15 of SLC6A5 [44]. None of the 6 SNPs we genotyped is located within the CNV. Although the frequency of the CNV in the Japanese population is unknown, SNP1 may be associated with the variant devoid of exon 15, which is a strong candidate of the susceptible allele. Therefore, it is necessary to test the association of the CNV with schizophenia in Japanese sample sets. The positive association observed in SLC6A5 also needs to be validated in different ethnic populations.
Conclusion
We conclude that at least one susceptibility locus for schizophrenia is located within or nearby SLC6A5, whereas SLC1A4 SLC1A5 and SLC6A9 are unlikely to be major susceptibility genes for schizophrenia in the Japanese population.
References
Gottesman PE: Schizophrenia genesis: The origins of madness. 1995, New York: Freeman
McGuffin P, Owen MJ, Farmer AE: Genetic basis of schizophrenia. Lancet. 1995, 346: 678-682. 10.1016/S0140-6736(95)92285-7.
Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R: Study of a new schizophrenomimetic drug, sernyl. AMA Arch Neurol Psychiatry. 1959, 81 (3): 363-369.
Javitt DC, Zukin SR: Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991, 148: 1301-1308.
Mohn AR, Gainetdinov RR, Caron MG, Koller BH: Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999, 98: 427-436. 10.1016/S0092-8674(00)81972-8.
Harrison PJ, Weinberger DR: Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005, 10: 40-68. 10.1038/sj.mp.4001558.
Riley B, Kendler KS: Molecular genetic studies of schizophrenia. Eur J Hum Genet. 2006, 14: 669-80. 10.1038/sj.ejhg.5201571.
de Bartolomeis A, Fiore G, Iasevoli F: Dopamine-glutamate interaction and antipsychotics mechanism of action: implication for new pharmacological strategies in psychosis. Curr Pharm Des. 2005, 11: 3561-94. 10.2174/138161205774414538.
Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH: Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am J Psychiat. 2001, 158: 1393-1399. 10.1176/appi.ajp.158.9.1393.
Matute C, Melone M, Vallejo-Illarramendi A, Conti F: Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia. 2005, 49: 451-455. 10.1002/glia.20119.
Ohnuma T, Tessler S, Arai H, Faull RL, McKenna PJ, Emson PC: Gene expression of metabotropic glutamate receptor 5 and excitatory amino acid transporter 2 in the schizophrenic hippocampus. Mol Brain Res. 2000, 85: 24-31. 10.1016/S0169-328X(00)00222-9.
McCullumsmith RE, Meador-Woodruff JH: Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology. 2002, 26: 368-375. 10.1016/S0893-133X(01)00370-0.
Deng X, Shibata H, Ninomiya H, Tashiro N, Iwata N, Ozaki N, Fukumaki Y: Association study of polymorphisms in the excitatory amino acid transporter 2 gene (SLC1A2) with schizophrenia. BMC Psychiatry. 2004, 4: 21-10.1186/1471-244X-4-21.
Deng X, Shibata H, Takeuchi N, Rachi S, Sakai M, Ninomiya H, Iwata N, Ozaki N, Fukumaki Y: Association study of polymorphisms in the glutamate transporter genes SLC1A1, SLC1A3, and SLC1A6 with schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2007, 144B (3): 271-8. 10.1002/ajmg.b.30351.
Arriza JL, Kavanaugh MP, Fairman WA, Wu YN, Murdoch GH, North RA, Amara SG: Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem. 1993, 268: 15329-15332.
Utsunomiya-Tate N, Endou H, Kanai Y: Cloning and functional characterization of a system ASC-like Na+dependent neutral amino acid transporter. J Biol Chem. 1996, 271: 14883-14890. 10.1074/jbc.271.25.14883.
Weiss MD, Derazi S, Kilberg MS, Anderson KJ: Ontogeny and localization of the neutral amino acid transporter ASCT1 in rat brain. Brain Res Dev Brain Res. 2001, 130: 183-90. 10.1016/S0165-3806(01)00250-4.
Sakai K, Shimizu H, Koike T, Furuya S, Watanabe M: Neutral amino acid transporter ASCT1 is preferentially expressed in L-Ser-synthetic/storing glial cells in the mouse brain with transient expression in developing capillaries. J Neurosci. 2003, 23: 550-60.
Yamamoto T, Nishizaki I, Nukada T, Kamegaya E, Furuya S, Hirabayashi Y, Ikeda K, Hata H, Kobayashi H, Sora I, Yamamoto H: Functional identification of ASCT1 neutral amino acid transporter as the predominant system for the uptake of L-serine in rat neurons in primary culture. Neurosci Res. 2004, 49: 101-11. 10.1016/j.neures.2004.02.004.
Broer A, Brookes N, Ganapathy V, Dimmer KS, Wagner CA, Lang F, Broer S: The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J Neurochem. 1999, 73: 2184-94.
Weis S, Llenos IC, Dulay JR, Verma N, Sabunciyan S, Yolken RH: Changes in region- and cell type-specific expression patterns of neutral amino acid transporter 1 (ASCT-1) in the anterior cingulate cortex and hippocampus in schizophrenia, bipolar disorder and major depression. J Neural Transm. 2007, 114: 261-71. 10.1007/s00702-006-0544-0.
Borowsky B, Mezey E, Hoffman BJ: Two glycine transporter variants with distinct localization in the CNS and peripheral tissues are encoded by a common gene. Neuron. 1993, 10: 851-63. 10.1016/0896-6273(93)90201-2.
Lechner SM: Glutamate-based therapeutic approaches: inhibitors of glycine transport. Curr Opin Pharmacol. 2006, 6: 75-81. 10.1016/j.coph.2005.11.002.
American Psychiatric Association: DSM-IV: Diagnostic and Statistical Manual of Mental Disorders. 1994, American Psychiatric Press, Washington, DC
Lahiri DK, Nurnberger JI: A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991, 19: 5444-10.1093/nar/19.19.5444.
dbSNP database. [http://www.ncbi.nlm.nih.gov/SNP]
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES: Linkage disquilibrium in the human genome. Nature. 2001, 411: 199-204. 10.1038/35075590.
Shibata H, Joo A, Fujii Y, Tani A, Makino C, Hirata N, Kikuta R, Ninomiya H, Tashiro N, Fukumaki Y: Association study of polymorphisms in the GluR5 kainate receptor gene (GRIK1) with schizophrenia. Psychiatr Genet. 2001, 11: 139-44. 10.1097/00041444-200109000-00005.
Nickerson DA, Tobe VO, Taylor SL: Polyphred: substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997, 25: 2745-2751. 10.1093/nar/25.14.2745.
Xie X, Ott J: Testing linkage disequilibrium between a disease gene and marker loci. Am J Hum Genet. 1993, 53: 1107-
Abecasis GR, Cookson WO: GOLD: graphical overview of linkage disequilibrium. Bioinformatics. 2000, 16: 182-183. 10.1093/bioinformatics/16.2.182.
Sham P: Statistics in human genetics. 1998, Oxford University Press, New York
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I: Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001, 125: 279-284. 10.1016/S0166-4328(01)00297-2.
Ohashi J, Tokunaga K: The power of genome-wide association studies of complex disease genes: statistical limitations of indirect approaches using SNP markers. J Hum Genet. 2001, 46: 478-482. 10.1007/s100380170048.
Coon H, Myles-Worsley M, Tiobech J, Hoff M, Rosenthal J, Bennett P, Reimherr F, Wender P, Dale P, Polloi A, Byerley W: Evidence for a chromosome 2p13-14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol Psychiatry. 1998, 3: 521-527. 10.1038/sj.mp.4000453.
Straub RE, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, O'Neill FA, Walsh D, Kendler KS: Genome-wide scans of three independent sets of 90 Irish multiplex schizophrenia families and follow-up of selected regions in all families provides evidence for multiple susceptibility genes. Mol Psychiatry. 2002, 7: 542-559. 10.1038/sj.mp.4001051.
Bennett PJ, Hoff M, Rosenthal J, Zhao M, Coon H, Myles-Worsley M, Byerley WF: Mutation screening of a neutral amino acid transporter, ASCT1, and its potential role in schizophrenia. Psychiatr Genet. 2000, 10: 79-82.
Skowronek MH, Georgi A, Jamra RA, Schumacher J, Becker T, Schmael C, Paul T, Deschner M, Hofels S, Wulff M, Schwarz M, Klopp N, Illig T, Propping P, Cichon S, Nothen MM, Schulze TG, Rietschel M: No association between genetic variants at the ASCT1 gene and schizophrenia or bipolar disorder in a German sample. Psychiatr Genet. 2006, 16: 233-4. 10.1097/01.ypg.0000218621.58009.d1.
Chen AC, Kalsi G, Brynjolfsson J, Sigmundsson T, Curtis D, Butler R, Read T, Murphy P, Barnard EA, Petursson H, Gurling HM: Exclusion of linkage between schizophrenia and the gene encoding a neutral amino acid glutamate/aspartate transporter, SLC1A5. Am J Med Genet. 1997, 74: 50-2. 10.1002/(SICI)1096-8628(19970221)74:1<50::AID-AJMG11>3.0.CO;2-Q.
Tsai SJ, Cheng CY, Hong CJ, Liao DL, Hou SJ, Yen FC, Liou YJ: Association study of polymorphisms in glycine transporter with schizophrenia. J Neural Transm. 2006, 113: 1545-9. 10.1007/s00702-006-0438-1.
Jamra RA, Villela AW, Klein K, Becker T, Schulze TG, Schmael C, Deschner M, Klopp N, Illig T, Propping P, Cichon S, Rietschel M, Nothen MM, Schumacher J: No association between genetic variants at the GLYT2 gene and bipolar affective disorder and schizophrenia. Psychiatr Genet. 2006, 16: 91-10.1097/01.ypg.0000199450.07786.ab.
HapMap database. [http://www.hapmap.org]
Ponce J, Poyatos I, Aragon C, Gimenez C, Zafra F: Characterization of the 5' region of the rat brain glycine transporter GLYT2 gene: identification of a novel isoform. Neurosci Lett. 1998, 242: 25-8. 10.1016/S0304-3940(98)00037-8.
Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE: An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006, 16: 1182-90. 10.1101/gr.4565806.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-244X/8/58/prepub
Acknowledgements
We are grateful to all the medical staff involved in collecting specimens, especially to Dr. Nobutada Tashiro, the emeritus professor of Kyushu University Graduate School of Medical Science for the initial support to our project. This work was supported by KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas "Applied Genomics" and other grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by The Naito Foundation Subsidy for Natural Science Researches.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XD carried out a portion of genotyping, statistical analyses and drafted the manuscript; NS, NT, MT carried out a portion of genotyping and statistical analyses; HN, NI and NO participated in collecting specimens and clinical data; HS participated in design of this study and statistical analyses; YF conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
Electronic supplementary material
12888_2007_502_MOESM1_ESM.xls
Additional file 1: PCR primers for genotyping of SNPs in the genes. The data provided the nucleotide sequences of primers, PCR conditions and genotyping methods for each SNP. (XLS 26 KB)
12888_2007_502_MOESM2_ESM.xls
Additional file 2: Genotype and allele frequencies of SNPs in the Full-size Sample Set (400 cases and 420 controls). The data show genotype and allele frequencies of SNPs in the Full-size Sample Set. (XLS 21 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Deng, X., Sagata, N., Takeuchi, N. et al. Association study of polymorphisms in the neutral amino acid transporter genes SLC1A4, SLC1A5 and the glycine transporter genes SLC6A5, SLC6A9with schizophrenia. BMC Psychiatry 8, 58 (2008). https://doi.org/10.1186/1471-244X-8-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-244X-8-58