
Abstract—
The involvement of the mitochondrial calcium-dependent NO synthase (mtNOS) in the regulation of mitochondrial respiration has not been sufficiently studied. Moreover, the possible functioning of a mitochondrial signaling system involving mtNOS/guanylate cyclase (GC)/protein kinase G (PKG) and the impact of this system (mtNOS/GC/PKG-SS) on mitochondrial respiration have not yet been analyzed. To investigate this issue, we performed experiments on isolated rat liver using specific inhibitors of NOS, GC, and PKG. The high rate of mitochondrial respiration was supported by pyruvate and glutamate or by succinate in the presence of hexokinase, glucose, and ADP. It was shown that L-arginine and the NO donor sodium nitroprusside (SNP) exert concentration-dependent effects on the mitochondrial respiration rate. At low concentrations, L‑arginine (up to 200 μM) and SNP (up to100 μM) activated mitochondrial respiration. The inhibitors of NOS, GC, and PKG eliminated this effect indicating that mtNOS/GC/PKG-SS is involved in the activation of respiration. At high concentrations, L-arginine and SNP, on the contrary, inhibited respiration. Under these conditions, the inhibitors of GC and PKG enhanced the inhibition of respiration, which indicates an opposite effect of the excess of NO and PKG on the mitochondrial respiration. The results suggest that the functioning of calcium-dependent mtNOS/GC/PKG-SS can ensure the activation of respiration at low concentrations of L-arginine or SNP in the medium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Studies of recent decades have convincingly shown that exogenous NO and NO produced by cytoplasmic NO synthases (NOS) lead to suppression of mitochondrial respiration by inhibiting cytochrome c oxidase (COX) and complexes I and II of the electron transport chain [1–6]. Inhibition of COX by NO is reversible and competitive with respect to oxygen [1]. In the experiments on HEK cells overexpressing inducible nitric oxide synthase-(iNOS) it was shown that an increase in the concentration of L-arginine in the medium from 1 to 10 µM leads to the reduction of cytochromes, accumulation of NO, and inhibition of oxygen consumption [6]. Mitochondrial isoform of NOS (mtNOS) localized on the inner membrane is another source of NO. According to the data obtained on liver and heart mitochondria, this isoform is related to brain (neuronal) isoform NOSα [7–9]. Activation of calcium-dependent mtNOS by its substrate L-arginine and Ca2+ also causes inhibition of mitochondrial respiration associated with a sharp increase in NO content [10]. Like cytoplasmic enzymes, mtNOS is an oxygen- and NADPH-dependent enzyme, so the production of mitochondrial NO is maximal at high membrane potential ΔΨm and low rates of mitochondrial respiration (at rest, in State 4) [10]. NO is a source of nitrosothiols, peroxynitrite, ROS, etc., the targets of which are numerous proteins of mitochondrial metabolism. Therefore, the mitochondrion is often regarded as a generator and a target of the NO effects [11] without any connection with the regulatory action of Ca2+. A complex action of Ca2+ on metabolic and signaling systems of mitochondria-includes activation of the proteins of the respiratory chain and Krebs cycle [12], activation of NO synthesis, and induction of mitochondrial permeability transition pore [13, 14]. This may indicate the existence of regulatory processes based on the NO and Ca2+ interplay that are insufficiently studied.
Taking this into consideration, we analyzed the influence of NO donors and L-arginine on the regulation of mitochondrial respiration. We assume that the regulation of the mitochondrial respiration with the participation of Ca2+-dependent mtNOS is not limited to the nitrosylation of mitochondrial proteins or the inhibition of the enzymes of respiratory chain by NO, but may also include the functioning of mitochondrial signaling systems with the participation of mtNOS, guanylate cyclase (GC), and protein kinase G (PKG). Effects of this system on the respiration of mitochondria have not been studied yet. The aim of this work was to show by means of inhibitory analysis that L-arginine and NO donors can participate in the regulation of mitochondrial respiration rate by activating Ca2+→mtNOS→NO→ mtGC→cGMP→mtPKG mitochondrial signaling system (mtNOS/GC/PKG-SS).
MATERIALS AND METHODS
All procedures were performed in accordance with the European Communities Council Directive (November 24, 1986; 86/609/EEC) and the Declaration on humane treatment of animals. The experimental protocol was approved by the Bioethics Committee of the Institute of Theoretical and Experimental Biophysics, RAS. Male Wistar rats were kept in the identical conditions for 6–8 weeks in air-conditioned and ventilated rooms at a temperature of 20–22°C (light/dark, 12 h/12 h). The experiments were performed at 26°C. Liver mitochondria were isolated using standard techniques of differential centrifugation in a medium containing 300 mM sucrose, 1 mM EGTA, and 10 mM Tris-HCl (pH 7.4). Mitochondrial preparations were twice washed with an EGTA-free isolation medium, resuspended in the medium of the same composition and stored on ice. The incubation medium for mitochondria contained 125 mM KCl, 3 mM KH2PO4, 10 mM HEPES (pH 7.4), and 1 mM MgCl2. The content of mitochondrial protein was determined by the Lowry method with bovine serum albumin as a standard. Oxygen consumption in the mitochondrial suspension containing 1.0–1.2 mg of protein was determined by a polarographic method with the Clark-type O2 electrode in a closed chamber with a volume of 1 mL under constant stirring. L-glutamate (10 mM) and pyruvate (0.5–1 mM) were used as respiration substrates to maintain the full turnover of the Krebs cycle and substrate-level phosphorylation in the cycle. In some experiments, respiration was supported by succinate (5 mM) in the presence of rotenone (1–2 µM) or pyruvate (1 mM) and L-malate (5 mM). All experiments were performed in the presence of 10 units of hexokinase, 5 mM glucose and 1 mM MgCl2. Subsequent additions of 0.75 mM ADP provided a high steady state respiration rate (VO2ss = d[O2]/dt) close to the maximal respiration rate in State 3 (80–85% of VO2max). To assess the statistical significance of the results, VO2ss was measured at a steady state at [O2] = 270–300 ng atom O/mL in linear parts of the corresponding polarographic curves in a series of four to six experiments. Electric potential difference (ΔΨm) at the inner mitochondrial membrane was determined by the redistribution of the lipophilic cation tetraphenylphosphonium (TPP+), the concentration of which in the medium [TPP+]out was recorded with a TPP+-selective electrode. The used reagents were from Sigma (USA). Statistical analysis of the experimental data was performed using the t-test of Sigma Plot 11 to compare pairs of data values. The data were expressed as mean ± SEM. The number of independent experiments was n = 4–6. The significance level was taken to be p < 0.05.
RESULTS AND DISCUSSION
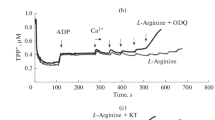
Activation and inhibition of mitochondrial respiration by sodium nitroprusside (SNP) and L-arginine.Figure 1 shows the polarographic curves characterizing oxygen consumption by rat liver mitochondria upon the activation of respiration by 0.75 mM ADP additions into the medium. Pyruvate (1 mM) and L‑glutamate (10 mM) were used as oxidation substrates. The steady state respiration rate was supported by the addition of hexokinase and glucose into the incubation medium. Figure 1 also shows the corresponding dynamics of the mitochondrial potential ΔΨm (TPP+). Preincubation of mitochondria with low concentrations of SNP (20 µM; dotted curve in Fig. 1a) or L-arginine (20 µM; dashed curve in Fig. 1b) caused an increase of the mitochondrial respiration rate characterized by an increase of the slope of the corresponding curves.

The effects of SNP (left) and L-arginine (right) on oxygen consumption (O2 traces) and mitochondrial membrane potential ΔΨm (TPP+ traces) depend on the concentrations of these sources of NO in the medium. Representative experiments are presented. Black curves correspond to the control experiments. Dashedcurves correspond to low concentrations of SNP (panel (a), 10 µM) and L-arginine (panel (b), 100 µM) in the incubation medium. Graylines correspond to high concentrations of SNP (panel (a), 500 µM) and L-arginine (panel (b), 1000 µM). Mitochondria (1.0–1.2 mg) were incubated in a closed chamber with a volume of 1 mL. The incubation medium included 1 mM pyruvate, 10 mM L-glutamate, glucose, and hexokinase. The dependencies of the mean steady state respiration rates (VO2ss) on the concentrations of SNP (c) and L-arginine (d) in the medium. The number of independent experiments was n = 5. The data are presented as mean ± SEM.
In contrast, high concentrations of SNP (400 µM; gray curve in Fig. 1a) and L-arginine (1000 µM; gray curve in Fig. 1b) caused inhibition of mitochondrial respiration below the control values. The observed inhibition of respiration induced by SNP and L-arginine is well known [1–6].
This inhibition of respiration may be caused by known inhibition of COX and other respiratory chain proteins by the excess of NO released from SNP or produced by mtNOS from L-arginine [1, 4, 6]. In contrast to the inhibitory effect, activation of respiration by low concentrations of NO donors or L-arginine was detected for the first time and observed for different combinations of substrates and on different types of mitochondria. Figures 2a and 2b (gray lines) shows that low concentrations of SNP and L-arginine (20 µM) also evoked an increase of the steady state respiration rate in rat liver mitochondria oxidizing succinate (5 mM) in the presence of rotenone (2 µM).

Activation of oxygen consumption upon incubation of mitochondria with low concentrations of SNP (panel (a), 5 µM) and L-arginine (panel (b), 20 µM). Elimination of this activation by the inhibitor of PKG, KT5823 (KT). Incubation medium contained 5 mM succinate, 2 µM rotenone and hexokinase. Other conditions were as in Fig. 1. Black lines correspond to the control experiments. Gray lines correspond to low (activating) SNP concentrations (panel (a), 5 µM) and L-arginine (panel (b), 20 µM). Dotted lines show the elimination of the activation effect of SNP and L-arginine by the PKG inhibitor KT (1.2 µM).
A similar effect was observed on rat heart mitochondria preincubated with low concentrations of SNP and L-arginine (2–10 µM) in the presence of pyruvate (1 mM) and L-glutamate (10 mM) as substrates. The addition of 5 µM L-arginine into the incubation medium caused an increase in the mitochondrial respiration rate by 20–25% as compared to the control. Higher concentrations of SNP and L-arginine also inhibited respiration (not shown).
The effect of mtNOS/GC/PKG-SS inhibitors on the activation of respiration by SNP and L-arginine. The detected activation of respiration by SNP and L-arginine (Figs. 1 and 2) may indicate the involvement of new signaling mechanisms unrelated to the direct effects of NO, nitrosothiols, peroxynitrite, etc. We assume that the mtNOS/GC/PKG-SS signaling system may participate in the regulation of mitochondrial respiration, similar to that of the mitochondrial signaling system involving adenylate cyclase and protein kinase A (AC/PKA) [15–17]. The results presented in Fig. 2 show that the activating effects of 5 µM SNP or 20 µM L-arginine (gray curves) are eliminated in the presence of 1.2 µM of the PKG inhibitor KT5823 (KT) in the incubation medium. This result indicates the participation of mtNOS/GC/PKG-SS and its final mediator PKG in the regulation of mitochondrial respiration rate.
The effect of mtNOS/GC/PKG-SS inhibitors on the respiration inhibition by high concentrations of SNP and L-arginine.Figure 3 shows that the inhibition of respiration and oxygen consumption caused by the preincubation of mitochondria with high concentrations of L-arginine (1 mM), on the contrary, was enhanced in the presence of the inhibitor of GC (dashed curve, [ODQ] = 50 µM) and PKG (dashed curve, [KT] = 1.2 µM). In the control, at respiratory State 4, the mitochondrial respiration rate was 14–15 ng atom O/min/mg protein. At the maximal steady state respiration rate driven by the ADP-hexokinase system the respiration rate increased up to 56 ng atom O/min/mg protein. In the presence of 1 mM L-arginine, the respiration rate decreased to 46 ng atom O/min/mg protein. Preincubation of mitochondria with the inhibitors of GC (ODQ) and PKG (KT) resulted in further inhibition of respiration to 28 and 16 ng atom O/min/mg protein, respectively. These results suggest that the protective effect of the mtNOS/GC/PKG-SS provided by the activation of respiratory chain proteins by its final mediator PKG, is eliminated in the presence of the inhibitors ODQ or KT, and the inhibition of respiration by the excess of NO produced by mtNOS becomes dominant.

Inhibitors of mtNOS/GC/PKG-SS, ODQ and KT, enhance the inhibition of oxygen consumption evoked by high concentration of L-arginine (1 mM). Representative experiment is presented. The experimental conditions were the same as in Fig. 3. Concentrations of the inhibitors ODQ and KT are given in µM.
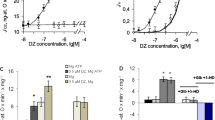
The effect of modulation of the activity of mtNOS/ GC/PKG-SS on the respiration rate of mitochondria. In a series of four to six experiments, the steady state mitochondrial respiration rate (VO2ss) was measured at [O] = 300–320 ng atom O/mL on the linear parts of the corresponding polarographic curves. The calculated average values of VO2ss are shown in Fig. 4 as columns. The left gray columns correspond to the control experiments with VO2ss = 91.8 ± 2.7 ng O/min/mg protein. The next three sets of columns illustrate the effects of various concentrations of SNP (Fig. 4a) and L-arginine (Fig. 4b) on the average values of VO2ss. These data show that the addition of 5–50 µM SNP into the incubation medium caused an increase in the mitochondrial respiration rate (VO2ss) in comparison with the control values. The maximal effect of 27–28% was observed at 10 µM SNP (Fig. 4a, the first and third columns; p < 0.05). Preincubation of mitochondria with the inhibitors ODQ and KT eliminated the activation of respiration caused by 10 µM SNP (Fig. 4a, p <0.05).

Effects of SNP and L-arginine and the inhibitors of mtNOS/GC/PKG-SS on the steady state oxygen consumption rate (VO2ss) during incubation of mitochondria with succinate (5 mM) and rotenone (2 µM). All other conditions were the same as in Fig. 1. The effects of SNP (a) and L-arginine (b) on VO2ss. The left columns correspond to control experiments. The next four and three columns in (a) and (b) characterize the dose-dependent effects of SNP (a) and L-arginine (b) on VO2ss, respectively. The right columns characterize the impact of the mtNOS/GC/PKG-SS inhibitors 7-NI, ODQ, KT5823 (KT) on VO2ss. Preincubation of mitochondria with the inhibitors 7-NI, ODQ, and KT abrogates the activation of respiration by SNP and L-arginine. Preincubation of mitochondria with 100 nM 1400W, a specific inhibitor of inducible iNOS, did not affect the studied effects (panel (b), right column). All concentrations are shown in µM. The data are presented as mean ± SEM. In (a), n = 4 for columns 1–5 and n = 6 for columns 6 and 7. In (b), n = 4 for columns 1–4, and n = 5 for columns 5–8. Student’s t-test was applied. The compared pairs of VO2ss values are marked by horizontal lines placed above the columns; symbol (*) indicates p < 0.05.
Similar to SNP, L-arginine (20 µM) increased VO2ss in comparison with the control value by 15–16% (Fig. 4b, the first and second columns; p <0.05). The inhibitors of NOS (7-NI), GC (ODQ), and PKG (KT) eliminated the activation of respiration caused by L-arginine (Fig. 4b, fifth, sixth, and seventh columns; p < 0.05), which indicated the functioning of all three proteins of the mtNOS/GC/PKG-SS. Preincubation of mitochondria with 1400W (100 nM), a specific inhibitor of calcium-independent inducible NOS, did not affect the mitochondrial respiration rate (Fig. 4b, column 8; p < 0.05). This implies that this enzyme is not involved in the production of NO by the mitochondrial mtNOS/GC/PKG-SS under the conditions of our experiments.
CONCLUSIONS
The results presented above demonstrate that mtNOS/GC/PKG-SS can participate in the regulation of oxidative phosphorylation. Probably, the mechanism providing the activation of mitochondrial respiration is based on phosphorylation of some mitochondrial proteins by PKG, similar to the mechanism realized by the mitochondrial AC/PKA signaling chain [15–17]. The activation of mtNOS/GC/PKG-SS by L-arginine can provide a bi-directional control of respiration, which may include activation of the respiratory chain proteins by PKG and inhibition of these proteins by the excess of NO. Cytoplasmic isoforms of GC are known to have approximately 10 times higher affinity to NO than COX [18]. Assuming comparable sensitivity of mitochondrial and cytosolic GCs to NO, we can speculate that the activation of respiration via PKG would dominate over well-known inhibitory effect of NO [1–6] only at low concentratios of L‑arginine (NO) and calcium ions in mitochondria.
Functioning of mtNOS/GC/PKG-SS is possible only in the presence and constant resynthesis of coenzymes NADPH and GTP in the mitochondrial matrix carried out with the participation of several enzymes. The absence of NADPH and GTP in the incubation medium of mitochondria excludes the possibility of functioning of this chain outside the mitochondria, for example, due to the contamination of the mitochondrial fraction with cytoplasmic proteins, including GC, PKG, dikinase, etc.
It is known that special mechanisms ensure transport of various kinases and phosphatases into the mitochondria, including, for example, protein kinase B [19, 20]. It is possible that GC and PKG are transported into mitochondria via such mechanisms. However, direct experimental data indicating the translocation of PKG and GC into the mitochondria are currently not available. This may be the issue of a further research. Nevertheless, the presented results obtained with the application of specific inhibitors of the proteins of mtN/GC/PKG-SS indicate the possibility of its functioning in mitochondria.
REFERENCES
Cleeter M.W., Cooper J.M., Darley-Usmar V.M.; Moncada S., Schapira A.H. 1994. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett.345 (1), 50–54. https://doi.org/10.1016/0014-5793(94)00424-2
Giulivi C. 1998. Functional implications of nitric oxide produced by mitochondria in mitochondrial metabolism. Biochem. J.332, 673–679.
Brown G.C. 2001. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys.Acta. 1504 (1), 46–57.https://doi.org/10.1016/S0005-2728(00)00238-3
Parihara M.S., Nazarewicza R.R., Kincaida E., Bringoldb U., Ghafourifara P. 2008. Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem. Biophys. Res. Commun.366 (1), 23–28. https://doi.org/10.1016/j.bbrc.2007.11.056
Giulivi C., Kato K., Cooper C.E. 2006. Nitric oxide regulation of mitochondrial oxygen consumption I: Cellular physiology. Am. J. Physiol. Cell. Physiol. 291 (6), C1225–C1231. https://doi.org/10.1152/ajpcell.00307.2006
Palacios-Callender M., Hollis V., Frakich N., Mateo J., Moncada S. 2007. Cytochrome c oxidase maintains mitochondrial respiration during partial inhibition by nitric oxide. J. Cell Sci.120 (Pt 1), 160–165. https://doi.org/10.1242/jcs.03308
Kanai A., Epperly M., Pearce L., Birder L., Zeidel M., Meyers S., Greenberger J., de Groat W., Apodaca G., Peterson J. 2004. Differing roles of mitochondrial nitric oxide synthase in cardiomyocytes and urothelial cells. Am. J. Physiol. Heart Circ. Physiol.286 (1), H13–H21. Erratum in: Am. J. Physiol. Heart Circ. Physiol.296 (2), H536.
Elfering S.L., Sarkela T.M., Giulivi C. 2004. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem.277, 38079–38086.
Kanai A.J., Pearce L.L, Clemens P.R., Birder L.A., VanBibber M.M., Choi S.Y., de Groat W.C., Peterson J. 2004. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA.98, 14126–14131.
Boveris A., Valdez L.B., Zaobornyj T., Bustamante J. 2006. Mitochondrial metabolic states regulate nitric oxide and hydrogen peroxide diffusion to the cytosol. Biochim. Biophys. Acta.1757 (5–6), 535–542.https://doi.org/10.1016/j.bbabio.2006.02.010
Giulivi C. 2007. Mitochondria as generators and targets of nitric oxide. Novartis Fdn. Symp.287, 92–100.
Traaseth N., Elfering S, Solien J., Haynes V., Giulivi C. 2004. Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta. 1658 (1–2), 64–71. https://doi.org/10.1016/j.bbabio.2004.04.015
Lemasters J.J., Tom P., Theruvath I., Zhong Z., Nieminen A.-L. 2009. Mitochondrial calcium and the permeability transition in cell death, Biochim. Biophys. Acta.1787, 1395–1401. https://doi.org/10.1016/j.bbabio.2009.06.009
Halestrap A.P. 2010. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38 (4), 841–860.https://doi.org/10.1042/BST0380841
Sardanelli A.M., Technikova-Dobrova Z., Scacco S.C., Speranza F., Papa S. 1995. Characterization of proteins phosphorylated by the cAMP-dependent protein kinase of bovine heart mitochondria. FEBS Lett. 377 (3), 470–474. https://doi.org/10.1016/0014-5793(95)01407-1
Di Benedetto G., Pendin D., Greotti E., Pizzo P., Pozzan T. 2014. Ca2+ and cAMP cross-talk in mitochondria. J. Physiol.592 (2), 305–312. https://doi.org/10.1113/jphysiol.2013.259135
Ould Amer Y., Hebert-Chatelain E. 2018. Mitochondrial cAMP-PKA signaling: What do we really know? Biochim. Biophys. Acta. 1859 (9), 868–877. https://doi.org/10.1016/j.bbabio.2018.04.005
Rodríguez-Juárez F., Aguirre E., Cadenas S. 2007. Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration. Biochem. J.405, 223–231.https://doi.org/10.1042/BJ20070033
Lim S., Smith K.R., Lim S.‑T. S., Tian R., Lu J., Tan M. 2016. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci.6 (25). eCollection 2016. https://doi.org/10.1186/s13578-016-0089-3
Su C.C., Yang J.Y., Leu H.B., Chen, Y., Wang P.H. 2012. Mitochondrial Akt-regulated mitochondrial apoptosis signaling in cardiac muscle cells. Am. J. Physiol. Heart. Circ. Physiol.302 (3), H716–H723. https://doi.org/10.1152/ajpheart.00455.2011
ACKNOWLEDGMENTS
The work was supported by the Russian Foundation for Basic Research (projects nos. 14-04-01695, VD, and 14-04-01597m, EG). The authors thank M.A. Simonova, M.H. Gali-mova, and A.I. Sergeeva for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
All procedures were performed in accordance with the European Communities Council Directive (November 24, 1986; 86/609/EEC) and the Declaration on humane treatment of animals. The experimental protocol was approved by the Bioethics Committee of the Institute of Theoretical and Experimental Biophysics, RAS.
Additional information
Translated by E. Puchkov
Abbreviations: MPTP, mitochondrial permeability transition pore; GC, guanylate cyclase; mtNOS, mitochondrial NO-synthase; PKG1, protein kinase G; COX, cytochrome-c-oxidase; SNP, sodium nitroprusside; cGMP, cyclic guanosine monophosphate; ODQ, selective inhibitor of soluble guanylate cyclase (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one); 7-NI, an inhibitor of endothelial and neuronal NO-synthases (7-nitroindasole; N‑[[3-(Aminomethyl)phenyl]methyl]-ethanimidamide dihydrochloride); 1400W, selective inhibitor of inducible NO-synthase; KT, selective protein kinase G inhibitor KT5823.
Rights and permissions
About this article

Cite this article
Dynnik, V.V., Grishina, E.V. & Fedotcheva, N.I. Implication of Mitochondrial NO/cGMP/PKG Signaling System in the Activation and Inhibition of Mitochondrial Respiration by L-Arginine and NO Donors. Biochem. Moscow Suppl. Ser. A 13, 334–340 (2019). https://doi.org/10.1134/S1990747819040056
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990747819040056