
Dedication
This article is dedicated to the memory of Sue Kim Hanson, a graduate student in the department of Pathology and Laboratory Medicine at Boston University School of Medicine, who perished in the terrorist attacks of September 11, 2001.
Abstract
Background
In cholinergic neurons, the hydrolysis of phosphatidylcholine (PC) by a phospholipase D (PLD)-type enzyme generates some of the precursor choline used for the synthesis of the neurotransmitter acetylcholine (ACh). We sought to determine the molecular identity of the relevant PLD using murine basal forebrain cholinergic SN56 cells in which the expression and activity of the two PLD isoforms, PLD1 and PLD2, were experimentally modified. ACh levels were examined in cells incubated in a choline-free medium, to ensure that their ACh was synthesized entirely from intracellular choline.
Results
PLD2, but not PLD1, mRNA and protein were detected in these cells and endogenous PLD activity and ACh synthesis were stimulated by phorbol 12-myristate 13-acetate (PMA). Introduction of a PLD2 antisense oligonucleotide into the cells reduced PLD2 mRNA and protein expression by approximately 30%. The PLD2 antisense oligomer similarly reduced basal- and PMA-stimulated PLD activity and ACh levels. Overexpression of mouse PLD2 by transient transfection increased basal- (by 74%) and PMA-stimulated (by 3.2-fold) PLD activity. Moreover, PLD2 transfection increased ACh levels by 26% in the absence of PMA and by 2.1-fold in the presence of PMA. Overexpression of human PLD1 by transient transfection increased PLD activity by 4.6-fold and ACh synthesis by 2.3-fold in the presence of PMA as compared to controls.
Conclusions
These data identify PLD2 as the endogenous enzyme that hydrolyzes PC to generate choline for ACh synthesis in cholinergic cells, and indicate that in a model system choline generated by PLD1 may also be used for this purpose.
Similar content being viewed by others


Background
The synthesis of the neurotransmitter acetylcholine (ACh)1 in cholinergic neurons is dependent on the availability of its precursor, choline, most of which is derived from the circulation (reviewed in [1]) and enters cholinergic neurons via a process catalyzed by a specific transporter [2, 3]. Some of the choline used for ACh synthesis is stored in membrane phosphatidylcholine (PC). Previous studies have shown that this choline is liberated from PC in a one-step reaction, thus indicating that it is produced by a phospholipase D (PLD, EC 3.1.4.4) -an enzyme that hydrolyzes the phospholipid into choline and phosphatidic acid (PA) [4]. However, the molecular identity of this PLD remains to be determined.
There are two distinct PC-specific mammalian PLDs, designated as PLD1 and PLD2 and encoded by separate genes [5, 6]. The two enzymes share 50% amino-acid identity, however, regulation of their activity and cellular localization are different. PLD1 is an extracellular signal-activated phospholipase; its activity rises when receptors for certain hormones, growth factors, neurotransmitters, cytokines, and other mediators are activated, and upon treatment of cells with phorbol esters (reviewed in [7, 8]). Studies using recombinant PLD1 tagged with green fluorescent protein or Flu-epitopes indicate that it is localized in the endoplasmic reticulum, Golgi apparatus, late endosomes, and plasma membrane ([6], reviewed in [7]). In vitro, basal PLD1 activity is usually low and can be stimulated by a variety of factors, including phosphatidylinositol 4, 5-bisphosphate (PIP2), protein kinase C (PKC), and several small GTPases, e.g. ADP ribosylation factors (ARF) and Rho (reviewed in [7, 8]). Regulation of PLD2 is less well understood. The enzyme exhibits a high constitutive activity in vitro, but it too can be activated by ARF (reviewed in [7]), and by phorbol esters, in intact cells [6, 9]. Several reports have suggested that PLD2 is mainly localized to the plasma membrane [6, 10], possibly in lipid rafts and caveolar membrane microdomains [11, 12]. Most studies on the physiological functions of PLDs have focused on the product PA, a second messenger involved in mitosis, cellular morphological changes and vesicular trafficking (reviewed in [7, 13]). However, the downstream events that occur as a consequence of PLD-generated PA production, and the identity of target proteins that are directly regulated by PA remains to be determined. PA can be further metabolized to diacylglycerol (DAG) and lysophosphatidic acid by PA phosphohydrolase and phospholipase A2, respectively. DAG is an activator of certain isozymes of PKC and lysophosphatidic acid acts as an intercellular messenger for many cell types.
The second product of PLD-catalyzed PC hydrolysis, choline, is used by all cells as a precursor of PC, and uniquely by cholinergic neurons as precursor of ACh. Previous studies have shown that choline derived from PC is indeed used for ACh synthesis [4, 14–16] and that this choline is generated by a PLD [4, 14]. This pathway may become particularly important when extracellular circulating choline concentrations are low (e.g. during dietary choline deficiency) or when ACh synthesis and release are accelerated by high neuronal activity, thus generating a high demand for choline [16].
In the present study, we used a cholinergic cell line, SN56 [17], which is derived from the fusion of septal neurons of postnatal day 21 mice with N18TG2 murine neuroblastoma cells, as a model for cholinergic neurons, in order to determine the possible role of PLD 1 and PLD2 in generating choline for ACh synthesis. We found that the cells express exclusively PLD2 and that downregulating PLD2 expression reduces ACh synthesis, while overexpressing and/or stimulating PLD2 activity increases the synthesis of the neurotransmitter, indicating that PLD2-catalyzed hydrolysis of PC generates choline for ACh synthesis in cholinergic cells.
Results
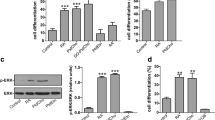
PLD2 mRNA was present in SN56 cells (Fig. 1A) while PLD1 message was undetectable either by Northern blot (Fig. 1A) or RT-PCR (data not shown). Consistent with these results an approximately 90 kDa PLD2 protein was found in SN56 cells by Western blot; however, no PLD1 protein could be detected (Fig. 1B). Positive controls for these studies showed that our PLD1 probe and antibody were robust in detecting this PLD isoform in rat brain (Fig. 1). These data show that SN56 cells express exclusively PLD2 and thus experimental manipulations of the activity of this PLD isoform were subsequently employed to determine if this enzyme is capable of producing choline for ACh synthesis in cholinergic cells. In order to reduce PLD2 activity in SN56 cells, we sought to inhibit its expression. The cells were treated with a 25-mer antisense oligonucleotide at different concentrations and PLD2 mRNA levels were measured. Northern blot showed that PLD2 mRNA expression was reduced by the antisense oligonucleotide in a concentration-dependent manner (Fig. 2A), and that, at the maximally-effective concentration of the oligo (200 nM), this inhibition reached 34% (Fig. 2B,C). In contrast, the control oligomer was inactive (Fig. 2B,C). Consistent with these findings. Western blot showed that the antisense oligomer decreased the PLD2 protein level by about 28%, whereas the control oligomer had no significant effect on PLD2 protein expression (Fig. 2D,E). Given the fact that the transfection efficiency obtained in these experiments was approximately 30% (as determined with fluorescent dye-labeled oligonucleotides; data not shown), the results indicate that, in cells that had taken up the antisense oligomer, the inhibition of PLD2 expression was complete.

SN56 cells express PLD2 but not PLD1 mRNA and protein. Total RNA or protein lysates were prepared and subjected to Northern or Western blot analyses as described in the Experimental Procedures. A. Northern blot. Twenty μg of total RNA was subjected to electrophoresis on a 1.0% agarose gel and hybridized respectively with a mouse PLD2 probe, a human PLD1 probe and a mouse G3PDH probe. B. Western blot. A polyclonal anti-mouse PLD1 antibody and a polyclonal anti-human PLD2 antibody were used as primary antibodies. Rat brain total RNA (A) and protein lysates (B) were used as positive controls.

PLD2 antisense oligonucleotides reduce endogenous PLD2 mRNA expression and protein levels in SN56 cells. SN56 cells were transfected with either control or antisense oligonucleotides as described in the Experimental Procedures. Two days later PLD2 gene expression was assessed by Northern blot analysis (A-C) as described in Fig. 1. G3PDH was used for normalization (A-C). For Western blots 40 μg of lysate protein was subjected to SDS-PAGE. A polyclonal anti-human PLD2 antibody was used first to detect PLD2, the blot was stripped and rehybridized with a monoclonal anti-mouse actin antibody for normalization (D,E). A. Concentration-response curve to the antisense oligonucleotide. B. Comparison of the effects of the antisense and control oligonucleotides on PLD2 expression. The cells were transfected with 200 nM of either control or antisense oligomer as indicated. C. Quantification of results presented in B. Intensity of the hybridization bands was normalized to that of G3PDH. The antisense oligomer reduced PLD2 expression by 34%, *p < 0.05. Data shown are from 3–6 experiments and expressed as average ± SEM. D. The antisense oligomer reduced PLD2 protein expression as compared to mock-treated and control oligomer-transfected cells. E. The intensity of the PLD2 bands was quantified using NIH Image 1.62 and normalized to that of actin. The antisense oligomer reduced the PLD2 protein level by 28% compared to controls, *p < 0.05. The results are from 3 experiments and expressed as average ± SEM.
Treatment of a variety of cell types with phorbol esters activates PLD2 [6, 12, 18, 19]. Thus we hypothesized that activation of PLD with PMA in SN56 cells would elevate intracellular concentrations of its product, choline, and result in increased ACh production. We tested the effects of PMA on PLD activity and ACh content in SN56 cells incubated in a choline-free medium. Under these conditions the choline precursor used for the synthesis of ACh is derived entirely from the intracellular pool, which includes the choline in PC [4, 15]. Consistent with previous studies, treatment of the cells with PMA activated PLD (Fig. 3A). The maximal effects of PMA (3.3 fold increase in the amount of its product [3H]phosphatidylpropanol relative to controls) was observed after 45 minutes (data not shown), a time period employed in subsequent studies. Free choline accumulation was also significantly increased by PMA. The magnitude of this effect tended to be small (approximately 15%), presumably because free choline is a rapidly turning over metabolic intermediate that does not accumulate in cells (Fig. 3B). Moreover, PMA stimulated ACh accumulation by 26% (Fig. 3C), in the cells. The fact that PMA increased both PLD activity and ACh content provided the initial evidence for a relationship between the endogenous PLD activity (i.e. PLD2) and ACh production. In control studies performed in the presence of choline (15 μM) in the medium PMA had no effect on ACh levels (data not shown), indicating that it did not stimulate the process of ACh synthesis but only the supply of choline.

PLD2 antisense oligonucleotides down-regulate PLD2 activity, and acetylcholine and choline levels in SN56 cells. SN56 cells were transfected with either control or antisense oligonucleotides as described in the Experimental Procedures. After 48 hours the cells were preincubated for 20 minutes in choline-free medium and then incubated in fresh choline-free medium containing 50 μM neostigmine with or without 100 nM PMA for 45 minutes. PLD activity in the intact cells (A) and the intracellular choline (B) and acetylcholine (C) content were measured. The data were analyzed by two-way ANOVA and Fisher's post hoc test and are expressed as average ± SEM. The asterisks indicate a significant difference between the antisense and control transfectants (A-C). A. Effect of antisense oligonucleotides on PLD activity. The effect of PMA treatment is statistically significant in all groups. B. Effect of antisense oligonucleotides on intracellular choline level. C. Effect of antisense oligonucleotides on intracellular ACh level. The effect of PMA treatment is statistically significant in all groups.
Next the effect of downregulation of PLD2 activity on ACh levels was studied in cells transfected with either control or antisense oligomer and treated with PMA. In the absence of PMA, the antisense oligomer decreased PLD2 activity by 19%. Although small, this change was statistically significant (p < 0.05). In the presence of PMA, antisense oligomer decreased PLD2 activity by 27% (Fig. 3A). Concomitantly the cellular choline content was decreased by 30% both in the presence and absence of PMA by the antisense oligomer (Fig. 3B). Moreover, the antisense oligomer decreased the cellular ACh content by approximately 30% compared to control oligomer treated cells both in the presence and absence of PMA (Fig. 3C). These results indicate that downregulation of PLD2 activity by antisense oligomers in SN56 cells decreases not only the endogenous PLD2 activity but also the intracellular choline and ACh content. These results are consistent with the hypothesis that PLD2 can generate choline for ACh synthesis in SN56 cells.
To further confirm this hypothesis, we transiently transfected the cells with a mouse PLD2 expression plasmid. Overexpression of PLD2 for 48 hours in the cells caused a 74% increase of basal PLD2 activity and this activity was further increased by PMA by 3.2-fold (Fig. 4A). To examine the ACh synthesis and choline turnover in these PLD2 transfectants, the cells and the media were collected and ACh and choline content were measured. In the absence of PMA, overexpression of PLD2 alone increased intracellular ACh levels by 26% and choline concentrations in the medium by 57%. In the presence of PMA, intracellular ACh levels and choline concentration in the medium rose further in the PLD2 transfectants reaching values more than two-fold higher than those in the control cells (Fig. 4B,C). Together with the data presented above, these results show that up-regulation of PLD2 activity by overexpression and/or stimulation with PMA increases intracellular ACh levels.

Overexpression of PLD2 in SN56 cells increases enzyme activity, acetylcholine synthesis and choline content in the medium. The cells were transfected with the expression plasmid as described in the Experimental Procedures. After 48 hours the cells were treated as described in Fig. 3 and PLD activity in intact cells (A), choline content of the medium (B), and intracellular ACh content (C), were measured. The data were analyzed by two-way ANOVA and Fisher's post hoc test and are expressed as average ± SEM. The asterisks indicate a significant difference between the PLD2 and mock transfectants (A-C). A. Effect of PLD2 overexpression on PLD activity. The effect of PMA treatment is statistically significant in all groups. B. Effect of PLD2 overexpression on media choline level. The effect of PMA treatment is statistically significant in PLD2 transfectants. C. Effect of PLD2 overexpression on intracellular ACh level. The effect of PMA treatment is statistically significant in PLD2 transfectants.
A recent immunohistochemical study of PLD1 expression in rat brain showed that this PLD isoform was expressed in neurons in medial septum, cranial motor nuclei and ventral spinal cord [20], all areas of the central nervous system enriched with cholinergic neurons. These data raised the possibility that certain populations of cholinergic neurons may express PLD1 in addition to, or instead of, PLD2. Therefore we transfected SN56 cells with PLD1 expression construct, in order to determine if choline generated by this enzyme could be used as ACh precursor. Transfection of PLD1 into SN56 cells resulted in increased PLD activity and ACh synthesis in a fashion similar to that observed in PLD2 transfectants (Fig. 5) indicating that choline generated by PLD1 was converted to ACh.

Overexpression of PLD1 in SN56 cells increases enzyme activity, acetylcholine synthesis and choline content in the medium. The cells were transfected with the hPLD1 expression plasmid as described in the Experimental Procedures. After 48 hours the cells were treated as described in Fig. 3 and PLD activity in intact cells (A), choline content of the medium (B), and intracellular ACh content (C), were measured. The data were analyzed by two-way ANOVA and Fisher's post hoc test and are expressed as average ± SEM. The asterisks indicate a significant difference between the PLD1 and mock transfectants (A-C). A. Effect of PLD 1 overexpression on PLD activity. The effect of PMA treatment is statistically significant in all groups. B. Effect of PLD1 overexpression on media choline level. The effect of PMA treatment is statistically significant in PLD1 transfectants. C. Effect of PLD1 overexpression on intracellular ACh level. The effect of PMA treatment is statistically significant in PLD1 transfectants.
Discussion
In the present study, we sought to establish which PLD isoform is the endogenous enzyme that generates the choline precursor for ACh synthesis in cholinergic neurons using, as a model, the SN56 basal forebrain cell line. We found that these cells expressed exclusively PLD2, a property that facilitated subsequent experiments. First, antisense oligonucleotides were used to down-regulate endogenous PLD2 activity. Although only 30% of the cells took up the oligonucleotides, PLD2 mRNA, protein expression, and activity were reduced proportionally to the transfection efficiency of the antisense oligomer, demonstrating high efficacy of this technique in the transfected cells. Significantly, intracellular choline and ACh content were similarly decreased by the antisense oligonucleotides, indicating that PLD2-catalyzed hydrolysis of PC provides choline for ACh synthesis. Next, PLD2 activity was up-regulated by either overexpressing a mouse PLD2 protein and/or by stimulation with PMA. Both PLD2 overexpression and stimulation with PMA increased PLD activity, choline production, and ACh accumulation. Moreover, PLD2 transfection and PMA treatment had a synergistic effect on PLD activity and ACh levels.
Our observation that PLD2 but not PLD1 was present in SN56 cells raised the possibility that the former might be preferentially expressed in cholinergic neurons. The data are consistent with the report that a variant of PC12 cells also express exclusively PLD2 [18] and this expression is stimulated by nerve growth factor [18], a treatment known to upregulate the cholinergic phenotype of PC12 cells [21, 22]. PLD2 mRNA is abundant in many regions of the adult rodent central nervous system [6, 23], including cerebellum, frontal cortex, hippocampus, hypothalamus, midbrain, brainstem, striatum, and spinal cord [24]. The latter three regions are enriched with cell bodies of cholinergic neurons. PLD1 is also highly expressed in brain tissue [6, 23, 25], notably in neurons in medial septum, cranial motor nuclei and ventral spinal cord [20], all of which are likely cholinergic. However, no data are currently available on the possible coexpression of PLD1 and/or PLD2 and cholinergic markers (e.g. choline acetyltransferase) in vivo. Transfection of PLD1 into SN56 cells resulted in increased PLD activity and ACh production suggesting that PLD1, like PLD2, could potentially be used to supply choline for ACh synthesis. Thus, while the catalytic activities of both PLD1 and PLD2 appear to be equivalent in generating choline for ACh synthesis in transfected cells, it is possible that the modes of regulation of their activity might specify which one is expressed in a particular population of cholinergic neurons in vivo. It will be important to determine the mechanisms that control the expression of the two PLD isoforms and their possible relationships with the mechanisms that regulate the cholinergic phenotype.
Stimulation of PLD activity by PMA and the resulting increase of ACh synthesis from PLD-generated choline indicates that this pathway is regulated by PMA-responsive PKC isozymes. In vivo, this form of regulation of ACh synthesis would be controlled by neurotransmitters released by neurons that innervate particular cholinergic cells. There are multiple neurotransmitters whose receptors are coupled to PKC-dependent activation of PLD [7], and thus this mode of regulating the availability of choline for ACh synthesis is likely to be widespread. Significantly, among the five subtypes of muscarinic ACh receptors (MAChRs), Ml and M3 receptors have been shown to efficiently activate PLD (and PKC) in various cell types [26–30], whereas activation of PLD by M2 and M4 receptors is less robust [26]. These observations suggest that in a cholinergic synapse, ACh released into the synaptic cleft might stimulate PLD activity leading to a release of choline that could be used for ACh synthesis. Since these MAChRs may be present on the pre- and postsynaptic neurons [31], this choline may be derived from PC residing in the membranes of either. However, while the presynaptically-derived choline released into the cytoplasm can be converted directly to ACh, the choline released by the postsynaptic cell must be initially taken up by the cholinergic nerve terminals. Our previous results in cholinergic human neuroblastoma LA-N-2 cells indicated that the choline moiety from PC breakdown was released directly into the cytoplasm [4] and a kinetic study using [3H]choline in BHK cells also showed that choline was initially released inside the cells by PMA-activated PLD and then rapidly appeared in the medium [32]. Consistent with the latter study, we also observed a rise in the medium choline levels in SN56 cells transfected with either PLD1 or PLD2 and treated with PMA.
Although it is clear that the PLD-generated choline is efficiently utilized by cholinergic cells (LA-N-2 and SN56) as a precursor of ACh, the quantitative significance of this pathway and mechanisms that regulate it remain to be fully determined. Studies on a variety of preparations indicate that most of the choline used for ACh synthesis is derived from the extracellular space (reviewed in [1]) and therefore the PLD pathway has to be considered as an accessory process. Interestingly, PLD activity is upregulated in the hippocampus of rats whose mothers consumed a choline-supplemented diet during pregnancy [33]. Given that prenatal choline supplementation improves memory and attention [34], two cognitive functions that require the hippocampal cholinergic system, it is possible that the upregulated PLD pathway underlies, at least in part, the cognitive enhancement observed in the prenatally choline-supplemented animals.
Abnormalities in the PLD-mediated release of choline would be expected to result in an impairment of cholinergic neurotransmission and/or in a generalized malfunction of cholinergic cells. Alzheimer's disease (AD) is characterized by such a malfunction together with degeneration of basal forebrain cholinergic neurons, and it is thought that memory loss in this disease is partly caused by this cholinergic lesion [35]. In vitro studies point to two novel mechanisms that might cause dysregulation of PLD2 activity in AD. The first is based on observations that PLD2 is inhibited by α-synuclein [36], a protein of unknown function that accumulates in the amyloid deposits in AD brain. Normally α-synuclein is present in the synaptic nerve terminals (including those of cholinergic neurons [37, 38]) but in AD its processing is altered, causing it to be abnormally deposited in plaques composed primarily of aggregates of β-amyloid peptide [39]. However, how this abnormal turnover of α-synuclein contributes to the pathophysiology of AD remains to be determined. Recent in vitro observations that α-synuclein is phosphorylated by G protein-coupled receptor kinases, and that this phosphorylation relieves its inhibitory action on PLD2 [40], suggest a novel mechanism by which neurotransmitter receptors (including muscarinic receptors) might modulate PLD2 activity. Since multiple receptor classes are affected by the pathophysiological processes of AD [41], abnormal phosphorylation of α-synuclein may contribute to both altered α-synuclein turnover and abnormal regulation of PLD2 activity. The second set of observations indicate that reactive oxygen species (ROS) activate PLD2 in a PKC-dependent manner [9]. There is much evidence that ROS generation is upregulated in AD brain [42]. Thus, it is possible that PLD2 may be activated in this disease resulting, perhaps, in dysregulation of PC metabolism. Indeed evidence of an accelerated PC turnover has been repeatedly observed in post-mortem samples of AD brain [43–47] as well as in an in vitro model of this disease characterized by the generation of ROS [48].
Conclusions
Most of the previous studies on the physiological functions PLDs have concentrated on the role of its product, PA [7]. Our data show that PLD has a dual function in cholinergic cells, as the second product of its activity, choline, is used as a metabolic precursor of ACh, and suggest that this pathway may be regulated by the type of PLD isoform expressed in a particular class of cholinergic neurons. In our basal forebrain-derived SN56 cells, this isoform is PLD2. However, our observations that PLD1 overexpression increases ACh production form endogenous sources suggest that PLD1 may serve the same function as PLD2 in other cholinergic cells. Moreover, the data showing that this pathway is stimulated by PMA, suggest that it is subject to regulation by PKC-activating neurotransmitters. The physiological, and possible pathophysiological significance in neurological diseases, of this pathway remains to be determined. It will also be important to determine how cholinergic cells regulate the metabolism of PA – a compound with multiple signaling and structural functions – when the PLD pathway is stimulated to meet the demand for choline.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and Lipofectamine Plus reagents were from Gibco BRL Life Technologies (Grand Island, NY). Phosphatidylpropanol (PP) was from Avanti Polar Lipids (Alabaster, AL). Silica LK6 Thin Layer Chromotography (TLC) plates were from Whatman. [3H]Myristic acid and [α-32P]dCTP were from Dupont-NEN (Boston, MA). Molecular biology reagents (except as noted) were from Sigma (St. Louis, MO) and phorbol 12-myristate 13-acetate was from Calbiochem (La Jolla, CA). Polyclonal anti-human PLD2- and anti-mouse PLD1 antibodies were kindly provided by Dr. Yoshinori Nozawa (Gifu International Institute of Biotechnology, Japan).
Cell culture
Mouse septal neuron x neuroblastoma hybrid SN56 cells were maintained at 37°C in an atmosphere of 95% air, 5% CO2 in DMEM supplemented with 10% FBS. The medium was changed every 2–3 days. Cells of passage 10 to 40 were used.
Transient transfection with plasmids or antisense oligonucleotides
The pCGN expression vector containing the cDNAs of mouse PLD2 (mPLD2) or human PLD1 (hPLD1) driven by the CMV promoter were described previously [5, 6]. An influenza-epitope (hemagglutinin-tag) is inserted in the pCGN vector and is expressed at the N-terminus of the PLDs. The SN56 cells were grown to about 50–70% confluence, then transiently transfected with these plasmids using Lipofectamine Plus reagent according to the manufacturer's instructions. The cells were assayed 48 hours after transfection. Additional cultures were transfected with a β-galactosidase expression plasmid and were stained on the day of the experiment (using staining kit from Invitrogen) to estimate transfection efficiency. The overall transfection efficiency was about 10–15%.
A mouse PLD2 antisense oligonucleotide was purchased from Sequitur, Inc. (Natick, MA). The sequence of the anti-mPLD2 oligonucleotide was 5'-CAUGUGCAACUGGCUGGAGUUCAGA-3' and corresponded to the sequence of mouse PLD2 cDNA (GeneBank accession number U87557) starting at position 180. A reversed control oligonucleotide was also used and its sequence was 5'-AGACUUGAGGUCGGUCAACGUGUAC-3'. SN56 cells were grown to 50–60% confluence and washed once with Opti-MEM (Gibco BRL). The oligonucleotides were diluted in 100 μl Opti-MEM to various concentrations, mixed with 100 μl of Opti-MEM containing 6 μl of Lipofectamine and incubated at room temperature for 20 minutes. The mixture was then added to 800 μl of serum-free DMEM and the cells were incubated in this medium for 4 hours at 37°C and then cultured in normal growth medium for two days before assay. A fluorescent-dye labeled oligonucleotide was also purchased from Sequitur Inc. and used as a control to monitor transfection efficiency. The transfection efficiency was about 30–40%.
Northern blot analysis
Northern blot analysis was carried out essentially as described previously [25] using 20 μg of total RNA per lane and 1% formaldehyde-agarose gel containing ethidium bromide. The RNA was size-fractionated by electrophoresis and transferred to a nylon membrane (HyBond, Amersham). The PLD probes were: PLD1 661 bp human PLD1 cDNA fragment (GenBank accession number U38545, pos. 951–1611) isolated from HeLa cell cDNA by PCR, and PLD2 (GenBank accession number U87557) excised from the pCGN-mPLD2 plasmid using endonuclease Xbal and Smal (3000 bp). DNA probes were labeled with [α-32P]dCTP with the random primer labeling kit (Pharmacia Biotech Inc., NJ). Prehybridization and hybridization steps were carried out at 65°C in Rapid-Hyb solution according to manufacturer's instructions. The blots were washed and exposed to an x-ray film (Kodak BioMax MS-2 film, Sigma) with an intensifying screen at -80°C for 4 days. Signal intensities were quantified with ImageQuant software (Molecular Dynamics) or NIH Image 1.62. Following Northern hybridization with these probes, the blots were stripped and re-probed with a mouse G3PDH probe for normalization purposes. For these experiments the results are expressed as units of band intensity normalized with G3PDH.
Western blotting of PLD isoforms
Protein lysates from whole cells and tissues were prepared with a buffer, which contained 50 mM Tris, pH 7.5, 150 mM NaCl, 1% (v/v) Nonidet P40 (NP40), 2 mM 4-(2-aminoethyl)benzenesulphonyl fluoride, 1 μg/ml leupeptin, 2 μg/ml aprotinin and 2 μg/ml pepstatin. Lysates were mixed vigorously, incubated on ice for 15 minutes and cleared by brief centrifugation. The extracts, normalized for total protein content, were separated by SDS/PAGE and transferred to a polyvinylidene difluoride membrane (Immobilon-P, Millipore Corp.). The membrane was blocked with 5% non-fat milk in Tris-buffered saline containing 0.15% Tween-20. The polyclonal anti-human PLD2 antibody and anti-mouse PLD1 antibody were used to detect PLD2 and PLD1, respectively. The membrane was then incubated with anti-rabbit IgG peroxidase conjugates, and proteins were visualized using the chemiluminescence method (Luminol reagent, Santa Cruz Biotechnology) and Kodak BioMax light film (Fisher Scientific). Some blots were stripped by incubating them at 55°C for 30 min in a solution containing Tris-HCl 62.5 mM, pH 6.7, 2% SDS and 100 mM β-mercaptoethanol, and then probed with anti-mouse actin antibody (Sigma, St. Louis, MO) for normalization purposes. Signal intensities of protein bands were quantified with NIH Image 1.62. Approximate molecular weights were estimated using prestained standards (NEN, Boston, MA).
In vivo assay of PLD activity
For in vivo assay, the cells were grown to subconfluence and prelabeled with 2 μCi/ml [3H]myristic acid in DMEM containing 10% FBS overnight. Labeled cells were incubated in 1 ml HEPES-buffered saline solution (HBS, composition in mM: NaCl, 145; KCl, 5; MgSO4, 1; CaCl2, 1.8; glucose, 10; HEPES, 10; pH 7.4) at 37°C for 30 min twice to wash out the unincorporated label. The labeled cells were then incubated in the presence or absence of 1.5% 1-propanol with or without 100 nM PMA at 37°C for 45 min. The reaction was terminated by adding ice-cold methanol and the lipids were extracted in a solution containing methanol/chloroform/water (1:1:1 v/v). The lipids were dried and separated by TLC in a mobile phase, composed of the upper phase of a mixture of ethyl acetate/isooctane/acetic acid/water (130:20:30:100 v/v), and the phospholipids were visualized with l,6-diphenyl-l,3,5,-hexatriene in hexane. The spots comigrating with the appropriate phospholipid standards were scraped off and their radioactivity was measured by liquid scintillation spectrophotometry and expressed as a percentage of the radioactivity present in the total lipids.
Acetylcholine measurements
The cells were preincubated for 20 minutes in a choline-free physiological salt solution (pH 7.4) consisting of 135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 0.75 mM MgCl2, 10 mM glucose and 10 mM HEPES. The cells were then incubated in the same buffer supplemented with 50 μM neostigmine (to inhibit acetylcholinesterase) with or without 100 nM PMA for 45 minutes. The media were collected for subsequent assays of ACh and choline. The cells were scraped into 1 ml of methanol and this mixture transferred to a tube containing 100 μl 1 M formic acid, vortex mixed and centrifuged. The pellet was collected for protein assay while ACh was extracted by mixing the supernatant fluid with chloroform and water (1:2:1 v/v). The samples were vortex mixed and centrifuged for 5 minutes at 3,000 rpm. The aqueous phase (containing ACh) was collected and dried under a vacuum. ACh content was determined by high performance liquid chromatography with an enzymatic reactor containing acetylcholinesterase and choline oxidase and an electrochemical detector using a commercial kit (Bioanalytical Systems, Inc.) based on the method of Potter et al. [49] as described previously [50]. Protein concentration was determined by the method of Smith et al. [51].
Statistical Analysis
Data presented in the figures were derived from 3–9 independent experiments and were analyzed using Systat 5.2.1 (SPSS Inc., Chicago, Ill) by t-test or analysis of variance as appropriate. Tukey or Fisher LSD were used for post hoc tests. Differences were considered to be significant at p < 0.05.
Abbreviations
- 1ACh:
-
acetylcholine
- ARF:
-
ADP-ribosylation factor
- DAG:
-
diacylglycerol
- G3PDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- MAChR:
-
muscarinic acetylcholine receptor
- PA:
-
phosphatidic acid
- PC:
-
phosphatidylcholine
- PIP2 :
-
phosphatidylinositol 4, 5-bisphosphate
- PKC:
-
protein kinase C
- PLD:
-
phospholipase D
- PMA:
-
phorbol 12-myristate 13-acetate
- PP:
-
phosphatidylpropanol
- ROS:
-
reactive oxygen species
- TLC:
-
thin layer chromatography.
References
Blusztajn JK, Wurtman RJ: Choline and cholinergic neurons. Science. 1983, 221: 614-620.
Apparsundaram S, Ferguson SM, George AL, Blakely RD: Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun. 2000, 276: 862-867. 10.1006/bbrc.2000.3561.
Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I: Identification and characterization of the high-affinity choline transporter. Nat Neurosci. 2000, 3: 120-125. 10.1038/72059.
Lee HC, Fellenz-Maloney MP, Liscovitch M, Blusztajn JK: Phospholipase D-catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proc Natl Acad Sci USA. 1993, 90: 10086-10090.
Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA: Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995, 270: 29640-29643. 10.1074/jbc.270.50.29640.
Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA: Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997, 7: 191-201. 10.1016/S0960-9822(97)70090-3.
Liscovitch M, Czarny M, Fiucci G, Tang X: Phospholipase D: molecular and cell biology of a novel gene family. Biochem J. 2000, 345: 401-415. 10.1042/0264-6021:3450401.
Exton JH: Regulation of phospholipase D. Biochim Biophys Acta. 1999, 1439: 121-133. 10.1016/S1388-1981(99)00089-X.
Oh SO, Hong JH, Kim YR, Yoo HS, Lee SH, Lim K, Hwang BD, Exton JH, Park SK: Regulation of phospholipase D2 by H2O2 in PC12 cells. J Neurochem. 2000, 75: 2445-2454. 10.1046/j.1471-4159.2000.0752445.x.
Slaaby R, Jensen T, Hansen HS, Frohman MA, Seedorf K: PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J Biol Chem. 1998, 273: 33722-33727. 10.1074/jbc.273.50.33722.
Czarny M, Lavie Y, Fiucci G, Liscovitch M: Localization of phospholipase D in detergent-insoluble, caveolin-rich membrane domains. Modulation by caveolin-1 expression and caveolin-182–101. J Biol Chem. 1999, 274: 2717-2724. 10.1074/jbc.274.5.2717.
Fiucci G, Czarny M, Lavie Y, Zhao D, Berse B, Blusztajn JK, Liscovitch M: Changes in phospholipase D isoform activity and expression in multidrug-resistant human cancer cells. Int J Cancer. 1999, 85: 882-888. 10.1002/(SICI)1097-0215(20000315)85:6<882::AID-IJC24>3.0.CO;2-E10.1002/(SICI)1097-0215(20000315)85:6<882::AID-IJC24>3.0.CO;2-E.
Exton JH: Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta. 1994, 1212: 26-42. 10.1016/0005-2760(94)90186-4.
Hattori H, Kanfer JN: Synaptosomal phospholipase D potential role in providing choline for acetylcholine synthesis. J Neurochem. 1985, 45: 1578-1584.
Blusztajn JK, Liscovitch M, Richardson UI: Synthesis of acetylcholine from choline derived from phosphatidylcholine in a human neuronal cell line. Proc Natl Acad Sci USA. 1987, 84: 5474-5477.
Ulus IH, Wurtman RJ, Mauron C, Blusztajn JK: Choline increases acetylcholine release and protects against the stimulation-induced decrease in phosphatide levels within membranes of rat corpus striatum. Brain Res. 1989, 484: 217-227. 10.1016/0006-8993(89)90364-8.
Blusztajn JK, Venturini A, Jackson DA, Lee HJ, Wainer BH: Acetylcholine synthesis and release is enhanced by dibutyryl cyclic AMP in a neuronal cell line derived from mouse septum. J Neurosci. 1992, 12: 793-799.
Gibbs TC, Meier KE: Expression and regulation of phospholipase D isoforms in mammalian cell lines. J Cell Physiol. 2000, 182: 77-87. 10.1002/(SICI)1097-4652(200001)182:1<77::AID-JCP9>3.0.CO;2-B.
Lee SD, Lee BD, Kim Y, Suh P, Ryu SH: Bradykinin activates phospholipase D2 via protein kinase cdelta in PC12 cells. Neurosci Lett. 2000, 294: 130-132. 10.1016/S0304-3940(00)01563-9.
Lee M, Jo Y, Chun M, Chung J, Kim M, Min D: Immunohistochemical localization of phospholipase D1 in rat central nervous system. Brain Res. 2000, 864: 52-59. 10.1016/S0006-8993(00)02134-X.
Schubert D, Klier FG: Storage and release of acetylcholine by a clonal cell line. Proc Natl Acad Sci USA. 1977, 74: 5184-5188.
Schubert D, Heinemann S, Kidokoro Y: Cholinergic metabolism and synapse formation by a rat nerve cell line. Proc Natl Acad Sci USA. 1977, 74: 2579-2583.
Saito S, Sakagami H, Kondo H: Localization of mRNAs for phospholipase D (PLD) type 1 and 2 in the brain of developing and mature rat. Dev Brain Res. 2000, 120: 41-47. 10.1016/S0165-3806(99)00189-3.
Peng JF, Rhodes PG: Developmental expression of phospholipase D2 mRNA in rat brain. Int J Dev Neurosci. 2000, 18: 585-589. 10.1016/S0736-5748(99)00112-4.
Zhao D, Berse B, Holler T, Cermak JM, Blusztajn JK: Developmental changes in phospholipase D activity and mRNA levels in rat brain. Dev Brain Res. 1998, 109: 121-127. 10.1016/S0165-3806(98)00071-6.
Sandmann J, Peralta EG, Wurtman RJ: Coupling of transfected muscarinic acetylcholine receptor subtypes to phospholipase D. J Biol Chem. 1991, 266: 6031-6034.
Schmidt M, Fasselt B, Rumenapp U, Bienek C, Wieland T, van Koppen CJ, Jakobs KH: Rapid and persistent desensitization of m3 muscarinic acetylcholine receptor-stimulated phospholipase D. Concomitant sensitization of phospholipase C. J Biol Chem. 1995, 270: 19949-19956. 10.1074/jbc.270.34.19949.
Schmidt M, Voss M, Weernink PA, Wetzel J, Amano M, Kaibuchi K, Jakobs KH: A role for rho-kinase in rho-controlled phospholipase D stimulation by the m3 muscarinic acetylcholine receptor. J Biol Chem. 1999, 274: 14648-14654. 10.1074/jbc.274.21.14648.
Boyano-Adanez MC, Lundqvist C, Larsson C, Gustavsson L: Characterization of phospholipase D activation by muscarinic receptors in human neuroblastoma SH-SY5Y cells. Neuropharmacology. 1997, 36: 295-304. 10.1016/S0028-3908(96)00178-5.
Gustavsson L, Boyano-Adanez MC, Larsson C, Aradottir S, Lundqvist C: Regulation of phospholipase D activity in neuroblastoma cells. J Lipid Mediat Cell Signal. 1996, 14: 229-235. 10.1016/0929-7855(96)00530-5.
Rouse ST, Marino MJ, Potter LT, Conn PJ, Levey AI: Muscarinic receptor subtypes involved in hippocampal circuits. Life Sci. 1999, 64: 501-509. 10.1016/S0024-3205(98)00594-3.
Decker C, Miro Obradors MJ, Sillence DJ, Allan D: Phorbol ester-sensitive phospholipase D is mainly localized in the endoplasmic reticulum of BHK cells. Biochem J. 1996, 320: 885-890.
Holler T, Cermak JM, Blusztajn JK: Dietary choline supplementation in pregnant rats increases hippocampal phospholipase D activity of the offspring. Faseb J. 1996, 10: 1653-1659.
Blusztajn JK: Choline, a vital amine. Science. 1998, 281: 794-795. 10.1126/science.281.5378.794.
Blusztajn JK, Berse B: The cholinergic neuronal phenotype in Alzheimer's disease. Metab Brain Dis. 2000, 15: 45-64. 10.1023/A:1011161718572.
Jenco JM, Rawlingson A, Daniels B, Morris AJ: Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998, 37: 4901-4909. 10.1021/bi972776r.
Hellstrom J, Arvidsson U, Elde R, Cullheim S, Meister B: Differential expression of nerve terminal protein isoforms in VAChT-containing varicosities of the spinal cord ventral horn. J Comp Neurol. 1999, 411: 578-590. 10.1002/(SICI)1096-9861(19990906)411:4<578::AID-CNE4>3.0.CO;2-L.
Maroteaux L, Campanelli JT, Scheller RH: Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988, 8: 2804-2815.
Iwai A: Properties of NACP/alpha-synuclein and its role in Alzheimer's disease. Biochim Biophys Acta. 2000, 1502: 95-109. 10.1016/S0925-4439(00)00036-3.
Pronin AN, Morris AJ, Surguchov A, Benovic JL: Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000, 275: 26515-26522. 10.1074/jbc.M003542200.
Pacheco MA, Jope RS: Phosphoinositide signaling in human brain. Prog Neurobiol. 1996, 50: 255-273. 10.1016/S0301-0082(96)00035-4.
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G: Oxidative stress in Alzheimer's disease. Biochim Biophys Acta. 2000, 1502: 139-144. 10.1016/S0925-4439(00)00040-5.
Barany M, Chang YC, Arus C, Rustan T, Frey WH: Increased glycerol-3-phosphorylcholine in post-mortem Alzheimer's brain. Lancet. 1985, 1: 517-10.1016/S0140-6736(85)92114-2.
Blusztajn JK, Lopez Gonzalez-Coviella I, Logue M, Growdon JH, Wurtman RJ: Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer's disease but not of Down's syndrome patients. Brain Res. 1990, 536: 240-244. 10.1016/0006-8993(90)90030-F.
Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH, Wurtman RJ: Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci USA. 1992, 89: 1671-1675.
Klunk WE, Panchalingam K, McClure RJ, Stanley JA, Pettegrew JW: Metabolic alterations in postmortem Alzheimer's disease brain are exaggerated by Apo-E4. Neurobiol Aging. 1998, 19: 511-515. 10.1016/S0197-4580(98)00105-5.
Klunk WE, Xu C, Panchalingam K, McClure RJ, Pettegrew JW: Quantitative 1H and 31P MRS of PCA extracts of postmortem Alzheimer's disease brain. Neurobiol Aging. 1996, 17: 349-357. 10.1016/0197-4580(96)00035-8.
Farber SA, Slack BE, Blusztajn JK: Acceleration of phosphatidylcholine synthesis and breakdown by inhibitors of mitochondrial function in neuronal cells: a model of the membrane defect of Alzheimer's disease. Faseb J. 2000, 14: 2198-2206. 10.1096/fj.99-0853.
Potter PE, Meek JL, Neff NH: Acetylcholine and choline in neuronal tissue measured by HPLC with electrochemical detection. J Neurochem. 1983, 41: 188-194.
Berse B, Blusztajn JK: Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor alpha, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J Biol Chem. 1995, 270: 22101-22104. 10.1074/jbc.270.38.22101.
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC: Measurement of protein using bicinchoninic acid. Anal Biochem. 1985, 150: 76-85. 10.1016/0003-2697(85)90442-7.
Acknowledgments
We thank Dr. Yoshinori Nozawa for generously providing the anti-PLD1 and anti-PLD2 antibodies. This work was supported by National Institute on Aging grant AG09525.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhao, D., Frohman, M.A. & Blusztajn, J.K. Generation of choline for acetylcholine synthesis by phospholipase D isoforms. BMC Neurosci 2, 16 (2001). https://doi.org/10.1186/1471-2202-2-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2202-2-16