
Abstract
The thermal decomposition of 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3-nitropyrazole (I) in isothermal and nonisothermal conditions is studied using the methods of thermal analysis, manometry, IR spectroscopy, mass spectrometry, and NMR 1H. The heat of the α → β polymorphic transition, the melting heat of β-I, and the heat of thermal decomposition I are 0.4, 6.2, and 79.2 kcal/mol (274 cal/g), respectively. The composition of the reaction products is analyzed. Among the products formed, N2O, H2O, NO2, NO, N2, CH3OH, and 3-nitropyrazole are identified. The kinetics of the thermal decomposition of I is complex and cannot be described by simple equations. The effective activation energies of the reaction are determined to be close to 34–35 kcal/mol and slightly increase with the progress of decomposition. It is concluded that the first stage of the reaction involves the elimination of the methoxydiazene oxide group through a five-membered cyclic transition state. The number of nitro groups attached to the pyrazole ring does not affect the kinetics of the thermal decomposition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Alkoxy-NNO-azoxy compounds (N-alkyl-N'-alkoxydiazene-N-oxides) are of considerable interest as effective hi-energy compounds [1–3]. Possessing a larger energy reserve than their related N-nitro compounds, they at the same time surpass them in the enthalpy of formation [4], as well as in thermal [5] and chemical stability [6–8].
In recent years, a number of substances have been synthesized, whose molecules, together with methoxydiazene oxide, contain other hi-energy groups [9–13], including nitropyrazole [9–14]. A number of nitropyrazole derivatives have been proposed as components of solid propellants [15–18]. The symbiosis of nitropyrazoles with methoxydiazene oxide groups makes it possible to obtain materials with a high hydrogen content while maintaining a positive enthalpy of formation, which is important for their use as components of gunpowders and solid propellants.
In this paper, thermal transformations of one of the representatives of this class of substances, 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3-nitropyrazole (I)

I
which can exist in the form of two polymorphic modifications, α and β, are studied [13].
Information on the thermal decomposition of alkoxy-NNO-azoxy compounds is very scarce. The decomposition of a number of alkoxy-NNO-azoxy compounds in the gas phase was studied in [5]. The reaction kinetics is described by a first order equation. It turned out that the structure of the molecules of the reacting substances noticeably affects the kinetics and mechanism of the process. With an N-alkyl substituent CH3 and (CH3)3CCH2, the reaction activation energies are 48.8–49.2 kcal/mol, the preexponential factors are 1014.5–1014.6 s–1, and its limiting step is the homolytic N–O bond cleavage. However, with an N‑alkyl substituent (CH3)3C, the presence of hydrogen atoms in the molecule in the β-position allows the reaction to proceed along an energetically more favorable channel, through a five-membered cyclic transition state. In this case, the activation energy is 35.4 kcal/mol, and the preexponential factor is 1013.3 s–1. Similar results were obtained in the study of the decomposition of alkoxy-NNO-azoxy compounds in the liquid phase [19]. In this case, the radical decomposition proceeds with activation energies of 43.5 to 44.5 kcal/mol and preexponential factors 1014.6–1015.5 s–1, and decay through a cyclic transition state, with activation energies of 31.9 to 38.8 kcal/mol and preexponential factors 1011.9–1014.0 s–1.
The thermal decomposition of nitropyrazoles has been studied in more detail [20–34]. It was shown in [27, 29] that an increase in the number of nitro groups, related to the pyrazole ring in the pyrazole molecule is accompanied by an acceleration of decomposition, although the opposite conclusion was made in [15]. In a number of works, the kinetic regularities of decomposition have been studied. In this case, two types of kinetic laws were observed: the first-order reaction equation [29, 31] and the autocatalysis equation [28, 29, 33]. As noted in [29], with an increase in the number of nitro groups related to the pyrazole ring, a gradual change in kinetic dependences is observed; i.e., a transition from a first-order reaction to kinetics described by the law of the first order of autocatalysis.
Despite the abundance of works on the thermal decomposition of nitropyrazoles, there is still no consensus on the nature of even the first stage of decomposition. Various options have been proposed: homolytic cleavage of C–NO2 [22, 23, 31], N–N [23], and C–N [23] bonds, transition of the nitro group to the aciform [29, 21, 22, 31], nitro-nitrite rearrangement [20], 1,5-hydrogen transfer [24, 33], and oxidation of the neighboring carbon atom by the nitro group [29].
Since the substance studied in this work contains a 3-nitropyrazole group, let us dwell on the data on the decomposition of 3-nitropyrazole in more detail. Judging by the shape of the kinetic curves given in [29], this substance decomposes, possibly with weak self-acceleration. Nevertheless, the kinetic data can be satisfactorily described by a first-order equation with an activation energy of 34.0 kcal/mol and a preexponential factor of 108.50 s–1. The first stage of the reaction, according to the authors, is the oxidation of the neighboring carbon atom by the nitro group. In [24], the thermal decomposition of 3-nitropyrazole was studied under nonisothermal conditions by thermogravimetry (TG) and differential thermal analysis (DTA), and the kinetic analysis was performed by the isoconversion method. The activation energies calculated by the Friedman and Flynn–Wall–Ozawa methods were found to be 31.6 and 27.5 kcal/mol, respectively. In both cases, it turned out that the calculated values of the effective activation energies strongly depend on the degree of conversion. In the same work, several possible schemes for the decomposition of 3‑nitropyrazole are given, but only for the mechanism starting from the 1,5-transfer of hydrogen, and a possible chain of further transformations is considered. In [26], the thermal decomposition of a number of nitropyrazoles (including 3-nitropyrazole) was studied by pulsed photoacoustic spectroscopy. The authors concluded that the reaction proceeds in multiple stages and may include tautomerization of the starting material. H2O, CO, CO2, NO2, NO, N2, N2O, HCN, and H2 were found among the gas-like decomposition products.
The authors of [35, 36] studied the thermal decomposition of substances whose molecules contain both alkoxy-NNO-azoxide and the nitropyrazole groups. In [35], the thermal decomposition of N-[2,2-bis(methoxy-NNO-azoxy)ethyl]-4-nitropyrazole in the nonisothermal mode was studied by DSC, TG, and IR spectroscopy, and the composition of the resulting products was determined. The Kissinger method was used to calculate the activation energy and the preexponential factor, the values of which were found to be 40.7 kcal/mol and 1013.4 s–1. The thermal decomposition of 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3,4-dinitro-1H-pyrazole was studied in [35, 36] in both isothermal and nonisothermal conditions. The liquid-phase decomposition of this substance proceeds with weak self-acceleration. In contrast to individual alkoxy-NNO-azoxy compounds and nitropyrazoles, the decomposition kinetics of a substance in which both these functional groups are simultaneously present could not be described either by a first-order equation or by an autocatalysis equation. Therefore, the isoconversion method was used to describe the process quantitatively. The effective activation energies estimated in this way vary depending on the degree of conversion, but are generally close to 30 kcal/mol. The results obtained in [35, 36] made it possible to conclude that the alkoxy-NNO-azoxy group is involved in the rate-limiting step of the reaction.
The structure of 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3-nitropyrazole studied in this study is very similar to the substances studied in [35, 36]; the difference lies only in the number and arrangement of nitro groups in the pyrazole ring. Therefore, this study seems to be useful in terms of establishing the role of the nitropyrazole group in the kinetics of thermal decomposition.
EXPERIMENTAL
As noted above, compound I can be in two crystalline modifications: α and β. Polymorph α-I was synthesized according to the known method [13]. The melting point (123.8–124.2°C), IR-spectrum, and spectra 1H-and 13C-NMR corresponded to the published data [13]. Before starting work, the substance is recrystallized from ethanol. Its purity is confirmed by the elemental analysis data. Found, %: C, 29.11; H, 3.87; N, 34.10; C7H11N7O6. Calculated, %: C, 29.07; H, 3.83; N, 33.90. Polymorph β-I obtained by recrystallization of the α-polymorph from toluene. Melting point (148.4–148.7°С), IR-spectrum and spectra 1H- and 13C-NMR also corresponded to the published data [13]. Found, %: C, 29.20; H, 3.95; N, 34.06; C7H11N7O6. Calculated, %: C, 29.07; H, 3.83; N, 33.90.
Thermal decomposition I in the nonisothermal mode were studied by differential scanning calorimetry and TG using an STA 449F5 synchronous thermal analyzer manufactured by Netzsch (Germany) coupled with a QMS 403C Aeolos quadrupole mass spectrometer in the temperature range 30–350°С when purging with argon at a rate of 40 mL/min, heating speed 5°C/min, and sample weight 1.3 mg. The mass spectrometric analysis of gas-like products of thermal decomposition I was carried out at an energy of ionizing electrons of 70 eV.
Under isothermal conditions, the decomposition kinetics was studied in two ways. Weight loss experiments were carried out in an open system using ATV-14M automatic thermobalances [37], and manometric measurements of gas release were carried out in a closed system using Bourdon glass membrane manometers. During thermogravimetric experiments, a sample weighing of ~8 mg was placed in a quartz cup and introduced into the thermostatically controlled part of the setup (electrically heated furnace). In the course of thermogravimetric experiments, white matter was observed to precipitate on the cold parts of the setup. As will be shown below, it was formed as a result of the sublimation of one of the thermal decomposition products of compound I.
The working part of the Bourdon manometer used in manometric measurements had a volume of 3.5 to 4 mL, and the sample of the test substance was 3.5–4 mg. Before the start of the experiment, the reaction vessel was evacuated to a residual pressure of ~0.1 Torr.
IR spectra of the starting material and condensed products of its decomposition were obtained using an ALPHA II IR Fourier spectrometer manufactured by Bruker Optics (Germany) in the range 360–3800 cm–1 at a resolution of 4 cm–1 and the number of scans was 16. Samples for recording IR spectra were prepared in the form of tablets with KBr. In each case, the spectrum of a similar tablet prepared from pure KBr was subtracted from the total spectrum.
The 1H-NMR spectra of the sublimations of decomposition products were recorded in 5% solution in CD3CN on a Bruker Avance III 500 MHz spectrometer (500.2 MHz); the internal standard was tetramethylsilane.
RESULTS AND DISCUSSION
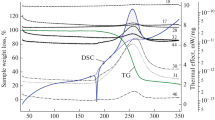
Figure 1 shows the data obtained by heating the test substance at a rate of 5°C/min. The DSC curves contain two endothermic peaks with minimums at temperatures of 121 and 148°C. The first one corresponds to the polymorphic transition α → β (observations in the capillary showed that this transition proceeds via by melting α-I); and the second one, by melting β-I. The heats of these transformations were found to be 0.4 and 6.2 kcal/mol, respectively. The exothermic peak on the DSC curve and the corresponding mass loss correspond to the process of thermal decomposition. When heated up to 300°C, the weight loss was ~90%, and the corresponding thermal effect was found to be 79.2 kcal/mol (274 cal/g). During thermal analysis, it turned out that the processes of thermal decomposition of the α- and β-polymorphs of compound I run proceed identically. Therefore, all subsequent measurements were performed with the α-polymorph of this compound.

DSC, TG curves for compound I at a heating rate of 5.0 deg/min.
Figure 2 shows the results of the mass spectrometric measurements carried out synchronously with the TG and DSC experiments. As a result, it was found that N2O, H2O, NO2, NO, N2, and CH3OH are formed as gas-like reaction products.

Mass spectrometric curves of the thermal decomposition of the compound I at a heating rate of 5°C/min; the numbers near the curves are the values m/z.
In the IR spectrum of the initial compound I (curve 1 in Fig. 3), there are bands of stretching vibrations of the C–H bonds of the pyrazole ring (at 3126 and 3143 cm–1), as well as methyl and methylene groups (in the range 2835–3050 cm–1). The band at 1543 cm–1 is related to the double bond C=N; and the intense band at 1054 cm–1, to the N–O–C fragment. The rest of the bands belong to mixed modes, including those with the participation of antisymmetric stretching modes (in the range of wave numbers 1450–1520 cm–1), antisymmetric valence (1240–1370 cm–1), and strain (750–830 cm–1) vibrations of nitro groups due to the resonance of these vibrations with vibrations of the triazole ring and N-oxide groups.

IR spectra of original compound I of α-modification (1), solid residue in the beaker after decomposition I at 173°C (2), sublimation of decomposition products on the cold wall above the glass (3), and the reference sample of 3-nitropyrazole (4).
The IR spectrum of the solid residue in the beaker after decomposition of compound I (curve 2 in Fig. 3) is a superposition of the spectra of crystalline (narrow bands) and amorphous (broad bands) substances; in this case, the broad bands in the wavenumber range 1100–1700 cm–1 belong to the polyconjugated system of bonds formed by carbon and nitrogen atoms, and most of the narrow bands coincide with bands of the sublimate on the cold wall above the cup (curve 3 in Fig. 3). In addition, the spectrum of the solid residue contains bands of water molecules (at 1615 and 3421 cm–1) adsorbed due to the contact of the product with the atmosphere. The band at 3158 cm–1 in the sublimation spectrum and the band at 3134 cm–1 in the spectrum of the solid residue cannot be attributed to vibrations of C–H bonds, since their large widths indicate the formation of hydrogen bonds of various strengths. At the same time, the positions and widths of these bands rule out their belonging to O–H bonds. Therefore, the bands in the wavenumber range 3130–3160 cm–1 of the IR spectra of the sublimate and the solid residue are related to vibrations with the participation of the N–H group, which forms a hydrogen bond.
In turn, the most intense bands in the IR spectrum of sublimation almost coincide with the strong bands of 3-nitropyrazole (curve 4 in Fig. 3). The additional peaks (relatively weak and narrow) in the sublimate spectrogram are probably related to a small admixture of crystals of a volatile reaction byproduct. The additional peaks (also relatively weak and narrow) in the spectrogram of 3-nitropyrazole used as the reference sample may be attributed to impurities, in particular to molecules of crystalline water (the band at 1683 cm–1 and increased absorption in the region of 3100 to 3300 cm–1). Apparently, the used sample of 3-nitropyrazole is wholly or partly a crystalline hydrate. In this case, the additional bands at 443 and 614 cm–1 in its spectrum can be related to the librational vibrations of water molecules.
It is interesting to note that not a single band of the pyrazole ring in the IR spectrum of 3-nitropyrazole coincides with any band in the spectrum of compound I. This can be explained by the resonant interactions of vibrations of different groups in the compound I and the formation of a hydrogen bond between the N–H group and the nitrogen atom in the pyrazole ring of 3‑nitropyrazole, which could lead to a redistribution of the electron density in the conjugated system.
Thus, based on the data of IR spectroscopy, it can be concluded that the main condensed products of the thermal decomposition of compound I are 3-nitropyrazole and an amorphous polymer containing a polyconjugated system of bonds formed by the carbon and nitrogen atoms. Note that the formation of such polymers is generally characteristic of the thermal decomposition of many hi-energy compounds [35, 38, 39].
According to the 1H-NMR data, the sublimate is a complex mixture with 3-nitropyrazole as the main component, identified for all three protons (δ, ppm: 6.92, doublet, 1H, 3J = 2.5 Hz, H-4; 7.71, doublet, 1H, 3J = 2.5 Hz, H-5; 11.8, 1H, broadened singlet, NH). AT spectrum 1H-NMR there are also several substances containing group CH3ON=N(O), which could not be identified (δ, ppm: 4.07, singlet, 3H, CH3O). The substrate contains the original compound I in a trace amount (less than 0.4 mol % in relation to 3-nitropyrazole), identified by the characteristic triplet (δ, ppm: 6.49, triplet, 1H, 3J = 6.8 Hz, CH2CH).
Figure 4 shows the results of kinetic measurements of the thermal decomposition of compound I in the isothermal mode, obtained by the TG method. As can be seen, the reaction proceeds with weakly pronounced self-acceleration at relatively low temperatures; however, as the temperature of the experiment increases, it gradually disappears.

Kinetic curves of thermal decomposition of compound I at different temperatures, °C: 1, 140; 2, 160; 3, 167; 4, 173; 5, 180; 6, 187; 7, 194; 8, 200.
Isothermal measurements were also carried out in a closed system using the manometric method. In general, the results of thermogravimetric and manometric measurements are in good agreement with each other, although there are some differences. Figure 5 compares the experimental results obtained at 200°C (a similar picture was observed at other temperatures). It can be seen from this figure that in a closed system the reaction proceeds somewhat faster, and self-acceleration is more pronounced.

Kinetic curves of thermal decomposition of compound I at 200°С: 1, open system; 2, closed system.
An attempt to quantitatively describe the kinetic dependences was not successful: these dependences could not be described by the equations usually used to analyze the thermal decomposition of hi-energy substances: a first-order equation or an autocatalysis equation. Therefore, the isoconversion method was used to determine the effective activation energies [40]. The activation energies were estimated by two methods: from the temperature dependence of the times for hte given conversion percentage and from the temperature dependence of the decomposition rates at the given degree of conversion. The results are shown in Table 1. It can be seen from it that both used calculation methods lead to almost identical results, and as the degree of conversion increases, the effective activation energy increases, and its average value is close to 34–35 kcal/mol.
It follows from the foregoing that the studied reaction proceeds according to a complex multistage mechanism and depends on a variety of factors. For example, from the fact that in a closed system (manometric measurements) the reaction is somewhat faster than in an open one (in thermogravimetric experiments), it can be concluded that it is affected by gas-like decomposition products. The composition of these products is listed above. Since nitrogen dioxide is the most active among them, it is natural to assume that one of the stages of the studied process is the oxidation of the initial material or its transformation products with nitrogen dioxide. This oxidation and the effect of gas-like products on thermal decomposition are characteristic of hi-energy substances [41].
Test compound I contains two types of reactive functional groups: 3-nitropyrazole and two geminal methoxydiazene oxide groups. Therefore, when analyzing the mechanism of its decomposition, we should first of all consider the possibility that the reaction begins with the decomposition of one of these groups. The thermal decomposition of nitropyrazoles was discussed above. 3-Nitropyrazole decomposes with an activation energy of 34.0 kcal/mol [29], and this value agrees closely with the values obtained in this study. However, the decay of 3-nitropyrazole is characterized by a very low value of the preexponential factor, 108.5 s–1, and in order for it to react at rates comparable to the decomposition rate of compound I, it would be necessary to increase the temperature by ~100°C. As for the methoxydiazene oxide group, if it decomposes through a cyclic transition state (and there is a possibility for this, since the molecule of compound I contains two hydrogen atoms in the β-position), then the value of the activation energy, as noted above, should be in the range 32–39 kcal/mol, and this is in good agreement with the data given in Table 1. Moreover, it was noted in the introduction that the preexponential factors for the decomposition reaction of diazene oxides according to the mechanism given above are several orders of magnitude higher than for 3-nitropyrazole. Therefore, the possibility of the decomposition of compound I according to the mechanism characteristic of alkoxydiazene oxides with hydrogen atoms in the β-position according to the following scheme seems entirely possible:

This scheme assumes that the thermal decomposition of compound I proceeds sequentially and includes the formation of an intermediate product II and its polymerization with the subsequent decomposition of polymer III with the formation of 3-nitropyrazole. Similar processes and effects were observed earlier in the synthesis of methoxy-NNO-azoxyethene by the method of the preparative pyrolysis of 1,1-bis(methoxy-NNO-azoxy)ethane [42, 43]. Since the effective activation energy increases in the course of decomposition, the second stage of the reaction may proceed more slowly than the first one.
Figure 6 compares the kinetic curves of the thermal decomposition of two analogs that are similar: 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3-nitropyrazole and 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3,4-dinitropyrazole (the data for the second substance were taken from [36]). It can be seen that they almost coincide. Thus, the nitropyrazole group as a substituent does not affect the limiting step of the reaction. This, in turn, means that an increase in the number of nitro groups in the pyrazole substituent (which leads to an increase in the oxygen balance and, accordingly, energy consumption) does not reduce the thermal stability of the substances of the studied class.

Kinetic curves of thermal decomposition at 200°C of compound I (1) and 1-[2,2-bis(methoxy-NNO-azoxy)ethyl]-3,4-dinitropyrazole (2).
REFERENCES
I. N. Zyuzin and D. B. Lempert, Bull. Acad. Sci. USSR, Div. Chem. Sci. 34, 753 (1985).https://doi.org/10.1007/BF00948052
I. N. Zyuzin, G. N. Nechiporenko, N. I. Golovina, R. F. Trofimova, and M. V. Loginova, Russ. Chem. Bull. 46, 1421 (1997).https://doi.org/10.1007/BF02505678
I. N. Zyuzin, N. I. Golovina, D. B. Lempert, G. N. Nechiporenko, and G. V. Shilov, Russ. Chem. Bull. 57, 632 (2008).https://doi.org/10.1007/s11172-008-0099-3
E. P. Kirpichev, I. N. Zyuzin, V. V. Avdonin, Yu. I. Rubtsov, and D. B. Lempert, Russ. J. Phys. Chem. A 80, 1359 (2006).https://doi.org/10.1134/S0036024406090019
I. N. Zyuzin, D. B. Lempert, and G. N. Nechiporenko, Bull. Acad. Sci. USSR, Div. Chem. Sci. 37, 1329 (1988). https://doi.org/10.1007/BF00962732
I. N. Zyuzin and D. B. Lempert, Russ. J. Gen. Chem. 80, 1792 (2010).https://doi.org/10.1134/S1070363210090124
I. N. Zyuzin and D. B. Lempert, Kinet. Catal. 52, 17 (2011).https://doi.org/10.1134/S0023158411010228
I. N. Zyuzin and D. B. Lempert, Russ. J. Gen. Chem. 82, 1105 (2012).https://doi.org/10.1134/S1070363212060114
I. N. Zyuzin, N. I. Golovina, B. S. Fedorov, G. V. Shilov, and G. N. Nechiporenko, Russ. Chem. Bull. 52, 761 (2003).https://doi.org/10.1023/A:1023952032749
I. N. Zyuzin and D. B. Lempert, Russ. Chem. Bull. 58, 2173 (2009).https://doi.org/10.1007/s11172-009-0297-7
I. N. Zyuzin, K. Yu. Suponitsky, and A. B. Sheremetev, J. Heterocycl. Chem. 49, 561 (2012). https://doi.org/10.1002/jhet.811
I. N. Zyuzin, Russ. J. Org. Chem. 49, 536 (2013).https://doi.org/10.1134/S1070428013040076
I. N. Zyuzin, K. Yu. Suponitskii, and I. L. Dalinger, Chem. Heterocycl. Compd. 53, 702 (2017).https://doi.org/10.1007/s10593-017-2112-y
I. N. Zyuzin, Russ. Chem. Bull. 69, 1949 (2020).https://doi.org/10.1007/s11172-020-2984-3
I. N. Zyuzin, A. I. Kazakov, D. B. Lempert, I. A. Vatsadze, L. S. Kurochkina, and A. V. Nabatova, Combust. Explos., Shock Waves 55, 327 (2019). https://doi.org/10.1134/S0010508219030109
I. N. Zyuzin, D. B. Lempert, A. V. Nabatova, and A. I. Kazakov, Combust. Explos., Shock Waves 56, 464 (2020). https://doi.org/10.1134/S0010508220040103
I. Yu. Gudkova, I. N. Zyuzin, and D. B. Lempert, Russ. J. Phys. Chem. B 14, 302 (2020). https://doi.org/10.1134/S1990793120020062
I. N. Zyuzin, I. Yu. Gudkova, and D. B. Lempert, Russ. J. Phys. Chem. B 14, 804 (2020). https://doi.org/10.1134/S1990793120050140
I. Zyuzin, D. Lempert, V. Prokudin, E. Kirpichev, and G. Manelis, in Energetic Materials. Performance and Safety, Proceedings of the 36th International Annual Conference (ICT, Karlsruhe, 2005), p. 162.
I. Dalinger, S. Shevelev, V. Korolev, et al., J. Therm. Anal. Calorim. 105, 509 (2011).https://doi.org/10.1007/s10973-010-1213-y
O. Gryzlova, A. Pivkina, I. Dalinger, et al., in Proceedings of the 15th Seminar on New Trends in Research of Energetic Materials, Pardubice, Czech Republic, 2012, Part 1, p. 173.
I. Dalinger, A. Pivkina, O. Gryzlova, et al., in Proceedings of the 38th International Pyrotechnics Seminar (Denver, Colorado, USA, 2012), p. 479.
P. Ravi, G. M. Goreb, A. K. Sikder, and S. P. Tewari, Thermochim. Acta 528, 53 (2012).
P. Ravi, A. A. Vargeese, and S. P. Tewari, Thermochim. Acta 550, 83 (2012).
Y. L. Wang, F. Q. Zhao, Y. P. Ji, et al., J. Anal. Appl. Pyrol. 98, 231 (2012).
F. Yehya and A. K. Chaudhary, Appl. Phys. B 110, 15 (2013).
A. Bragin, A. Pivkina, N. Muravyev, et al., Phys. Proc. 72, 358 (2015).
V. V. Dubikhin, G. M. Nazin, V. G. Prokudin, et al., Russ. Chem. Bull. 64, 127 (2015).https://doi.org/10.1007/s11172-015-0830-9
V. V. Dubikhin, G. M. Nazin, V. G. Prokudin, I. L. Dalinger, and S. A. Shevelev, Russ. J. Phys. Chem. B 9, 211 (2015).
N. V. Muravyev, A. A. Bragin, K. A. Monogarov, et al., Propell. Explos. Pyrotech. 41, 999 (2016).https://doi.org/10.1002/prep.201600068
V. P. Sinditskii, S. P. Smirnov, V. Yu. Egorshev, et al., Thermochim. Acta 651, 83 (2017).
V. P. Sinditskii, T. H. Hoang, A. D. Smirnova, et al., Thermochim. Acta 667, 1 (2018).
M. V. Gorn, K. A. Monogarov, I. L. Dalinger, et al., Thermochim. Acta 690, 178697 (2020).
V. P. Sinditskii, A. D. Smirnova, V. V. Serushkin, et al., Thermochim. Acta 698, 178876 (2021).
V. V. Zakharov, N. V. Chukanov, I. N. Zyuzin, V. V. Nedel’ko, and B. L. Korsunskii, Russ. J. Phys. Chem. B 13, 62 (2019).https://doi.org/10.1134/S1990793119010305
B. L. Korsunskii, V. V. Zakharov, T. S. Larikova, et al., Khim. Fiz. 41 (7), 32 (2014).
L. N. Gal’perin, Yu. R. Kolesov, and N. A. Zelenov, Izmerit. Tekh., No. 4, 23 (1981).
V. V. Nedel’ko, B. L. Korsunskii, T. S. Larikova, Yu. M. Mikhailov, S. V. Chapyshev, and N. V. Chukanov, Russ. J. Phys. Chem. B 5, 244 (2011).
V. V. Nedel’ko, N. V. Chukanov, B. L. Korsunskiy, T. S. Larikova, S. V. Chapyshev, and V. V. Zakharov, Russ. J. Phys. Chem. B 12, 997 (2018).
S. Vyazovkin, Isoconversional Kinetics of Thermally Stimulated Processes (Springer, Heidelberg, 2015).
G. B. Manelis, G. M. Nazin, Yu. I. Rubtsov, and V. A. Strunin, Thermal Decomposition and Combustion of Explosives and Gunpowders (Nauka, Moscow, 1996) [in Russian].
I. N. Zyuzin, Russ. J. Appl. Chem. 82, 1799 (2009).https://doi.org/10.1134/S1070427209100152
V. P. Grachev, I. N. Zyuzin, S. V. Kurmaz, et al., Russ. Chem. Bull. 69, 2312 (2020).https://doi.org/10.1007/s11172-020-3037-7
Funding
The study was carried out according to the theme of a state task for Institute of Problems of Chemical Physics, Russian Academy of Sciences (IPCP RAS) (registration number AAAA-A19-119101690058-9) and on the topic of a state task for Semenov Institute of Chemical Physics, Russian Academy of Sciences (registration number AAAA-A17-117040610346-5).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zakharov, V.V., Korsunsky, B.L., Larikova, T.S. et al. Thermal Decomposition of 1-[2,2-bis(Methoxy-NNO-azoxy)ethyl]-3-nitropyrazole. Russ. J. Phys. Chem. B 16, 846–853 (2022). https://doi.org/10.1134/S1990793122050128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990793122050128