
Abstract
Cerium dioxide (CeO2), zirconium dioxide (ZrO2) and tin dioxide (SnO2) also known as ceria, are fascinating materials with a wide range of applications due to their unique properties. To investigate the doping response of La in of XO2 (X = Ce, Zr, Sn) generalized gradient approximation (GGA-PBEsol) based on CASTEP (Cambridge Serial Total Energy Package) code with ultra-soft pseudo-potential (USP) plane wave method is used. The incorporation of La in pure material shows a significant reduction of band gap and enhances the conductivity of the material. The bandgap narrowing primes to the maximum optical absorption in the visible light region. To assess the mechanical stability, the elastic responses, including the bulk Poisson's ratios, and Young's modulus calculate the significant hardness and ductile nature of the materials. The nature of the materials for optoelectronic applications, such as solar cells, is evident in the optical characteristics, including real and fictitious components of the dielectric constant, refractive index, absorption, and energy loss function. The computation of the structural, electronic, mechanical, and optical responses validates its significance for optoelectronic applications.
Graphical abstract

Similar content being viewed by others
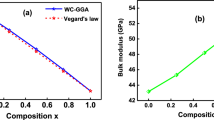

Avoid common mistakes on your manuscript.
1 Introduction
Cerium dioxide (CeO2) is a versatile oxide material that belongs to the family of rare earth oxides. It has a fluorite crystal structure and exhibits a wide range of properties, including excellent oxygen storage capacity, redox activity, and catalytic behavior. Due to its unique characteristics, CeO2 has found applications in diverse areas, such as catalysis, solid oxide fuel cells, oxygen sensors, and environmental remediation. Zirconium dioxide, commonly known as zirconia (ZrO2), is a metal oxide that exhibits exceptional properties, making it an essential material in numerous technological applications. From being a critical component in solid oxide fuel cells (SOFCs) to playing a significant role in dental ceramics and high-temperature coatings, ZrO2's properties have sparked interest among researchers, leading to in-depth investigations using theoretical methods like density functional theory. SnO2 is an inorganic compound composed of tin (Sn) and oxygen (O) atoms. Its chemical formula is often written as SnO2. The compound appears as a white or off-white powder in its pure form and is insoluble in water. It has a high melting point and is stable under normal atmospheric conditions. One of the key properties of SnO2 is its high electrical conductivity, which makes it an important material for various electronic and optoelectronic applications. Additionally, it is a wide-band gap semiconductor, meaning it has potential in optoelectronics and solar energy-related devices. In solid oxide fuel cells (SOFCs), CeO2 is used as an electrolyte material due to its high oxygen ion conductivity. This allows the efficient transfer of oxygen ions through the cell, contributing to the cell's overall performance and stability. Additionally, CeO2's redox properties have made it essential in oxygen sensors, which are widely used in automotive applications to monitor and regulate the air-to-fuel ratio, leading to better combustion and lower emissions. One of the most significant responsibilities assigned to modern research has taken curious attention for researchers in many industrial applications involving modern catalytic, solar cells, energy storage devices, optoelectronic devices and biomedical uses [1,2,3]. Due to its large type of applications in catalysts, solid oxide fuel cells and high oxygen performance storage capacity [4]. As a very prominent field in renewable energy sources, solar engineering has attracted huge interest from many scientists all over the world. CeO2 and ZrO2 are fluorite structure and crystallizes in the cubic space group. Ce+4 and Zr+4 are bonded in a body-centered geometry to eight similar O−2 atoms. All (Ce–O) and (Zr-O) bond lengths are 2.37 Å or 2.23 Å. It has been found that La-doping can improve the electronic and optical response of cubic CeO2, SnO2 and ZrO2. The aforementioned explanations supply large knowledge and employ a wide range of applications. The optical response of doped- XO2 (X = Ce, Zr, Sn) have been done employing DFT computations through CASTEP code. The computed data represent increased optical response with the incorporation of La in cubic CeO2, SnO2 and ZrO2. Stannic oxide (SnO2) also has comprehensive employment for the electron transport layer application due to its response namely, electronic, band gap tuning and optical transparency. Lanthanum (La) concentration utilizing structural modification has been proceeding to optimize the different responses. Zirconia (ZrO2) has awesome technological excellence outstanding to its exceptional mechanical, elastic, and electronic engineering, towering dielectric value as well as large band gap tuning [5]. The impact of La on the physical responses of CeO2, SnO2, and ZrO2 was calculated and investigated the increment of other optical parameters was in detail. Hence, the research described in this manuscript is aimed at a complete understanding of the structural, electronic, elastic, mechanical and optical response. Moreover, it was also verified that CeO2, SnO2 and ZrO2 have excellent candidates for photocatalytic and could be also employed in environmental applications owing to their nontoxicity [6].
2 Theoretical method
First principle calculations have been performed using the software CASTEP (Cambridge Serial Total Energy Package) code with the ultrasoft pseudo-potential (USP) plane wave method, which was created using the plane-wave technique and is dependent on the density functional theory, to examine the characteristics of the oxides [7]. DFT was designed by Hohenberg, Kohn, and Sham to solve the enormous Kohn–Sham equations using an iterative matrix diagonalization approach [8, 9]. Due to the exchange–correlation technique of La-doped CeO2, SnO2, and ZrO2 were adopted by the generalized gradient approximation (GGA + PBEsol) functional
Well-converged plane-wave energy cutoff and K-mesh are the most significant aspects of any SCF computation for correctly showing the attributes of the electronic structure [11]. According to the reported works, the CeO2, SnO2, and ZrO2 belong to the cubic structures while the determined lattice parameters of ZrO2 are a = 5.47 Å, 4.89 Å and 5.15 Å. In XO2 (X = Ce, Zr, Sn), the Zr, Sn and Ce atoms and O atoms occupy the Wyckoff positions a (0, 0, 0), b (1/2, 1/2, 1/2) and (3/4, ¼, ¾) sites. To study the inspiration of evolution metal on the electronic and optical responses of CeO2, SnO2 and ZrO2, two Zr, Sn and Ce atoms in XO2 (X = Ce, Zr, Sn) are replaced by the La atom founded on the symmetrical structure. To study the effect of La concentration and electronic and optical responses of XO2, we built a 2 × 2 × 1 supercell. The doped concentration in XO2 is 13.5 at%. After convergence check, the electronic wave function of the La-doped XO2 (X = Ce, Zr, Sn) was 480 eV. The k-meshes of 16 × 16 × 16 were implemented for the XO2 (X = Ce, Zr, Sn) and La-doped concentrations. All structural coefficients of XO2 (X = Ce, Zr, Sn) and La-doped XO2 were fully unperturbed during the process of structural optimizations. The convergence thresholds of total energy were smaller than 1 × 10−6 eV/atom and the maximum ionic displacement was within 0.001 Å [12].
3 Results and discussions
3.1 Structural property
CeO2, SnO2 and ZrO2 have a cubic crystal structure with fluorite-family compound and have three phases space groups cubic (Fm \(\overline{3 }\) m). For the best sympathetic, our work based on cubic CeO2, SnO2 and ZrO2 are also attributed to fluorite oxides. So, the researcher is anxious about cubic fluorite-type crystal structures and employs some responses of cubic XO2 (X = Ce, Zr, Sn)-doped La. The geometry optimization structure of the cubic XO2 (X = Ce, Zr, Sn) has been shown in Fig. 1. After generating a 2 × 22 × 1 supercell, the geometrical parameters for original CeO2, SnO2 and ZrO2 were obtained. Geometry optimization, also known as molecular optimization or molecular geometry optimization, is a computational technique used in various scientific fields, particularly in chemistry and materials science. It involves finding the most stable or lowest-energy arrangement of atoms within a molecule or a solid material. geometry optimization is a powerful computational tool that plays a crucial role in understanding molecular structures, properties, and interactions, as well as aiding in the design of new materials and drugs. The ground state energy of the crystal structures CeO2, SnO2 and ZrO2 are computed with the help of Birch Murnaghan's approach of using the GGA-PBE function.
V volume of the unit cell.

Optimized structures of a CeO2, b SnO2 and c ZrO2
Table 1 shows the unit volume change with the effect of La-doping and its impact also depends on bond lengths. Additionally, the computational details and lattice parameters are acceptable for an explanation of structural, electronic, mechanical and optical responses of Ce1–xLaxO2, Sn1–xLaxO2 and Zr1–xLaxO2. Computed the equilibrium lattice parameters and volumes for CeO2, SnO2 and ZrO2 are 5.47 Å, 4.89 Å and 5.15 Å as well as 1.63 Å3, 80.58 Å3 and 136.59 Å3, respectively. The founded lattice parameters are 158.3 Å3, 71.49 Å3 and 131.87 Å3. Our computed lattice parameters value and cell volume are discussing above table strongly agree with other computed values of the lattice parameters are 164.5 Å3 and 1.35 Å3 and bond length 2.366 Å. After doping concentration of La atoms we noted that the computed value of lattice parameter and cell volume values are 193.18 Å3, 74.17 Å3 and 151.41 Å3 respectively. Concurrently, from Table 1 we can noticeably see that the lattice parameters enhance in the case of CeO2 and ZrO2 as well as decreases in the case of SnO2 with reasonable impurities of La-doping atoms is in good comparable with other computed data as well as X-ray diffraction (XRD) data. Due to this reason, the difference of covalent radius Zr, Sn and Ce in comparison with Ce–La, Sn–La and Zr–La are (1.85 Å), (1.4 Å) as well as (1.6 Å) [18].
3.2 Electronic properties
Some key electronic properties of CeO2 include: cerium atoms in CeO2 can exist in both Ce(IV) and Ce(III) oxidation states, resulting in mixed valence behavior. This property makes CeO2 a good candidate for catalytic applications and solid oxide fuel cells (SOFCs). It can easily form oxygen vacancies, which can participate in redox reactions with oxygen molecules. This property contributes to its excellent oxygen storage and release capability, making it a crucial component in catalytic converters. CeO2 exhibits high oxygen-ion conductivity at elevated temperatures, making it applicable in oxygen-ion conducting solid electrolytes used in fuel cells and oxygen sensors. Tin dioxide, also known as stannic oxide or tin (IV) oxide, is a semiconductor material with a rutile crystal structure. It has various applications due to its unique electronic properties: SnO2 is an n-type semiconductor, meaning it has an excess of electrons and can conduct electricity. It is commonly used as a transparent conducting oxide (TCO) material in applications such as touch screens, solar cells, and gas sensors. Zirconium dioxide, or zirconia, is a metal oxide with multiple crystal phases, including monoclinic, tetragonal, and cubic. It has several electronic properties that are advantageous for various applications: ZrO2 exhibits exceptional thermal stability, and it can maintain its crystalline structure at high temperatures. This property makes it a vital component in high-temperature ceramics and refractories. Similar to CeO2, zirconia has excellent oxygen ion conductivity, especially in its yttria-stabilized form (YSZ). YSZ is widely used as an electrolyte in solid oxide fuel cells (SOFCs) due to its ability to conduct oxygen ions at elevated temperatures. The computed electronic band gaps of pure and La-doped (CeO2, SnO2 and ZrO2) are compared to experimental calculations. Among them, PBEsol obtained better band gaps but below value the experiment values of CeO2, SnO2 and ZrO2 are (2.5 eV), (1.69 eV) and (3.4 eV). Doping material and its impacts on electronic band gap tuning decreased trends due to the transitions of valance electrons into the conduction region and maximum holes left in the valance band. The nature of the electronic band gap is a famous basis for understanding optoelectronic semiconductor materials. Above the valance band and below the conduction band the Fermi levels are obtained at zero electron volt energy (Ef = 0 eV) and its band energy expands along a high symmetric point of BZ. Computed band gaps tuning of CeO2, SnO2 and ZrO2 and doping effects on it are shown in Fig. 2 (see Table 2).

Band structure with GGA (PBEsol); of; a pure CeO2 b La-doped CeO2 c pure SnO2 d La-doped SnO2, e pure ZrO2 and f La-doped ZrO2
The charge density rearrangement before and after concentration Ce and Sn, as well as Zr with La atoms in CeO2, SnO2 and ZrO2, is under the condition of an electron exchange approach. It is considerable to compute that suitable addition of impurity La has changed the Mulliken charge (e) of Oxygen and Ce, Sn as well as Zr atoms. Additionally, the structure of CeO2, SnO2 and ZrO2 with enhanced La concentrations and we noted that the charge of oxygen atoms is decreased as well as the charge of CeO2, SnO2 and ZrO2 atoms are enhanced because they are reverse La atoms are extra suitable to contribute electrons to oxygen as compare to CeO2, SnO2 and ZrO2atoms. Before and after the doping concentration of La the charge of CeO2, SnO2 and ZrO2 atoms change from – 0.60 to – 0.78e, 1.85e to 1.94e and 1.53 to 1.36e as well as the charge of oxygen atoms also revolution from – 0.64 to – 0.73e. Especially, obtained result with enhanced La doping, it is attention-grabbing to compute that the charge of CeO2, SnO2 and ZrO2 atoms will be reasonably enhanced, the charge of La atoms are considerably enhanced and decrease the main charge of oxygen atoms as well as we compute that the crinum negative charge and Sn, Zr are compute positive change in Ce1–xLaxO2, Sn1–xLaxO2 and Zr1–xLaxO2 are greater than in all pure cranium oxide, tin oxide and zirconium oxide. The only zirconium atom positive charge is small as compared to pure zirconium oxide [21]. This suggests that La doping concentration will strongly attention to the weakened covalent behavior of Ce–O, Sn–O and Zr–O bonds. The exact strength of the bond that links a pair of atoms is employed to compute the bond orders. With the aid of an atomic basis expansion, it can be determined from the background of overlap integrals between the Bloch functions [24, 25]. For a crystal structure of CeO2, SnO2, and ZrO2, the overall bond order can be calculated by multiplying each bond order by the exact number of bonds per molecular unit. The computed bond energy in both cases of pure CeO2, SnO2, and ZrO2 and La-doped atoms noted are 535.2 eV, 545.1 eV and 530.7 eV as well as 548.4 eV, 556.9 eV and 542.7 eV, respectively. From Fig. 2a, b, the top of VB pinpoint at the space between Z point and the minima below of CB located at G point in the BZ. Calculated the electronic band gap tuning of CeO2 due to the difference between the VB to CB, it forms an indirect band gap semiconductor with a band gap of (1.86 eV). After the addition of the concentration of La-CeO2, the transitions of electrons occur between VB to CB and its impact on the fermi level more shifted near the conduction band. The computed band gap enhanced from 1.86 to 2.16 eV, which is consistent with the data in the literature. Figure 2c, d computed the electronic band gap tuning of pure and doped SnO2. Effect of La-concentration of SnO2 The band gap trends go on decreasing from 1.90 to 1.82 eV and the fermi level shifted more into the conduction band and show p-type characteristics. Namely, La-doping is beneficial to promote the electronic jump in comparison to SnO2 and CeO2 doping. Based on the band structure tuning, the variation of electronic responses of La-doped SnO2 and CeO2 are directly related to the maximum contribution Sn-4d and Ce-4d state. As mentioned above the overlapping band gap impact of phenomena develops for all contributions in Ce–La and Sn–La, especially band gap trends continually reduced and enhanced due to the taking part of the electron on Ce-5d, Ce-4f and Sn-5d, Sn-4f states were a contribution, leading to the states Ce-5d and Sn-d were enhanced. Figure 2e–d calculated the electronic band gaps tuning of pure and La-doped ZrO2. Before and after the suitable concentration of La–ZrO2 the electron band gap reduced from 3.7 to 3.6 eV. We noted that the upper VB of CeO2, SnO2 and ZrO2 lies in the region above 2 eV, and 3.3 eV are mainly O-2p and O-3p states with Ce-5d, Sn-3p and Zr-5d states admixture. The bottom of CB is mainly contributed to by the Ce-4d, Sn-3p and Zr-4d-states of the CeO2, SnO2 and ZrO2. When the Ce, Sn and Zr atoms are substituted by the La atoms in CeO2, SnO2 and ZrO2 their p-states and d-states behaviors are extra understandable at the upper CB and it transfers up in energy levels and intersections with the band region 1.5–3 eV. The range of the CB energy becomes large as compared to the undoped crystal structure system. After the doped system as declared upper the intersecting band processes occur for suitable concentrations in Ce1–xLaxO2, Sn1–xLaxO2 and Zr1–xLaxO2 as well as gaps nearly disappear with doping La atoms in CeO2, SnO2 and ZrO2. The main strong contribution of the electron on La-3d states, a new band created between Ce-5d, Sn-4d and Zr-4d-states was involved leading to the states of cerium (Ce-5d) and zirconium (Zr-4d) were enhanced. The most important thing and interesting to compute is that for Ce1–xLaxO2 the BG enhanced is absorbed as compared with pure CeO2. To explain the charge of band structure and its impacts on it the reasons under zero stress can be seen from the study in the above figs. Under combination, the DOS, and CB of CeO2, SnO2 and ZrO2 are the contribution of Ce-4d, Zr-5f and O-2p. The addition of La-concentration enhancement led to the occupation of Ce, Sn and Zr-4f electrons and their impact’s in CB increased, and the maximum contribution of O-2p electrons enhanced in VB. At the same impact, because of the energy of Ce-4d, the Zr-5f ground state is larger than that of the O-2p orbit and its 4f required for the transition is relatively increased as compared with that of the O-2p state. Due to this reason, the electronic band gap tuning curves become dense as well as generally move to the fermi levels. After computing the electronic band structure, we calculate the TPDOS/EPDOS and TDOS of pure and doped CB of CeO2, SnO2 and ZrO2. We also computed that the thicknesses of O-2p and O-3p states as well as BG increase with La impurities enhancement. This is mostly because La-doping enhanced the interaction between the Sn-4p, Ce-5d, Zr-5d and Ce-3p as well as O-2p states, relating to that in pure SnO2 CeO2, and ZrO2. And the most notable peak value of the thin band located above the Fermi level decreased with La-doping impurities also enhanced. This is perhaps the main explanation that the involvement of the electron on La-4d states a new band between the Ce-5d, Sn-4d and Zr-3p-states were involvement leading to the states of cerium (Ce-5d), tin (Sn-4d) and zirconium (Zr-3p)-states were enhanced. The calculated density of stats, EPDOS, TPDOS and TDOS profile of La-doped CeO2, SnO2 and ZrO2 as shown in Fig. 3.

With GGA (PBEsol) a TPDOS of CeO2 b PDOS of Ce c PDOS of O in case of CeO2 d TPDOS of SnO2 e PDOS of Sn f PDOS of O g TPDOS of ZrO2 h PDOS of Zn and i TDOS
Figure 3 represents TPDOS/EPDOS and the TDOS profile is composed of O-2p, Ce-4p, Sn-5p and Zr-4p states, respectively. From Fig. 3a–c, the total TPDOS in the case of CeO2 is shifted between – 4.4 eV and 0.4 eV, and the maximum contribution of p-states is 58.28 at – 0.89 eV. In the case of elemental PDOS of pure and doped Ce is transferred from the point 6.5–13.2 eV and the valuable contribution of d-states 37.9 at 6.5 eV and fermi level more shifted into CB. From Fig (d, e, f) the TPDOS/EPDOS of pure and La-doped SnO2 prominent peaks are shifted from the points – 4.9 to 0.3 eV, 3.4–4.8 eV and – 4.9 to 4.5 eV, the maximum contribution of p-states, s-states are 73.6 at – 0.4 eV, 67.5 eV at 4.2 and 73.1 at – 0.5 eV. from fig (g, h) in case of pure and La-doped ZrO2 the energy peaks of both TPDOS and EPDOS shifted from – 5.02 to 0.09 eV, 3.3–11.3 eV and the maximum contribution is p-states and d-states (ZrO2) peaks 52.7 at – 1.2 eV, 33.1 at 5.2 eV. Doping effects on DOS it is worth noticing that CB of cubic CeO2, SnO2 and ZrO2 is mainly contributed by Sn-5p and Zr-4p states. The comparison in the case of TDOS of CeO2, SnO2 and ZrO2 maximum contribution of CeO2 is 258.8 at 2.3 eV. Especially, in particular, in the band gap in both cases of pure and doped perovskite materials, the fermi level separates the bonding orbit and anti-bonding orbits. Due to this reason, the electronic band gaps separate increases the main difficulty of free electron jumps between VB and CB.
4 Elastic and mechanical properties
Elastic properties responses are more valuable in phase to compute compounds' mechanical stability such as stiffness, brightness and ductility [26]. The elastic modulus of CeO2 varies depending on its crystal structure and temperature. At room temperature, the elastic modulus typically ranges from 200 to 400 GPa. The hardness of CeO2 is around 5.5–6.5 on the Mohs scale. The fracture toughness of CeO2 is relatively low, typically in the range of 1.5–3.5 MPa√m. Young's modulus of CeO2 is approximately 300–350 GPa. The Poisson's ratio for CeO2 is around 0.25–0.30. The elastic modulus of SnO2 is typically around 200 GPa. The hardness of SnO2 is approximately 6–7 on the Mohs scale and the fracture toughness of SnO2 is relatively low, similar to CeO2, and falls within the range of 1.5–3.5 MPa√m. Young's modulus of SnO2 is around 180–200 GPa. The Poisson's ratio for SnO2 is roughly 0.28–0.33. The elastic modulus of ZrO2 is higher than the previous two materials, ranging from 200 to 380 GPa, depending on the phase and temperature and the hardness of ZrO2 is approximately 7.5 on the Mohs scale. ZrO2 exhibits high fracture toughness, typically in the range of 7–10 MPa√m. Young's modulus of ZrO2 varies with its crystal structure and temperature, ranging from 200 to 400 GPa. Poisson's ratio for ZrO2 is around 0.25–0.31. The mechanical responses of Ce1–xLaxO2, Sn1–xLaxO2 and Zr1–xLaxO2 depend on their elastic parameters which justify the response to the addition of La-doping effects. Therefore, the elastic constants of CeO2, SnO2 and ZrO2 are important parameters of the materials and provide considerable important information about the mechanical stability, brittleness, stiffness anisotropic and stiffness character. From the perspective of materials of CeO2, SnO2 and ZrO2 some of the excellent knowledge of the materials can be gained from elastic parameters Cij, which can be achieved from the ground state required total energy calculations. If the crystal structure XO2 (X = Ce, Zr, Sn)-La-doped unless its elastic parameters depend on certain relationships and it cannot exist in a metastable phase and stable phase. The impacts of the crystal structure CeO2, SnO2 and ZrO2 to external forces as characterized by the bulk modulus (B), Poisson ratio (V), shear modulus (G) and Young’s modulus (E) are computed by Cij and add the valuable role in calculating the strength of the materials. A cubic crystal structure XO2 (X = Ce, Zr, Sn)-doped La have three important elastic coefficients C11, C12 and C44. Under these conditions, the stress is compulsory for the existence of strain in the configurations indicated by the data of the elastic parameters. Especially, the large value of elastic constants the force needed to reduce crystal structure strain. The Voigt–Reuss–Hill computations have been employed to achieve the cubic crystal lattice isotropic bulk, Young’s, Passions ratio and shear modulus from the crystal parameters as described below equations [27].
The shear modulus is obtained by;
The value of the Poisson ratio \((\sigma )\) is determined by the following equation.
The anisotropy factor A can be deduced by using this formula [28].
Additionally, determine the plasticity, hardness and connecting features of the crystal structure CeO2, SnO2 and ZrO2, information of these quantities (see Table 3).
Comparison of large stiffness of the compound XO2 (X = Ce, Zr, Sn)-doped La, understanding the information of these materials have an anisotropic value of larger than A > 1 are obtained to be the elastically anisotropic response [31]. Another hand if it has a value smaller than A < 1 is obtained by its elastically isotropic nature [32]. Here C11 elastic coefficient justifies the material's flexibility regarding the arrangement and C12, and C44 obtained these materials' flexibility as the consultation of phase transformation. Under the study of the ductile and brittle response of XO2 (X = Ce, Zr, Sn)-doped La, after calculating the required data of elastic constants. Pugh’s ratio (B/G) and also Poisson ratio (V) provide more knowledge to verify the nature of different materials to achieve the brightness and ductility of our materials [33, 34]. The B/G ratio is used to classify the brittleness and ductility of various materials. The value 1.75 is the critical number that identifies the brittleness and ductility characteristics of materials, according to Pugh, When the B/G ratio is greater than the critical value, the material is said to be ductile. When the B/G number is less than the critical value, however, the material is termed as brittle. As indicated above Table, the B/G ratio pure and Ce1–xLaxO2, Sn1–xLaxO2 and Zr1–xLaxO2 materials computations are 2.14, 1.15 & 1.79 and 2.16, 1.51 and 0.81, respectively which is greater than in case of CeO2 and smaller in case of SnO2 and ZrO2 the critical value 1.75, for PBEsole and confirm the brutality of CeO2 and ductility of SnO2 and ZrO2. In addition, bulk modulus and shear modulus can be employed to compute the material hardness and softness. The nature of CeO2 is (brittle), SnO2 (ductile) and ZrO2 brittle as well after the doping effect on these materials and the nature of CeO2 is (brittle), SnO2 (ductile) and ZrO2 (ductile) [38, 39].
5 Optical properties
CeO2 is generally transparent to visible light and absorbs light in the ultraviolet (UV) region. The refractive index of CeO2 varies with wavelength, but it typically falls within the range of 2.1–2.3 for visible light. CeO2 is known for its interesting electronic structure, where the bandgap can be tailored by doping or changing the crystal structure. The bandgap of pure CeO2 is around 3.2–3.7 eV, depending on the crystal phase. Tin dioxide, also known as stannic oxide or tin(IV) oxide, is a compound made up of tin and oxygen. It is widely used in gas sensors, transparent conducting films, and as a polishing material. SnO2 is transparent in the visible light range and has good optical clarity. The refractive index of SnO2 ranges from approximately 2.0 to 2.3 for visible light, depending on the wavelength and film thickness. Zirconium dioxide, commonly known as zirconia, is a versatile material with applications in ceramics, dental implants, and oxygen sensors. ZrO2 is transparent to visible light and exhibits good optical transparency. The refractive index of ZrO2 varies with the crystal phase and ranges from approximately 1.8–2.3 for visible light. The bandgap of ZrO2 depends on its crystal structure and typically falls within the range of 5 to 7 eV. Under the electronic transitions, the optical responses of La-doped XO2 (X = Ce, Zr, Sn) can be computed in the CASTP code. We take the ionic polarization into attention as they impact the optical responses of materials and the optical polarization is strongly acceptable to affect the dielectric constant [40, 41]. The optical properties can be identified to understand by its absorption capacity of light which is the change that occurs between the VB band and CB. Completely described the optical responses we discuss of various optical parameters like dielectric constants (real, complex), absorption, energy loss function, reflectivity and refractive index.
Here \(\varepsilon_{1} \left( \omega \right) \) and \(\varepsilon_{2} \left( \omega \right) \) are real and complex parts of the dielectric constant [42], respectively
\(n\left( \omega \right) = n\left( \omega \right) + ik\left( \omega \right)\)
where n(ω) is called a real part of the refractive index and k(ω) represents the extinction coefficient [43, 44].
The comparison between optical responses CeO2, SnO2 and ZrO2, employ GGA Before and after La-doping as shown in Fig. 4.

Before and after doped inoculation: a, b real and imaginary parts of DF; c absorption; d energy loss function; e reflectivity; f real part of refractive index and g Conductivity
From Fig. (a, b) the dielectric constants both real and complex parts in the low energy range are high and with very small increments it again reduces un the large frequency range. Due to this favourable profile of dielectric functions is the most excellent candidate for microelectronic and integrated circuits devices today [45, 46]. The complex part of the dielectric function impact can be through as indicating the real transitions between the completed and uncompleted orbitals [46, 47]. A specific, limit of 0.92 eV for scissors and 0.3 eV for is smearing employed to simulate the broad effects [48]. From the above fig, the read dielectric constant estimates the energies 0 eV up to 40 eV. It represents a sharp enhancement at the first peak of the optical transition at point 0.6 eV. The maximum contribution prominent peak observed value is 34.4 at energy 0.61 eV. The second, third and fourth prominent peaks in the case of both materials are situated at 9.9 eV 22.1 eV and 31.1 eV which are comparable with already reported experimental and theoretical data. Due to the interbond transitions from VB to the CB orbital concerning the complex part peaks. In the case of the complex dielectric function, the first prominent peak identifies at the energy point of 2.8 eV and the maximum contribution is 14.0 at 1.6 eV. After the addition of La concentrations, the value change will be noted as compared to pure XO2 (X = Ce, Zr, Sn) perovskite materials. Understanding, to quantum mechanical approach the dipolar-based rule obtained from the quantum mechanical angular momentum conservation method. La-doped CeO2, SnO2 and ZrO2 strongly represent the first peak in the imaginary part of a dielectric function at 2.7 eV for Ce-5d/Sn-4f, Ce-5d/Ce-3f and Zr-4d/Zr-5f transitions done, while the 2nd prominent peak occurs the transition of Ce-4d/Sn-4f, Ce-5d/Ce-3f and Zr-5d/Zr-5f orbits. From fig (c) the optical response on absorption coefficient understand the analysis of the electronic nature of our materials. The absorption cure edge does not start from zero photon energy for La-doped and pure XO2 (X = Ce, Zr, Sn). In this case, the prominent peak noted the point at energy 12.6 eV and the maximum peak contribution at the highest peak point at 25.2 eV. The second, third and fourth prominent peaks we strongly noted at points energies 12.5 eV, 22.8 eV, and 31.8 eV in both pure and La-doped materials. Computed result for optical absorption is most important and its impact on the absorption peak takes place at the UV region [49, 50]. It is representing the strong UV absorber of the analysis of La-doped CeO2, SnO2 and ZrO2 materials. A comparison of the absorption at the UV region between our three materials CeO2, SnO2 and ZrO2, strongly noted that the SnO2 La-doped represents better performance obtained. From Fig. 4d Energy loss functions of optical response represent energy loss, due to fast-moving electrons in the crystal structure of XO2 (X = Ce, Sn, Zr). The effect of pressure and its impact on loss due to scattering, heating as well as dispersion. This response direct relates to Plasmon resonance which is also called plasma frequency. The energy loss function prominent peaks absorbed at points first, second, third and fourth are points 1.8 eV, 24.2 eV, 32.3 eV, and 35.9 eV. From Fig. 4 (e), calculate the reflectivity for the pure and LA-doped CeO2, SnO2 and ZrO2 crystal structure. The computed data of the reflectivity for XO2 (X = Ce, Zr, Sn) start at 16.7% (SnO2), 17.9% (CeO2) and 19.5% (ZrO2) as well as have large data roughly 52% at noted energy value [51]. The calculated present value of reflectivity comparable to experimental data starts from 20% and other hand computations have a maximum value of 65% at energy value. Strongly noted the prominent peaks first, second, third and fourth are at energies points 1.8 eV, 13.3 eV, 27.4 eV, and 35.3 eV, as well as heights contribution, noted the point 36.5 at energy 1.58 eV. From Fig. 4f, the word refractive index is explained as the speed of light in a vacuum and the speed of light in a medium. Additionally, it identifies the accurate capacity of CeO2, SnO2 and ZrO2 and how much light passes from them. In the case of refractive index and the spectrum of CeO2, SnO2 and ZrO2 can be indicated in two main three absorption bands created around the energy points 5.9 eV, 12.9 eV and 29.7 eV. Due to these energy points conditions, the first and second peaks can be noted to the O-2p/Sn-5d, O-2p/ Zr-4f and O-2p/Ce-5d electrons transition of pure and La-doped materials, respectively. Before and after the La-doped refractive index first band ranges shifted from point 3.8–7.4 eV and the second strongly shifted from energy point 12.1–13.5 eV. From fig (g) the prominent peaks are noted at points 9.9 eV, 22.03 eV and 31.2 eV in the case of La-doped CeO2 showing maximum conductivity is 34.4 at 0.6 eV. The optical responses of CeO2, SnO2, and ZrO2 as well as the lattice parameters and cell volume estimated results are all in excellent agreement with the experimental findings and other theoretical values. When the Ce, Sn, and Zr atoms in CeO2, SnO2, and ZrO2 are replaced with La atoms, the lattice parameter, cell volume, and bond length are all linearly reduced with increasing doping concentration x. The argument that the chemical interactions between CeO2, SnO2, and ZrO2 in Ce1–xLaxO2, Sn1–xLaxO2, and Zr1–xLaxO2, respectively, are weaker than those between CeO2, SnO2, and pure ZrO2, was supported by the evidence provided. The vibrational relationship between the cation O-d distances and the cation charge leads to the following conclusion the smaller the cation charge, the longer the oxygen bond length of the metal. Additionally, in La-doped CeO2, SnO2, and ZrO2 structures, the CB energy range is larger than in undoped CeO2, SnO2, and ZrO2 structures. In particular, for the structure's second BG, the overlapping band process occurs for all concentrations of Ce1–xLaxO2, Sn1–xLaxO2, and Zr1–xLaxO2, especially for the structure's second BG almost vanishes. Based on the obtained dielectric constants, it appears that when the La doping concentration increased, the refractive index n0 and the value of dielectric constant ε0 both significantly dropped. The effect of La-concentration enhanced, all bands and overlapped into one with two main distinct maximum contributions. It can be observed from the found data that La-doped concentration shows the main role in optical responses and have potential applications.
6 Conclusion
In this work, we symmetrically explored structural, electronic, mechanical, and optical responses of pure CeO2, SnO2 and ZrO2 and La-doped materials analyzed in detail using GGA PBEsol functional. Compared with the doped and undoped CeO2, SnO2 and ZrO2 when the (Sn Ce, Zr) atoms are interchanged by La atoms in materials and the bond length of Ce–O, Sn–O and Zr-O is reduced linearly with doping concentration enhanced. The computed results represent the electronic band gaps of exact cubic crystal CeO2, SnO2 and ZrO2 and La-doped materials are in excellent agreement with experimental and theoretical data, respectively. Calculated band gap tuning acknowledges that the semiconductor aspect of CeO2, SnO2 and ZrO2 are attributed to the electronic band gap separation between Ce-5d, Sn-4d, Ce-5d states and O-2p states near the Fermi level. Essentially, the addition of the La atom can improve the electronic response due to the electronic jump between the VB and CB near the Fermi level point. Elemental partial density of states (EPDOS) is utilized to indicate the reduced band gap after La-doping in detail. The optical response of dielectric constants (real, imaginary), absorption, conductivity, refractive index and energy loss functions have also been computed and analyzed concerning the electronic structure of CeO2, SnO2 and ZrO2 determinations. Absorption spectra represent a redshift, and a considerable change is observed there. The refractive index of all materials is inversely proportional concerning the pressures and energy loss function as well as absorption are both dependable. It is worth mentioning that the high flexibility and large transmission of visible light make ZnO2 the best applicant to be applied in flexible optoelectronic devices.
Data availability
This manuscript has no associated data or the data will not be deposited. Authors’ comment: this is a theoretical study and no experimental data has been listed.
Code availability
Not applicable.
References
D. Rohilla, S. Chaudhary, A. Umar, An overview of advanced nanomaterials for sensor applications. Eng. Sci. 16, 47–70 (2021)
D. Sriparna, M. D. Ayano Shanko, A.P. Pashupulla Parsanta, A.K. Srivastava, S. K. Yadav, S. Saxena. Enzyme based Glucose Biosensor-an overview. J Pharmaceutical Negative Results pp. 2494-2502 (2022)
O.B. Fadil, A. Hashim, Fabrication and tailored optical characteristics of CeO2/SiO2 nanostructures doped PMMA for electronics and optics fields. SILICON 14, 1–8 (2022)
P. Knauth, H.L. Tuller, Solid-state ionics: roots, status, and future prospects. J. Am. Ceram. Soc. 85(7), 1654–1680 (2002)
V. Rani, A. Sharma, A. Kumar, P. Singh, S. Thakur, A. Singh, Q.V. Le, V.H. Nguyen, Raizada, ZrO2-based photocatalysts for wastewater treatment: from novel modification strategies to mechanistic insights. Catalysts 12(11), 1418 (2022)
V. Madaan, B. Mohan, V. Bhankar, R. Ranga, P. Kumari, P. Singh, M. Sillanpää, A. Kumar, A.A. Solovev, K. Kumar, Metal-decorated CeO2 nanomaterials for photocatalytic degradation of organic pollutants. Inorg. Chem. Commun. 146, 110099 (2022)
R. Hernandez-Bravo, Study of dolomite surface stability by DFT approach considering defects. J. Porous Media 20(11), 1031–1041 (2017)
Y. Zhou, J.R. Chelikowsky, Y. Saad, Chebyshev-filtered subspace iteration method free of sparse diagonalization for solving the Kohn-Sham equation. J. Comput. Phys. 274, 770–782 (2014)
C. Ricca, Combined Theoretical and Experimental Study of the Ionic Conduction in Oxide-Carbonate Composite Materials as ELECTROLYTES for Solid Oxide Fuel Cells (SOFC) (Université Pierre et Marie Curie-Paris VI, 2016)
A. Sharma, P. Suryanarayana, Real-space density functional theory adapted to cyclic and helical symmetry: application to torsional deformation of carbon nanotubes. Phys. Rev. B 103(3), 035101 (2021)
R. Smith, A DFT Investigation of Al-Based Atomically Precise Epitaxy (UCL (University College London), 2021)
H. Hu, G.H. Peslherbe, Accurate mechanical and electronic properties of spinel nitrides from density functional theory. J. Phys. Chem. C 125(17), 8927–8937 (2021)
B. Meredig, Data-Driven Computational Methods for Materials Characterization, Classification, and Discovery (Northwestern University, 2012)
N.V. Skorodumova, R. Ahuja, S.I. Simak, I.A. Abrikosov, B. Johansson, B.I. Lundqvist, Electronic, bonding, and optical properties of CeO2 and Ce2O3 from first principles. Phys. Rev. B 64(11), 115108 (2001)
L. Eyring, The binary rare earth oxides. Handb. Phys. Chem. Rare Earths 3, 337–399 (1979)
A.A. Bolzan, C. Fong, B.J. Kennedy, C.J. Howard, Structural studies of rutile-type metal dioxides. Acta Crystallogr. B 53(3), 373–380 (1997)
J.C. Garcia, L.M.R. Scolfaro, A.T. Lino, V.N. Freire, G.A. Farias, C.C. Silva, E. da Silva Jr, F, Structural, electronic, and optical properties of ZrO2 from ab initio calculations. J. Appl. Phys. 100(10), 104103 (2006)
S.R. Dailey, Geochemistry of the Fluorine-and Beryllium-Rich Spor Mountain Rhyolite, Western Utah (Brigham Young University, 2016)
J.S. Niu, I.P. Liu, K.H. Chen, J.H. Tsai, W.C. Hsu, W.C. Liu, Ammonia sensing characteristics of a cerium oxide thin film coated with platinum nanoparticles. Sens. Actuators, B Chem. 369, 132241 (2022)
K. Reimann, M. Steube, Experimental determination of the electronic band structure of SnO2. Solid State Commun. 105(10), 649–652 (1998)
N. Beloufa, Y. Cherchab, S. Louhibi-Fasla, S. Daoud, H. Rekab-Djabri, A. Chahed, Theoretical investigation of structural, electronic and optical properties of Sc-doped SnO2. Comput. Condens. Matter 30, e00642 (2022)
D. Tian, C. Zeng, H. Wang, H. Luo, X. Cheng, C. Xiang, X. Zhu, Performance of cubic ZrO2 doped CeO2: first-principles investigation on elastic, electronic and optical properties of Ce1− x ZrxO2. J. Alloy. Compd. 671, 208–219 (2016)
Y.L. Yang, X.L. Fan, C. Liu, R.X. Ran, First principles study of structural and electronic properties of cubic phase of ZrO2 and HfO2. Physica B 434, 7–13 (2014)
R. Arora, U.V. Waghmare, C. Rao, Metavalent bonding origins of unusual properties of group IV chalcogenides. Adv. Mater. 35(7), 2208724 (2023)
Hashemi, A., Modeling Raman and photoluminescence spectra of defective materials. (2023)
M.A. Volosova, A.A. Okunkova, S.V. Fedorov, K. Hamdy, M.A. Mikhailova, Electrical discharge machining non-conductive ceramics: combination of materials. Technologies 8(2), 32 (2020)
H. Ullah, F.S. Kayani, R. Khenata, Insight into the mechanical, thermal, electronic and magnetic properties of cubic lanthanide built perovskites oxides PrXO3 (X= Al, Ga). Mater. Res. Express 6(12), 126105 (2019)
B. Mayer, H. Anton, E. Bott, M. Methfessel, J. Sticht, J. Harris, P.C. Schmidt, Ab-initio calculation of the elastic constants and thermal expansion coefficients of Laves phases. Intermetallics 11(1), 23–32 (2003)
A. Nakajima, A. Yoshihara, M. Ishigame, Defect-induced Raman spectra in doped CeO2. Phys. Rev. B 50(18), 13297 (1994)
X.S. Zhao, S.L. Shang, Z.K. Liu, J.Y. Shen, Elastic properties of cubic, tetragonal and monoclinic ZrO2 from first-principles calculations. J. Nucl. Mater. 415(1), 13–17 (2011)
F. Fakhera, N.A. Shahed, S. Khanom, M.K. Hossain, F. Ahmed, The first-principle study on structural, mechanical, electronic and optical properties of half-metallic CaMSi2O6 (M= Co, Fe, Mn) clinopyroxenes. Comput. Condens. Matter 33, e00749 (2022)
M. Sajjad, U.A. Khan, H. Ullah, A. Alhodaib, M. Amami, V. Tirth, A. Zaman, Structural, electronic, magnetic and elastic properties of xenon-based fluoroperovskites XeMF 3 (M= Ti, V, Zr, Nb) via DFT studies. RSC Adv. 12(42), 27508–27516 (2022)
G.N. Greaves, A.L. Greer, R.S. Lakes, Rouxel, T, Poisson’s ratio and modern materials. Nat. Mater. 10(11), 823–837 (2011)
Z.E. Biskri, H. Rached, M. Bouchear, D. Rached, M.S. Aida, A comparative study of structural stability and mechanical and optical properties of fluorapatite (Ca5 (PO4) 3F) and lithium disilicate (Li2Si2O5) components forming dental glass–ceramics: first principles study. J. Electron. Mater. 45(10), 5082–5095 (2016)
Q. Long, X. Nie, S.L. Shang, J. Wang, Y. Du, Z. Jin, Z.K. Liu, C15 NbCr2 Laves phase with mechanical properties beyond Pugh’s criterion. Comput. Mater. Sci. 121, 167–173 (2016)
Z.Q. Wang, M.S. Wu, G. Liu, X.L. Lei, B. Xu, C.Y. Ouyang, Elastic properties of new solid state electrolyte material Li10GeP2S12: a study from first-principles calculations. Int. J. Electrochem. Sci. 9(2), 562–568 (2014)
X. Li, M. Ren, Y. Zhang, Ab-initio prediction on the micro-mechanical properties of low-alloy steel. Mater. Today Commun. 34, 105035 (2022)
M.H.K. Rubel, S.K. Mitro, M.K. Hossain, K.M. Hossain, M.M. Rahaman, J. Hossain, A.K.M.A. Islam, First-principles calculations to investigate physical properties of single-cubic (Ba0 82K0 18)(Bi0 53Pb0 47) O3 novel perovskite superconductor. Mater. Today Commun. 33, 104302 (2022)
C. Kurkcu, S. Al, C. Yamcicier, Investigation of mechanical properties of KCaH3 and KSrH3 orthorhombic perovskite hydrides under high pressure for hydrogen storage applications. Eur. Phys. J. B. 95(11), 1–11 (2022)
C.F. Wang, C. Shi, A. Zheng, Y. Wu, L. Ye, N. Wang, H.Y. Ye, M.G. Ju, P. Duan, J. Wang, Y. Zhang, Achieving circularly polarized luminescence and large piezoelectric response in hybrid rare-earth double perovskite by a chirality induction strategy. Mater. Horiz. 9(9), 2450–2459 (2022)
S. Rahimi, R. Ebrahimi-Jaberi, F. Jalali-Asadabadi, L. Mollabashi, S. Jalali-Asadabadi, Influence of (Ba, F) multidoping on structural, magnetic, optical, and electrical properties as well as performance enhancement of multiferroic BiFeO3. Phys. Rev. B 106(11), 115205 (2022)
T.J. Kim, J.S. Byun, N. Barange, H.G. Park, Y.R. Kang, J.C. Park, Y. Kim, D, Parametrization of the optical constants of AlAs x Sb 1–x alloys in the range 0.74–6.0 eV. J. Opt. Soc. Korea 18(4), 359–364 (2014)
L. Sohrabi, A. Boochani, A. Taghavi, The effect of substituting Cobalt into graphene-like InAs on its optical properties: a DFT study. Optik 271, 170194 (2022)
M. Usman, J. ur Rehman, M.B. Tahir, A. Hussain, Structural, electronics, magnetic, optical, mechanical and hydrogen storage properties of Ga-based hydride-perovskites X GaH3 (X= K, Li). Int. J. Energy Res. 46(11), 15617–15626 (2022)
J. Robertson, High dielectric constant oxides. Eur. Phys. J.-Appl. Phys. 28(3), 265–291 (2004)
M. Dawber, K. Rabe, J. Scott, Physics of thin-film ferroelectric oxides. Rev. Mod. Phys. 77(4), 1083 (2005)
Q. Zhou, S. Zhu, Z. Ma, Y. Liu, L. Liu, L. Gao, Experimental and first-principles study on the effect of oxygen vacancy on infrared emissivity of CeO2. Ceram. Int. 48(8), 11313–11319 (2022)
Jarolimek, K., Atomistic models of amorphous semiconductors. Proefschrift, Technische Universiteit Delft, (2011)
E.D. Sherly, J.J. Vijaya, L.J. Kennedy, A. Meenakshisundaram, M. Lavanya, A comparative study of the effects of CuO, NiO, ZrO2 and CeO2 coupling on the photocatalytic activity and characteristics of ZnO. Korean J. Chem. Eng. 33(4), 1431–1440 (2016)
Y. Hao, L. Li, D. Liu, H. Yu, Q. Zhou, The synergy of SPR effect and Z-scheme of Ag on enhanced photocatalytic performance of 3DOM Ag/CeO2-ZrO2 composite. Mol. Catal. 447, 37–46 (2018)
G.C. Cruz, de Almeida, Franchising (Forense, Rio de Janeiro, 1993)
Author information
Authors and Affiliations
Contributions
MM, AW researched about the material and worked on this project theoretically. MB, AA and RB assisted in graphical representations and edited figures and tables of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no competing interest.
Ethics approval
The study does not require any ethical clearances.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
The participants have considered to the submission of the case report to the journal.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moin, M., Anwar, A.W., Babar, M. et al. Computational determination of La substitution in promising XO2 (X = Ce, Zr, Sn): emerging materials for optoelectronic applications. Eur. Phys. J. B 97, 20 (2024). https://doi.org/10.1140/epjb/s10051-024-00657-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjb/s10051-024-00657-9