
Abstract—Protein misfolding and accumulation of protein aggregates is a distinctive feature of most neurodegenerative diseases. They lead to disruption of cellular homeostasis, loss of synaptic connections, and therefore cellular apoptosis. It has been demonstrated that some innate immune responses play an important role in the emergence and progression of neurodegenerative diseases. Inflammasomes are components of innate immunity that play a major role in the maintenance of chronic inflammation. Inflammasomes function as intracellular sensors, detecting both exogenous and endogenous stimuli. They also take part in caspase-1 activation and the synthesis of pro-inflammatory cytokines. In the central nervous system (CNS), inflammasomes are predominantly expressed by microglia, the key cells of innate immunity responsible for activation and maintenance of inflammation. In addition to microglia, inflammasomes can be expressed and activated by astrocytes and neurons, as well as infiltrating myeloid cells. Understanding the mechanisms of activation and functioning of inflammasomes is crucial for the development of novel drugs targeted at modulation of the immune response associated with their excessive activation. This review provides up-to-date information on the inflammasome structure and mechanisms of action, the role of protein misfolding, aggregation and the influence of these factors on inflammasome activation, as well as potential therapeutic targets in neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
GENERAL MECHANISMS OF ACCUMULATION AND AGGREGATION OF PATHOLOGICAL PROTEINS
A necessary condition for the normal functioning of a protein is acquisition of a tertiary structure which is formed as it folds. Folding is a complex, multisystem process in which chaperone proteins play a key role, ensuring proper folding and a stable conformation. In addition to folding, other molecular processes, such as transcription, translation, post-translational modifications, degradation mediated by the ubiquitin-proteasome system, and autophagy, are also important for the correct folding of protein chains.
The acquisition of the correct conformation by a protein is ensured by a special class of proteins—chaperones. They bind to peptides even before the process of translation from mRNA is completed, and participate in the folding process, protecting growing peptide chains from the effects of particles of the cellular environment, thereby facilitating the formation of a stable conformation [1]. If protein folding occurs incorrectly, chaperone proteins correct the unfolded protein, and in case of failure, they act as signaling molecules and, depending on the specific cause, can activate various cellular programs to take radical measures to eliminate the protein with an incorrect structure, up to complete degradation [2]. These cellular programs are: unfolded protein response (UPR), heat shock response (HSR), ubiquitin-proteasome system (UPS), and endoplasmic-reticulum-associated degradation (ERAD). The UPR and HSR programs form a network of cellular proteostasis to create prohomeostatic transcriptional and posttranscriptional programs [3].
There are several hypotheses explaining the disruption of the protein folding process. First, in the case of correct translation using the putative amino acid sequences, an alternative stable conformation of the protein can be found, which will lead to folding failure. Secondly, genetic mutations also lead to disruption of protein folding and, accordingly, function; in this case, even one erroneous amino acid can lead to incorrect folding, protein aggregation, and possible cell death.
Some genes can produce multiple variants of proteins. In these cases, certain exons of the primary transcript may be included or excluded from the final synthesized mRNA. This process is called alternative splicing. Proteins resulting from translation from such mRNAs will differ both in their amino acid sequences and often in their biological functions. An error during alternative splicing can lead to the synthesis of a protein with an incorrect amino acid sequence and, ultimately, a distorted tertiary structure.
Finally, folding may be impaired due to mutations in the genes of chaperone proteins, as a result of which the predominance of alternative protein conformations is observed. At the same time, mutations in the genes responsible for the degradation of pathological proteins will help the products of impaired folding to avoid elimination, further aggregation, and the formation of fibrils. Any of the above mutations can cause cell death, however, if a cell manages to avoid apoptosis, this can lead to the development of a neurodegenerative process [4, 5].
In addition to mutations in genes, impaired protein folding can also be associated with cellular pathology, namely, mitochondrial dysfunction, calcium-induced folding disorder, and inflammation [4]. Disruption of the normal functioning of mitochondria leads to an increase in the amount of reactive oxygen species due to disruption of the processes of oxidative phosphorylation, which can in turn lead to damage to the structure of proteins [6]. Calcium-induced disruption of protein folding often accompanies pathological conditions characterized by an excess of glutamate in the synaptic cleft, leading to hyperstimulation of NMDA receptors and subsequent calcium influx. An excess of cytosolic calcium can also lead to the generation of reactive oxygen species and nitrosative stress, which disrupts the mechanisms of control of protein synthesis, thereby contributing to the accumulation of proteins with a disturbed conformation [7]. Misfolded proteins can form transmembrane pores, which further increase calcium influx, leading to a vicious cycle of cytotoxicity [8].
The main mechanism for the development of most neurodegenerative diseases is a disruption of protein homeostasis (proteostasis), leading to the formation of pathological conformation of proteins, their aggregation, accumulation, and development of neurotoxicity. Clinical manifestations of neurodegenerative diseases usually depend on the involvement of a certain population of neurons in the pathological process [9].
Diseases that develop as a result of improper folding and aggregation of proteins include Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and transmissible spongiform encephalopathies [9].
At least three mechanisms have been proposed by which protein misfolding and aggregation lead to the development of misfolding diseases. Thus, one of the key links in the pathogenesis of neurodegenerative diseases may be the loss of normal activity of a protein, the amount of which is depleted due to improper folding and aggregation. According to the second and more widely accepted hypothesis, misfolding and aggregation lead to the acquisition of neurotoxic properties by proteins with a disturbed tertiary structure, which manifest themselves in the ability of these proteins and their aggregates to activate proapoptotic signaling pathways, form ion channels, and induce oxidative stress processes. Finally, some authors explain the pathogenesis of the neurodegenerative process in terms of neuroinflammation, suggesting that abnormal protein aggregates act as antigens and cause a chronic inflammatory response that leads to cell death, probably by activating innate immunity processes, in particular, those mediated by various types of inflammasomes [10]. This review highlights the key issues of the structure and functioning of inflammasomes, as well as current data on their role in a number of neurodegenerative diseases.
THE ROLE OF INFLAMMATION IN NEURODEGENERATION
Neuroinflammation is a protective mechanism, which is primarily designed to eliminate various pathogens that disrupt homeostasis [11]. The inflammatory response that occurs under pathological conditions has beneficial effects, contributing to the removal of cellular debris and the restoration of the integrity and homeostasis of tissues, but chronicity of the inflammatory response is detrimental and, on the contrary, prevents regeneration processes [12]. The stimuli that support the inflammatory response can be either endogenous (for example, genetic mutation and protein aggregation) or come from the environment in the form of infection, traumatic injury, or toxic effects, including drugs [13].
Innate immunity is the first line of defense against not only infectious agents. Its mechanisms also play a key role in tissue repair and removal of apoptotic bodies and cellular debris. The key cells of innate immunity in the CNS are microglia and astrocytes, macrophages, natural killer (NK), and mast cells. Oligodendrocytes and neurons also contribute to the innate immune response processes in the CNS. Pathogen-associated molecular patterns (PAMP) and damage-associated (endogenous) molecular patterns (DAMP) include misfolded and aggregated proteins, such as in Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). Cellular receptors that recognize PAMP and DAMP are Toll-like receptors, C-type lectins, oxidized lipoprotein detectors, and NLR receptors, which play a key role in inflammasome assembly [14].
Microglial cells are the main resident macrophages in the CNS. In the ontogeny of the nervous system, they are involved in the formation of neural circuits, synaptogenesis, and also regulate cell death and elimination of waste products in conditions of inflammation or CNS damage. Differential activation of microglia is often referred to as classical (M1) or alternative (M2) based on in vivo expression of chemokines and cytokines [15]. Switching between these microglial phenotypes is necessary for the processes of regeneration and remyelination. Microglia synthesize both pro-inflammatory and anti-inflammatory factors that can favorably influence or, conversely, exacerbate the course of neurodegenerative diseases [16].
Similar to the M1 and M2 phenotypes of macrophages and microglia, astrocyte subpopulations are reported to produce pro-inflammatory mediators (A1) and immunoregulatory mediators (A2). A1 astrocytes secrete IL-1α, tumor necrosis factor alpha (TNFα), and complement component C1q. This phenotype is considered pro-inflammatory and promotes damage to neurons and oligodendrocytes in vitro, and also induces apoptosis by suppressing the activation and proliferation of T-helper cells. On the contrary, astrocytes of the A2 phenotype have a neuroprotective effect, promoting neuronal survival and synaptic plasticity processes. The process of astrogliosis is observed in many neurodegenerative diseases, including AD, PD, and ALS [17]. It has been suggested that reactive astrocytes of the A1 phenotype have a toxic effect in ALS, PD, AD, schizophrenia, and normal aging [18].
In addition to these cell types, oligodendrocytes also participate in innate immune responses by expressing receptors and producing immunomodulatory cytokines and chemokines. When the CNS is damaged, oligodendrocytes can promote both protective and regenerative processes and neurodegeneration due to disruption of remyelination processes [19].
The role of adaptive immunity in neurodegenerative disorders is supported by changes in T and B cell subpopulations and antibody levels in the blood, cerebrospinal fluid, and brain tissues. Thus, the role of adaptive immunity reactions in PD and ALS is actively being studied; in AD, inflammation is mainly due to resident microglia. In the early stages of Parkinson’s disease, there is an increased number of Th17 cells in the blood, some of which recognize α-synuclein [20]. In mouse models with ALS, a decrease in the number of Treg cells was accompanied by a more pronounced rate of motor neuron death and lower animal survival, while the transfer of Treg cells suppressed neuroinflammation and led to an increase in the lifespan of mice. It is reported that patients with ALS have disturbances in the functioning of Treg cells, but the direct impact of their dysfunction on the progression of neurodegeneration in humans remains unstudied [21].
STRUCTURE, ACTIVATION, AND FUNCTIONS OF INFLAMMASOME
The term “inflammasome” was introduced by J. Tschopp et al. in 2002 to describe a high molecular weight complex present in the cytosol of activated immune cells that mediates the activation of pro-inflammatory caspases [22]. Subsequently, several different inflammasomes have been identified, each of which is assembled by a unique pattern-recognition receptor (PRR) in response to pathogen-associated molecular patterns (PAMP) or endogenous stimuli in the cytosol of the host cell (damage-associated molecular patterns, DAMP) [23].
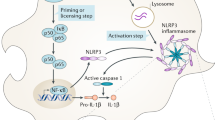
Recognition of the pro-inflammatory ligand leads to inflammasome activation, oligomerization, and recruitment of an adapter protein known as ASC (adaptor molecule apoptosis-associated speck-like protein), which consists of two domains: the pyrin domain (PYD) and the caspase activation and recruitment domain (CARD). These domains allow the adapter protein to connect the inflammasome sensor molecule to caspase-1. Autoprocessing leads to the formation of a catalytically active caspase-1, which initiates subsequent responses, including the release of interleukin-1β (IL-1β) and IL-18, and causes pyroptosis, programmed cell death mediated by gasdermin D; influx of sodium and water ions, resulting in cell swelling and membrane rupture; and spontaneous release of cytosolic contents into the extracellular space. When activated by inflammasomes, caspase-1 and other non-canonical caspases (caspase-4, caspase-5, and caspase-11) activate gasdermin D, which subsequently forms pores in the cell membrane. Due to these pores, IL-1β and IL-18 are secreted into the extracellular space, and a simultaneous influx of Na+ and water occurs, causing cell swelling and membrane rupture [24].
Inflammasomes play a key role in protecting the body from pathogens; however, their hyperactivation is associated with the development of oncological, autoimmune, metabolic, and neurodegenerative diseases.
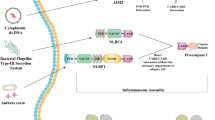
The classification of inflammasomes is based mainly on the sensory molecule, the main trigger involved in their activation. Various inflammasome sensory molecules have a similar structure and may belong to a group of pattern recognition receptors capable of responding to cytosolic pathogen-associated molecular structures (PAMP) or endogenous stimuli (DAMP). These receptors include the nucleotide-binding domain and the family of leucine-rich repeat containing receptors (NLR), inflammasomes may also include AIM2 (absent in melanoma 2-like), and pyrin receptors.
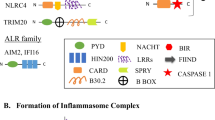
The structure of an inflammasome usually has three main components: a cytosolic pattern-recognizing receptor, caspase-1, and an adapter protein that mediates the interaction between them. As mentioned above, the receptor can either belong to the NLR family of proteins, or contain pyrin or AIM2 HIN (hematopoietic interferon-inducible nuclear protein)-domains. NLRs in humans are encoded by a family of 22 genes and contain a carboxy-terminal domain rich in leucine repeats (leucine-rich repeat, LRR), a conserved central domain NACHT (domain present in NAIP, CIITA, HET-E, TP-1), which is required for binding nucleotides and oligomerization of proteins involved in the formation of inflammasomes, and a variable amino-terminal domain which determines the NLR subfamily [25].
NLR inflammasomes can be grouped into two main subfamilies, NLRP and NLRC, depending on whether the N-terminal domain is represented by pyrin or the N-terminal caspase activation and recruitment domain (CARD) [26]. In humans, 14 NLRP and 5 NLRC genes have been identified; the most studied of which are NLPR1 and NLPR3. Both subfamilies share common features: a leucine-rich C-terminal repeat domain and a central NACHT domain responsible for oligomerization [27].
After activation and oligomerization, NLRP recruit the adapter protein ASC, which is the second component of most inflammasomes. As already mentioned, ASC consists of two domains: pyrin (PYD) and CARD. It acts as a link between the PYD of the corresponding NLRP receptor protein and the CARD of procaspase-1, which is the third component of the inflammasome. The exceptions are NLRC4 and NLRP1, since they can directly interact with procaspase 1 through their own CARD domains [28].
Given the significant role of NLRP3 inflammasomes in the mechanisms of neurodegeneration, let us consider the process of inflammasome activation using it as an example.
Activation of the NLRP3 inflammasome can be mediated in 3 ways: canonical, non-canonical, and alternative. The canonical pathway is a classic two-step model in which two signals are needed to activate the NLRP3 inflammasome. The first signal, or priming, is necessary for binding toll-like receptors (TLRs) with pathogen-associated molecular structures (PAMPs). It induces transcription of NLRP3, pro-IL-1β, and pro-IL-18 via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) [29]. The first signal not only contributes to the activation of transcription but also induces a number of post-translational modifications that allow NLRP3 to rearrange into its active conformation [30]. The second signal is triggered by various stimuli, including PAMP, DAMP, and other particles that are detected by NLRP3 through yet to be determined mechanisms. The second signal leads to the formation of an active inflammasome complex and autoproteolytic cleavage of caspase-1. A feature of the NLRP3 inflammasome is the ability to respond to a wide range of signals, such as extracellular adenosine triphosphate (ATP), microbial toxins, crystals, protein aggregates, and viral particles. The exact molecular mechanism that triggers NLRP3 activation in response to such a wide range of signals is still not fully understood. Many NLRP3 activators induce an K+ efflux from the cell, which was initially considered to be a general trigger for NLRP3 inflammasome activation [31]. There is mounting evidence that, along with K+ efflux, other mechanisms may contribute to NLRP3 activation, such as Cl– efflux, Ca2+ signaling, dysfunction of mitochondria with reactive oxygen species, and lysosome rupture [32]. Given the variety of possible activating signals, it is likely that NLRP3 responds to a common activation mechanism induced in the cytosolic environment by intracellular processes rather than directly interacting with all activator molecules [32].
Non-canonical activation is triggered by caspase-4 in humans and caspase-11 in mice in response to intracellular infection with Gram-negative bacteria (e.g., Escherichia coli) [33]. It is believed that caspase-11 and caspase-4 are activated by intracellular lipopolysaccharide (LPS) via direct binding of LPS to the CARD domain. In addition, caspase-4 and caspase-11 can also be activated by other components of gram-negative bacteria, as well as exogenous drugs, for example, methamphetamine [34]. Caspase-11 and caspase-4 activation mediated by intracellular LPS may promote K+ efflux from the cell either through gasdermin D cleavage and subsequent pyroptosis or through other currently unknown mechanisms leading to membrane instability. As a result, due to the K+ efflux from the cell, the NLRP3 inflammasome is activated [35].
Alternative inflammasome activation is a new specific pathway for NLRP3 inflammasome activation. It is present in human and porcine peripheral blood mononuclear cells but absent in mice [36]. Within this activation mechanism, the presence of lipopolysaccharide is sufficient to induce activation of the NLRP3 inflammasome, followed by activation of caspase-1 and IL-1β processing and secretion. Assembly of the inflammasome occurs after activation of the toll-like receptor 4 (TLR4) by lipopolysaccharide, which triggers the caspase-8 signaling cascade, which in turn leads to the activation of the NLRP3 inflammasome. This activation pathway does not depend on the outflow of K+ from the cell. Pyroptosis does not occur, so IL-1β is released gradually, in contrast to the all-or-nothing response characteristic of canonical activation [37].
The recruitment of procaspase-1 to the inflammasome is believed to cause autoproteolytic conversion of the proenzyme to active caspase-1. Caspase-1 activation leads to cleavage and subsequent release of IL-1β and IL-18, primarily from innate immune cells. The central nervous system is particularly sensitive to IL-1β and IL-18 signaling due to the fact that several cell types in the CNS express receptors to these cytokines [38]. Signaling cascades induced by cytokines exert their effects both at the systemic level (pathological activation of the hypothalamic-pituitary-adrenal axis) and at the local level of damage (proliferation and activation of microglia and astrocytes) [39]. Caspase-1 activation, subsequent cleavage and release of cytokines contribute to the development of immunopathological conditions that lead to neuronal death. In addition to inducing cytokine release, caspase-1 activation can mediate pyroptosis. There is evidence that caspase-1 plays a direct role in the initiation of neuron death [40].
The study of the functioning of inflammasomes during neuroinflammation processes allows us to see the theory of the role of infectious diseases in the provocation of autoimmune and degenerative diseases of the nervous system in a new light. For example, the role of NLRP3 inflammasome in the development and more severe course of acute respiratory distress syndrome, which is also observed in the novel coronavirus infection COVID-19, has been proven; thus, the ORF3a protein of coronaviruses leads to the induction of the activity of the NLRP3 inflammasome [41]. In numerous studies of the cytokine profile in the blood of patients with COVID-19 infection, an increase in the levels of IL-1ß and IL-18 was convincingly demonstrated, which can serve as another argument in favor of NLRP3 hyperactivation by inflammasomes in this disease [42]. Based on these data, some authors do not exclude the possibility of provocation or aggravation of the course of neurodegenerative diseases in patients who have undergone COVID-19 infection [43].
INFLAMMASOME ACTIVATION IN VARIOUS NEURODEGENERATIVE DISEASES. PATHOLOGICAL PROTEINS-INDUCERS OF INFLAMMASOME ACTIVATION
Most neurodegenerative diseases have common pathogenetic mechanisms, the most important of which is disruption of protein folding. A protein with a disturbed conformation is repeatedly synthesized and subjected to incorrect folding, avoiding both the mechanisms of correct folding and degradation of pathological proteins, which leads to the formation of aggregates that subsequently form fibrillar structures. When localized extracellularly, these fibrillar structures are called “amyloid fibrils”. Pathogenetic features of the neurodegenerative process depend on a number of factors: whether aggregated proteins are formed inside or outside the cells, which part of the cells produces proteins with abnormal conformation, and the area of the brain in which these aggregates are localized [4].
The data available to date indicate that, despite differences in amino acid sequences, proteins involved in neurodegenerative processes have a similar structure in aggregated forms [44]. The structural homology of proteins involved in the neurodegenerative process in the native form may be insignificant or absent, but the secondary structure of their aggregates may be similar. Most of the aggregates are rich in β-pleated sheets, while their normal functional forms are mainly α-helices and globular structures [10].
Alzheimer’s disease. Alzheimer’s disease (AD) is the most common neurodegenerative disease and the most common form of dementia. The most significant risk factor for developing AD is age, with prevalence rates doubling every 5 years after the age of 65 [45].
The classical clinical picture of the disease includes progressive amnestic and emotional-affective disorders, disorders of abstract thinking, concentration of attention, and reduced criticism of one’s own state. In the terminal stage of the disease, dementia is associated with weight loss, seizures, increased drowsiness and lack of control over the functions of the pelvic organs, and secondary infectious processes.
Structural changes in the brain in AD include diffuse atrophy of brain matter, especially the frontal lobes and hippocampus, with degeneration of cholinergic neurons. There is an increase in ventricular spaces, granulo-vacuolar degeneration, and widespread synaptic collapse. However, cerebral plaques formed by proteins with a disturbed tertiary structure are a key characteristic of AD. Hyperphosphorylated tau protein aggregates in the form of neurofibrillary tangles (NFT), while β-amyloid (Aβ) forms β-amyloid plaques. Cerebral plaque formation and atrophy begin predominantly in the hippocampus and medial temporal lobe. The accumulation of proteins with disturbed tertiary structure leads to oxidative stress and the development of an inflammatory response, which further exacerbates the progression of neurodegeneration. Aggregates of α-synuclein may also be found in patients with advanced AD, but this finding is usually secondary and is limited to the amygdala. In addition, progressive degeneration of neurons in the basal ganglia, locus coeruleus, and raphe nuclei is observed in AD, which corresponds to the loss of cholinergic, noradrenergic, and serotonergic neurons [46].
Most AD cases are associated with abnormalities in the amyloid precursor protein (APP), presenilin-1, presenilin-2, and apolipoprotein E4 (ApoE-ε4) genes [47]. For AD, the areas of the brain with predominant accumulation of pathological protein aggregates are the temporal and parietal lobes, as well as areas of the frontal cortex and cingulate gyrus [48].
It is assumed that the deposition of Aβ in the brain is a key link in the pathogenesis of AD. In the brain of patients with Alzheimer’s disease, an increase in the level of various pro-inflammatory cytokines, such as TNF-α, IFN-γ, and interleukins was noted [49]. An increase in the concentration of pro-inflammatory cytokines is noted in the blood and cerebrospinal fluid of patients with Alzheimer’s disease. It is known that interleukins, in particular, IL-1β and IL-18, are one of the causes for the development of inflammation processes in the central nervous system, which induce the expression of other pro-inflammatory genes [50]. IL-1β can be produced by many cell types, including macrophages, microglia, and neurons. Many types of inflammasomes, including NLRP1, NLRC4, and NLRP3, were shown to be involved in inflammation-mediated release of IL-1β in the CNS [51]. Aβ was the first protein with disturbed tertiary structure, for which the ability to activate inflammation in the CNS was proven. In particular, Aβ activates lipopolysaccharide-primed microglial caspase-1, which leads to the release of IL-1β, and this reaction depends on the activation of the NLRP3 inflammasome. Phagocytosis of Aβ fibrils can cause rupture of the endosome followed by the release of cathepsin B into the cytosol, which is also an important endogenous signal for the activation of the NLRP3 inflammasome [52].
A link between neuroinflammation and AD progression has been proven. Higher levels of IL-1β in the CNS may exacerbate the pathogenesis of AD and affect synaptic plasticity and long-term potentiation. Thus, inhibition of IL-1β leads to a positive effect in the form of inhibition of disease progression in mouse models with AD [53]. Activation of the NLRP3 inflammasome by Aβ in the CNS is required for cleavage of caspase-1, release of IL-1β, and development of the subsequent inflammatory response, but the ultimate role of NLRP3 activation in AD in vivo is still not completely clear. A recent study in the APP/PS1 mouse model with the clinical picture of AD showed that activation of the NLRP3 inflammasome plays a critical role in the pathogenesis of AD. Thus, APP/PS1/NLRP3 and APP/PS1/caspase-1 knockout mice showed significantly less pronounced signs of spatial memory impairment and other manifestations of AD compared with APP/PS1 mice. NLRP3 deficiency reduces caspase-1 activation and IL-1β secretion and increases Aβ clearance. In addition, deficiency of NLRP3 or caspase-1 leads to a shift in microglial activation towards the M2 phenotype, which has anti-inflammatory properties [54]. This is consistent with the results of another study showing that inhibition of the NLRP3 inflammasome by cytochalasin D reduces classical microglial activation upon exposure to Aβ [55]. Activation of the NLRP3 inflammasome was shown to induce the acquisition of a pro-inflammatory M1 phenotype by microglia and lead to clearance of Aβ; in the case of the M2 phenotype, Aβ deposition decreases and favorable conditions for synaptogenesis are created [56].
It should be noted that IL-1β cleavage is only one aspect of NLRP3 inflammasome activation. In particular, the NLRP1 inflammasome is one of the key pathways responsible for Aβ neurotoxicity. It has been shown that NLRP1 expression is upregulated in APP/PS1 mice, and this increase in NLRP1 levels in neurons is associated with Aβ accumulation. In addition, an increase in NLRP1 expression activates the caspase-1 signaling cascade and leads to neuronal pyroptosis and the release of pro-inflammatory cytokines [57]. Thus, despite the extensive data in favor of the involvement of inflammasomes in the pathogenesis of AD, the nature of the relationship between the NLRP3 inflammasome and other signaling pathways involved in the pathogenesis of AD requires clarification.
Parkinson’s disease. Parkinson’s disease (PD) is the second most common neurodegenerative disease after AD, affecting about 10 million people worldwide. PD, according to many studies, is the result of the death of dopaminergic neurons in the substantia nigra. The classic motor symptoms of PD include bradykinesia, rigidity, tremors, and postural unsteadiness. PD also has many non-motor symptoms, including dementia, which occurs in 40% of cases [58].
Two histopathological features of PD are accumulation of α-synuclein protein aggregates in Lewy bodies and loss of dopaminergic neurons in the substantia nigra [59]. α-Synuclein is a 140 amino acid cytoplasmic protein encoded by the SNCA gene. It is found in large quantities in the human brain, namely in the neurons of the neocortex, hippocampus, substantia nigra, thalamus, and cerebellum. At lower concentrations, α-synuclein is also present in glial cells. It is assumed that in presynaptic nerve terminals it interacts with SNARE proteins (soluble N-ethylmaleimide-sensitive-factor attachment receptor) and participates in the exocytosis of neurotransmitters. Cytosolic α-synuclein is initially in an unfolded state, and upon binding to membranes or vesicles acquires an α-helical structure. The interaction of α-synuclein with lipid membranes causes its conformational changes, and it specifically interacts with lipid rafts rich in cholesterol and sphingolipids [60]. In the folding disorder, α‑synuclein adopts a conformation rich in β-sheets and begins to oligomerize with other α-synuclein molecules, which can subsequently form fibrils and insoluble Lewy bodies [61].
There is a group of related neurodegenerative diseases associated with misfolding and aggregation of α‑synuclein called α-synucleinopathies. These include PD, dementia with Lewy bodies, and multiple system atrophy. Aggregates of α-synuclein were also found in brain tissue samples in AD [61].
Phosphorylation is the most common post-translational modification of α-synuclein, especially at its serine and tyrosine residues. Phosphorylation of α‑synuclein is believed to be involved in the initiation of abnormal folding of α-synuclein. Ubiquitination is the second most common process of post-translational modification and occurs at lysine residues, leading to disruption of its localization in the cell and avoidance of its degradation. Nitration is another form of post-translational modification in which tyrosine residues are targets for the attachment of nitro groups; this process leads to mitochondrial dysfunction and cell apoptosis [60].
Oxidative stress may be another factor in α-synuclein aggregation. When dopamine is oxidized, semiquinone radicals are formed from its catechol part, and the products of these processes oxidize α-synuclein on the surface of synaptic vesicles, leading to its accumulation. Moreover, α-synuclein can form transmembrane channels leading to intracellular calcium excess and excitotoxicity [62].
It is well established that the inflammatory process plays a decisive role in the pathogenesis and progression of PD. Extracellular α-synuclein was shown to be taken up by neuronal and microglial cells in culture, although the nature of this mechanism is still unclear. When α‑synuclein is released from the cell in the early stages of the disease it acts as an endogenous signal and activates microglia, which is followed by the release of pro-inflammatory molecules such as TNF-α and IL-1β, which negatively affect dopaminergic neurons [63].
There is evidence in favor of the involvement of inflammasome NLRP3 activation in α-synuclein-mediated activation of microglia. In particular, fibrillar α-synuclein induces the synthesis of IL-1β through a TLR2-dependent pathway, and its phagocytosis causes the production of reactive oxygen species and the release of cathepsin B into the cytosol, which leads to the activation of the NLRP3 inflammasome [64]. Aβ fibrillar forms of α-synuclein increase the release of monocyte and microglial IL-1β mediated by caspase-1 activation [65].
Inflammasome activation by classical stimuli directly leads to partial degradation of α-synuclein via caspase-1 cleavage. During this process, the propensity of α-synuclein to aggregation and, accordingly, neurotoxicity increases. At the same time, when caspase-1 is inhibited, the rate of neuron death decreases. The activation of inflammasomes induced by α-synuclein and inflammasome-mediated degradation and subsequent aggregation of α-synuclein can lead to a vicious circle, which ultimately causes an increase in the concentration of pro-inflammatory cytokines, an increase in the amount of aggregated α-synuclein, and promotion of neuronal death [66]. Confirmation of the neuroprotective effect of inflammasome inhibition in PD is the decrease caused by phenolic flavonoid baicalein of inflammasome activation and apoptosis in the dopaminergic system of the substantia nigra in rats [67].
Amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease caused by damage to motor neurons in the motor cortex, brainstem, and spinal cord. ALS is characterized by rapidly progressing weakness, muscle atrophy and fasciculations, spasticity, dysarthria, dysphagia, and respiratory disorders [68]. The steadily progressive clinical picture is a consequence of degeneration of the upper and lower motor neurons. In the process of disease progression, there is an increase in the severity of muscle hypotrophy, loss of motor functions; up until the late stages of the disease, the urinary tract sphincters and oculomotor muscles remain relatively intact [69]. In some patients with ALS, cognitive impairments of varying severity are noted: from minor impairments in executive functions and emotional and affective disorders to dementia in patients with the “ALS-frontotemporal dementia” phenotype [68, 69]. As in many other neurodegenerative diseases, the processes of excitotoxicity, mitochondrial dysfunction, oxidative stress, impaired energy metabolism, and neuroinflammation play key roles [70].
The protein responsible for the most common form of familial ALS is superoxide dismutase (SOD1), and the most common mutation is a mutation variant in the SOD1 gene known as A4V [69]. Superoxide dismutase is a Cu/Zn metalloenzyme that serves as an antioxidant to convert superoxide radicals into O2 and H2O2. As with the APP gene in AD, the SOD1 gene is also located on chromosome 21. Studies in mouse models show that SOD1 has proapoptotic functions, and the development of the neurodegenerative process is caused by an increase in the toxic properties of the mutant SOD1 protein, rather than loss of SOD1 function [68, 69]. However, the exact role of SOD1 in the pathogenesis of ALS is still not completely defined, although mutations in the gene lead to misfolding of the translated protein and its subsequent aggregation. SOD1 protein aggregation is a common pathological feature of both familial and sporadic forms of ALS. The mutant SOD1 protein is inherently unstable and forms cytoplasmic aggregates, which are believed to accumulate and damage mitochondria, proteasomes, chaperones, and other proteins [68, 69]. The presence of mutant SOD1 is associated with dysfunction of the excitatory amino acid transporter 2 (EAAT2), located on the presynaptic membrane and responsible for the elimination of glutamate from the synaptic cleft. A decrease in the amount of EAAT2 was also noted in autopsy tissue samples of the brain and spinal cord of patients with ALS [71]. Thus, disruption of the normal functioning of EAAT2 leads to an increase in the level of glutamate in the synaptic cleft, and, as a consequence, excitotoxicity.
Ubiquitin inclusions (UIs) are the most common type of inclusions found in the brains of nearly 100% of ALS patients. UIs are found in the motor neurons of the brainstem and spinal cord, as well as in the motor neurons of the temporal and frontal lobes of the neocortex. UIs consist of ubiquitin, peripherin, Cu/Zn superoxide dismutase, and dorphin. DNA-binding protein-43 (TDP-43), which is a nuclear protein involved in RNA processing, was also found in UIs [72]. The FUS protein and Bunina bodies are also found in the motor neurons of the brainstem and spinal cord. FUS is also a nuclear protein involved in RNA processing. Inclusions can be found in neurons of the motor cortex, predominantly consisting of intermediate filaments, including hyperphosphorylated neurofilaments and peripherin. It is believed that in addition to the above compounds, the following proteins can play a role in the pathogenesis of ALS: senataxin helicase (senataxin, SETX), a protein involved in RNA processing; alsin (ALS2), a guanine nucleotide exchange factor involved in the movement of endosomes; and dynactin, part of the dynein motor complex that binds microtubules and participates in cell transport [69]. It was shown that the mechanisms of innate immunity may be involved in the activation of microglia [73]. Activation of these innate immune cells leads to the production of pro-inflammatory neurotoxic cytokines such as IL-1β and IL-18, which further contribute to the death of motor neurons [74].
Mutations in the SOD1 gene were the first identified mutations in familial forms of ALS and to date are the most well studied [75]. The SOD1G93A mutation is used to create transgenic SOD1G93A mice; it was shown to reduce the stability of SOD1 protein folding and cause the formation of protein aggregates [69]. As mentioned above, protein aggregates in ALS patients are also characterized by the presence of the TDP-43 protein, which is believed to be translocated from the cell nucleus to the cytoplasm [76]. Mutations in the TDP-43 gene (for example, TDP-43Q331K) lead to the development of familial forms of ALS [77], and in transgenic TDP-43Q331K mice, microglial activation and degeneration of motor neurons increase [78].
Protein aggregates in ALS are powerful triggers of the microglia-mediated immune response [74]. The NLRP3 inflammasome is a key component of the innate immune system activated by protein aggregates [64]. Activation of inflammasomes and an increase in the concentration of their components are observed in patients with ALS, as well as in animal models of ALS [79], while caspase-1 and IL-1β play an important role in the pathogenesis of the disease [74]. Despite this, Johann et al. showed that microglia of SOD1G93A mice and ALS patients do not express the NLRP3 inflammasome [79]. In addition, it was demonstrated in mouse models that SOD1G93A-mediated activation of caspase-1 and production of IL-1β in microglia occur independently of the NLRP3 inflammasome [74].
It is assumed that in microglial cells, the mutant SOD1-G93A gene activates caspase-1 via the ASC-adapter protein and NLRP3, thereby leading to the subsequent release of IL-1β and provoking an inflammatory response. Free oxygen species and peroxynitrite can also contribute to the development of this signaling cascade [80].
In the study of transgenic animals with genetic ablation of caspase-1 and IL-1β, an increase in their survival rates, a decrease in the activation of astrocytes and microglia, and a decrease in the rate of death of motor neurons in the anterior horns of the spinal cord were noted. In the same in vivo model, the use of anti-IL-1 antibodies had a positive effect on animal survival rates [74].
In the study of the cytokine profile of the cerebrospinal fluid of patients with sporadic ALS, an increase in the level of total IL-18, its inhibitor IL-18-binding protein (IL-18BP), and free IL-18 was revealed, which is probably associated with the activation of inflammasomes involved in the maturation of these interleukins [81]. This hypothesis is confirmed by the increased content of NLRP3, ASC, caspase-1, and mature IL-18 in the spinal cord tissue in sporadic ALS [79].
INFLAMMASOME AS A TARGET FOR THERAPEUTIC INTERVENTION
The relationship of inflammasomes, in particular, the NLRP3 inflammasome, with many pathological processes in the CNS is of considerable interest in the context of developing effective methods for controlling their activity. Taking into account the complex signaling cascade of NLRP3 inflammasomes, a wide range of targets for their inhibition can be assumed. For example, the following strategies might be suggested:
— suppression of activating signals;
— blockage of inflammasome assembly;
— inhibition of caspase-1 activation;
— blockade of the cleavage of the pore-forming protein gasdermin D;
— neutralization of pro-inflammatory cytokines produced by the NLRP3 inflammasome;
— P2X receptor inhibition;
— inhibition of K+ outflow from the cell and the ATP-binding domain of NLRP3 [82].
Small molecules, NLRP3 inhibitors. Compounds with a sulfonylurea fragment can specifically inhibit the activation of the NLRP3 inflammasome in the activation phase without affecting the priming stage dependent on NF-κB signaling [83]. Glyburide was the first identified drug containing a sulfonylurea fragment that exhibited inhibitory activity against NLRP3 in vitro, but the dose required to achieve a therapeutic effect in vivo leads to the development of severe hypoglycemia. The small molecule compound MCC950, which is similar to sulfonylurea, was shown to block NLRP3-induced ASC oligomerization, making it a highly effective and selective inhibitor of NLRP3. This substance led to a decrease in the severity of the inflammatory response in mouse models of experimental allergic encephalomyelitis and ex vivo human samples, but its effect in other neurological pathologies has not been studied [83].
In addition, one of the intermediate substrates in glyburide synthesis, 16673-34-0, has no effect on glucose metabolism and was shown to improve reperfusion in models of myocardial ischemia by inhibiting the formation of the NLRP3 inflammasome [84].
The ketone metabolite β-hydroxybutyrate (BHB) inhibits NLRP3 inflammasome activation by inhibiting NLRP3-ASC oligomerization. It was experimentally shown that BHB reduces the K+ efflux from the cell and the reaction of the endoplasmic reticulum [85]. In addition, BHB can penetrate the blood-brain barrier into the brain parenchyma and have a neuroprotective effect in some pathological conditions [86]. The leukotriene receptor antagonist cysteinyl was found to prevent caspase-1 activation by directly inhibiting ASC oligomerization [83].
INF4E is a recently synthesized compound that directly inhibits NLRP3 ATPase and specifically inhibits inflammasome NLRP3 activation, which has also been demonstrated in mouse models of myocardial ischemia, but requires further study in the context of neurological pathology [87].
3,4-methylenedioxy-β-nitrostyrene (MNS) is a new tyrosine kinase inhibitor that specifically and effectively inhibits the NLRP3 inflammasome, directly affecting the NOD and LRR domains [88]. According to a recent study, MNS prevented wound progression and improved healing in an experimental burn model [89]. The pronounced positive effect and low cytotoxicity make MNS an attractive candidate for research in the treatment of neurological diseases; however, there are no data in the literature regarding its effect in the CNS [90].
Anti-IL-1 therapy. Currently, anti-IL-1 therapies, including interleukin-1 receptor (IL-1Ra) antagonists such as anakinra, and specific monoclonal antibodies such as canakinumab, are approved for use in patients with autoimmune diseases [91]. Administration of IL-1R antagonists reduces ischemic brain damage in a mouse model of stroke, but the long-term benefit of this therapy is questionable. The IL-1 receptor antagonist anakinra is believed to be predominantly effective in the neurological manifestations of cryopyrin-associated periodic syndrome due to good BBB penetration. Despite their effectiveness, anti-IL-1 drugs cannot effectively inhibit all processes associated with inflammasome activation. However, caspase-1 mediated pathways such as pyroptosis also contribute to disease progression; for this reason, direct blockade of inflammasome activation may be a more effective method for controlling inflammation processes than neutralization of the products of their activity [92].
Other compounds that act on specific pathways. The antimalarial drug artemisinin exerts its anti-inflammatory effects by inhibiting the NF-κB signaling pathway. In a transgenic mouse model with AD, artemisinin therapy has been shown to reduce the activity of NLRP3 inflammasomes. However, artemisinin has a number of side effects associated with its neurotoxicity, cardiotoxicity, and embryotoxicity; its long-term use may lead to the development of allergic reactions [93].
ATP-dependent receptor is also involved in the activation of the NLRP3 inflammasome [94]. The use of its antagonist brilliant blue G (BBG) reduced the severity of inflammation and neurological symptoms in rodent models of subarachnoid hemorrhage [95]. BBG can cross the BBB at relatively low blood concentrations. The use of P2X7R antagonists is controversial because these receptors are localized in various cell types and can cause undesirable effects outside the target [96].
Probenecid, a drug used to treat gout and hyperuricemia, is a specific blocker of pannexin-1 channels [97]. It is able to suppress NLRP3 activation in cultured neurons and astrocytes, providing a high extracellular K+ concentration and reducing caspase-1 expression in the rat brain [98].
Non-steroidal anti-inflammatory drugs (NSAIDs), including flufenamic and mefenamic acids, are neuroprotective in rodent models of AD. They selectively inhibit the NLRP3 inflammasome by blocking VRAC channels in macrophages [99]. These NSAIDs are aimed at both VRAC/NLRP3 and cyclooxygenase, making them more effective than drugs that inhibit only one pro-inflammatory pathway.
Post-translational modifications of NLRP3 proteins and other components of the NLRP3 inflammasome are considered important links in the process of its maturation. It can be assumed that a promising direction of research in the field of therapy of neurodegenerative diseases is the development of methods for modulating post-translational modifications in order to inhibit inflammasome activity.
CONCLUSIONS
For a long time, it was believed that the processes of neuroinflammation play a key role only in the pathogenesis of autoimmune diseases of the nervous system. However, sufficient experimental, epidemiological, genetic, and epigenetic data have been accumulated to suggest the direct involvement of innate immunity mechanisms in the development of neurodegenerative diseases. An important part of these mechanisms is the activation of inflammasomes, which are involved in the emergence, maintenance, and chronicity of the immune response. Study of the features of inflammasome activation in models of AD, PD, ALS, and other neurodegenerative diseases suggests potential therapeutic targets for effective inhibition of this immune signaling mechanism.
REFERENCES
Dobson, C.M., Semin. Cell Dev. Biol., 2004, vol. 15, no. 1, pp. 3–16.
Lee, S. and Tsai, F.V., J. Biochem. Mol. Biol., 2005, vol. 38, no. 3, pp. 259–265.
Hekmatimoghaddam, S., Zare-Khormizi, M.R., and Pourrajab, F., Biofactors, 2017, vol. 43, no. 6, pp. 737–759.
Gandhi, J., Antonelli, A.P., Afridi, A., Vatsia, S., Joshi, G., Romanov, V., Murray, I.V.J., and Khan, S.A., Rev. Neurosci., 2019, vol. 30, no. 4, pp. 339–358.
Hartl, F.U., Annu. Rev. Biochem., 2017, vol. 86, pp. 21–26.
Karbowski, M. and Neutzner, A., Acta Neuropathol., 2012, vol. 123, no. 2, pp. 157–171.
Gu, Z., Nakamura, V., and Lipton, S.A., Mol. Neurobiol., 2010, vol. 41, nos 2-3, pp. 55–72.
Torres, M., Encina, G., Soto, P., and Hetz, P., Commun. Integr. Biol., 2011, vol. 4, no. 3, pp. 258–261.
Vaquer-Alicea, J. and Diamond, M.I., Annu. Rev. Biochem., 2019, vol. 88, pp. 785—810.
Soto, P., Nat. Rev. Neurosci., 2003, vol. 4, no. 1, pp. 49–60.
Wyss-Coray, V. and Mucke, L., Neuron, 2002, vol. 35, no. 3, pp. 419–432.
Kempuraj, D., Thangavel, R., Natteru, P.A., Selvakumar, G.P., Saeed, D., Zahoor, H., Zaheer, S., Iyer, S.S., and Zaheer, A., J. Neurol. Neurosurg. Spine, 2016, vol. 1, no. 1.
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S., Immunology, 2018, vol. 154, no. 2, pp. 204–219.
Lill, P.M., Rengmark, A., Pihlstrom, L., Fogh, I., Shatunov, A., Sleiman, P.M., Wang, L.S., Liu, V., Lassen, P.F., Meissner, E., Alexopoulos, P., Calvo, A., Chio, A., Dizdar, N., Faltraco, F., Forsgren, L., Kirchheiner, J., Kurz, A., Larsen, J.P., Liebsch, M., Linder, J., Morrison, K.E., Nissbrandt, H., Otto, M., Pahnke, J., Partch, A., Restagno, G., Rujescu, D., Schnack, P., Shaw, P.E., Shaw, P.J., Tumani, H., Tysnes, O.B., Valladares, O., Silani, V., Berg, L.H., van Rheenen, W., Veldink, J.H., Lindenberger, U., Steinhagen-Thiessen, E., Teipel, S., Perneczky, R., Hakonarson, H., Hampel, H., von Arnim, P.A.F., Olsen, J.H., Van Deerlin, V.M., Al-Chalabi, A., Toft, M., Ritz, B., and Bertram, L., Alzheimers Demen., vol. 11, no. 12, pp. 1407–1416.
Sica, A. and Mantovani, A., J. Clin. Inves., vol. 122, no. 3, pp. 787–795.
Wes, P.D., Holtman, I.R., Boddeke, E.W., Moller, V., and Eggen, B.J., Glia, 2016, vol. 64, no. 2, pp. 197–213.
Liddelow, S.A. and Barres, B.A., Immunity, 2017, vol. 46, no. 6, pp. 957–967.
Vilalta, A. and Brown, G.P., FEBS J., 2018, vol. 285, no. 19, pp. 3566–3575.
Peferoen, L., Kipp, M., van der Valk, P., van Noort, J.M., and Amor, S., Immunology, 2014, vol. 141, no. 3, pp. 302–313.
Sulzer, D., Alcalay, R.N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., Liong, P., McMurtrey, P., Hildebrand, W.H., Mao, X., Dawson, V.L., Dawson, V.M., Oseroff, P., Pham, J., Sidney, J., Dillon, M.B., Carpenter, P., Weiskopf, D., Phillips, E., Mallal, S., Peters, B., Frazier, A., Lindestam Arlehamn, P.S., and Sette, A., Nature, 2017, vol. 546, no. 7660, pp. 656–661.
Beers, D.R., Zhao, W., Wang, J., Zhang, X., Wen, S., Neal, D., Thonhoff, J.R., Alsuliman, A.S., Shpall, E.J., Rezvani, K., and Appel, S.H., JCI Insigh., vol. 2, no. 5.
Martinon, F., Burns, K., and Tschopp, J., Molecular cell, 2002, vol. 10, no. 2, pp. 417–426.
Broz, P. and Dixit, V.M., Nat. Rev. Immunol., 2016, vol. 16, no. 7, pp. 407–420.
Lieberman, J., Wu, H., and Kagan, J.P., Science immunology, 2019, vol. 4, no. 39.
Walsh, J.G., Muruve, D.A., and Power, P., Nat. Rev. Neurosci., 2014, vol. 15, no. 2, pp. 84–97.
Guo, H., Callaway, J.B., and Ting, J.P., Nature medicine, 2015, vol. 21, no. 7, pp. 677–687.
Minkiewicz, J., de Rivero, Vaccari, J., and Keane, R., J. Immunol., 2012, V. 188, 1 Supplement, 54.12.
Faustin, B., Lartigue, L., Bruey, J.-M., Luciano, F., Sergienko, E., Bailly-Maitre, B., Volkmann, N., Hanein, D., Rouiller, I., and Reed, J.P., Molecular Cell, 2007, vol. 25, no. 5, pp. 713–724.
Bauernfeind, F.G., Horvath, G., Stutz, A., Alnemri, E.S., Macdonald k., Speert, D., Fernandes-Alnemri, V., Wu, J., Monks, B.G., and Fitzgerald, K.A., The Journal of Immunology, 2009, vol. 183, no. 2, pp. 787–791.
Yang, J., Liu, Z., and Xiao, V.S., Cellular & molecular immunology, 2017, vol. 14, no. 1, pp. 65–79.
Munoz-Planillo, R., Kuffa, P., Martinez-Colon, G., Smith, B.L., Rajendiran, V.M., and Nunez, G., Immunity, 2013, vol. 38, no. 6, pp. 1142–1153.
Mangan, M.S., Olhava, E.J., Roush, W.R., Seidel, H.M., Glick, G.D., and Latz, E., Nat. Rev. Drug discovery, 2018, vol. 17, no. 8, pp. 588–606.
Casson, P.N., Copenhaver, A.M., Zwack, E.E., Nguyen, H.V., Strowig, V., Javdan, B., Bradley, W.P., Fung, V.P., Flavell, R.A., and Brodsky, I.E., PLoS pathogens, 2013, vol. 9, no. 6.
Chu, L.H., Indramohan, M., Ratsimandresy, R.A., Gangopadhyay, A., Morris, E.P., Monack, D.M., Dorfleutner, A., and Stehlik, P., Nat. Communi., 2018, vol. 9, no. 1, pp. 1–16.
Schmid-Burgk, J.L., Gaidt, M.M., Schmidt, V., Ebert, V.S., Bartok, E., and Hornung, V., Eur. J. Immunol., 2015, vol. 45, no. 10, pp. 2911—2917.
Oroz, J., Barrera-Vilarmau, S., Alfonso, P., Rivas, G., and de Alba, E., Journal of Biological Chemistry, 2016, vol. 291, no. 37, pp. 19487—19501.
Gaidt, M.M., Ebert, V.S., Chauhan, D., Schmidt, V., Schmid-Burgk, J.L., Rapino, F., Robertson, A.A., Cooper, M.A., Graf, V., and Hornung, V., Immunity, 2016, vol. 44, no. 4, pp. 833—846.
Alboni, S., Cervia, D., Sugama, S., and Conti, B., Journal of neuroinflammation, 2010, vol. 7, no. 1, pp. 1—12.
John, G.R., Lee, S.P., Song, X., Rivieccio, M., and Brosnan, P.F., Glia, 2005, vol. 49, no. 2, pp. 161—176.
Bergsbaken, V., Fink, S.L., and Cookson, B.V., Nat. Rev. Microbiol., 2009, vol. 7, no. 2, pp. 99–109.
Siu, K.L., Yuen, K.S., Castano-Rodriguez, P., Ye, Z.W., Yeung, M.L., Fung, S.Y., Yuan, S., Chan, P.P., Yuen, K.Y., Enjuanes, L., and Jin, D.Y., FASEB J., 2019, vol. 33, no. 8, pp. 8865–8877.
Satış, H., Özger H.S., Aysert Yıldız, P., Hızel, K., Gulbahar, Ö., Erbaş, G., Aygencel, G., Guzel Tunccan, O., Öztürk, M.A., Dizbay, M., and Tufan, A., Cytokine, 2021, vol. 137, p. 155302.
Heneka, M.V., Golenbock, D., Latz, E., Morgan, D., and Brown, R., Alzheimer’s research & therapy, 2020, vol. 12, no. 1, pp. 1–3.
Kayed, R., Head, E., Thompson, J.L., McIntire, V.M., Milton, S.P., Cotman, P.W., and Glabe, P.G., Science, 2003, vol. 300, no. 5618, pp. 486–489.
Ziegler-Graham, K., Brookmeyer, R., Johnson, E., and Arrighi, H.M., Alzheimers Demen, 2008, vol. 4, no. 5, pp. 316–323.
DeTure, M.A. and Dickson, D.W., Mol. Neurodegener., 2019, vol. 14, no. 1, p. 32.
Lanoiselee, H.-M., Nicolas, G., Wallon, D., Rovelet-Lecrux, A., Lacour, M., Rousseau, S., Richard, A.-P., Pasquier, F., Rollin-Sillaire, A., and Martinaud, O., PLoS medicine, 2017, vol. 14, no. 3.
Wenk, G.L., Journal of Clinical Psychiatry, 2003, vol. 64, pp. 7–10.
Johnston, H., Boutin, H., and Allan, S.M., Biochem. Soc. Trans., 2011, vol. 39, no. 4, pp. 886–8890.
Rubio-Perez, J.M. and Morillas-Ruiz, J.M., Scientific World Journal, 2012, vol. 2012, p. 756357.
Trendelenburg, G., J. Cereb. Blood Flow Metab., 2008, vol. 28, no. 5, pp. 867–881.
Halle, A., Hornung, V., Petzold, G.P., Stewart, P.R., Monks, B.G., Reinheckel, V., Fitzgerald, K.A., Latz, E., Moore, K.J., and Golenbock, D.V., Nat. Immunol., 2008, vol. 9, no. 8, pp. 857–865.
Kitazawa, M., Cheng, D., Tsukamoto, M.R., Koike, M.A., Wes, P.D., Vasilevko, V., Cribbs, D.H., and LaFerla, F.M., J. Immunol., 2011, vol. 187, no. 12, pp. 6539–6549.
Heneka, M.V., Kummer, M.P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., Griep, A., Axt, D., Remus, A., Tzeng, V.P., Gelpi, E., Halle, A., Korte, M., Latz, E., and Golenbock, D.V., Nature, 2013, vol. 493, no. 7434, pp. 674–678.
Shi, F., Yang, L., Kouadir, M., Yang, Y., Wang, J., Zhou, X., Yin, X., and Zhao, D., J. Neuroinflammation, 2012, vol. 9, p. 73.
Shi, F., Yang, L., Wang, J., Kouadir, M., Yang, Y., Fu, Y., Zhou, X., Yin, X., and Zhao, D., Acta Biochim. Biophys. Sin (Shanghai), 2013, vol. 45, no. 11, pp. 973–978.
Tan, M.S., Tan, L., Jiang, V., Zhu, X.P., Wang, H.F., Jia, P.D., and Yu, J.V., Cell Death D., vol. 5, no. 8.
Shulman, J.M., De Jager, P.L., and Feany, M.B., Annu. Rev. Pathol., 2011, vol. 6, pp. 193–222.
Gibb, W.R. and Lees, A.J., J. Neurol. Neurosurg. Psychiatry, 1988, vol. 51, no. 6, pp. 745–752.
Kim, W.S., Kagedal, K., and Halliday, G.M., Alzheimers Res. Ther., 2014, vol. 6, no. 5, p. 73.
Sulzer, D., Mov. Disord., 2010, vol. 25 S.
Tsigelny, I.F., Sharikov, Y., Wrasidlo, W., Gonzalez, V., Desplats, P.A., Crews, L., Spencer, B., and Masliah, E., FEBS J., 2012, vol. 279, no. 6, pp. 1000–1013.
Litteljohn, D., Mangano, E., Clarke, M., Bobyn, J., Moloney, K., and Hayley, S., Parkinsons D., vol. 2011, no. is. 2010, p. 713517.
Codolo, G., Plotegher, N., Pozzobon, V., Brucale, M., Tessari, I., Bubacco, L., and de Bernard, M., PLoS One, 2013, vol. 8, no. 1.
Gustot, A., Gallea, J.I., Sarroukh, R., Celej, M.S., Ruysschaert, J.M., and Raussens, V., Biochem. J., 2015, vol. 471, no. 3, pp. 323–333.
Wang, W., Nguyen, L.V., Burlak, P., Chegini, F., Guo, F., Chataway, V., Ju, S., Fisher, O.S., Miller, D.W., Datta, D., Wu, F., Wu, P.X., Landeru, A., Wells, J.A., Cookson, M.R., Boxer, M.B., Thomas, P.J., Gai, W.P., Ringe, D., Petsko, G.A., and Hoang, Q.Q., Proc. Natl. Acad. Sci. U S A, 2016, vol. 113, no. 34, pp. 9587–9592.
Hung, K.P., Huang, H.J., Wang, Y.V., and Lin, A.M., J. Ethnopharmacol., 2016, vol. 194, pp. 522–529.
Kiernan, M.P., Vucic, S., Cheah, B.P., Turner, M.R., Eisen, A., Hardiman, O., Burrell, J.R., and Zoing, M.P., Lancet, vol. 377, no. 9769, pp. 942–955.
Pasinelli, P. and Brown, R.H., Nat. Rev. Neurosci., 2006, vol. 7, no. 9, pp. 710–723.
Turner, M.R., Bowser, R., Bruijn, L., Dupuis, L., Ludolph, A., McGrath, M., Manfredi, G., Maragakis, N., Miller, R.G., Pullman, S.L., Rutkove, S.B., Shaw, P.J., Shefner, J., and Fischbeck, K.H., Amyotroph. Lateral Scler. Frontotemporal Degener., 2013, vol. 14 S.
Rothstein, J.D., Van Kammen, M., Levey, A.I., Martin, L.J., and Kuncl, R.W., Ann. Neurol., 1995, vol. 38, no. 1, pp. 73–84.
Gordon, P.H., Aging D., vol. 4, no. 5, pp. 295–310.
Brites, D. and Vaz, A.R., Front Cell Neurosci., 2014, vol. 8, p. 117.
Meissner, F., Molawi, K., and Zychlinsky, A., Proc. Natl. Acad. Sci. U S A, 2010, vol. 107, no. 29, pp. 13046–13050.
Rosen, D.R., Siddique, V., Patterson, D., Figlewicz, D.A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, J.P., Deng, H.X., et al., Nature, 1993, vol. 362, no. 6415, pp. 59–62.
Barmada, S.J. and Finkbeiner, S., Rev. Neurosci., 2010, vol. 21, no. 4, pp. 251–272.
Sreedharan, J., Blair, I.P., Tripathi, V.B., Hu, X., Vance, P., Rogelj, B., Ackerley, S., Durnall, J.P., Williams, K.L., Buratti, E., Baralle, F., de Belleroche, J., Mitchell, J.D., Leigh, P.N., Al-Chalabi, A., Miller, P.P., Nicholson, G., and Shaw, P.E., Science, 2008, vol. 319, no. 5870, pp. 1668–1672.
Lee, J.D., Levin, S.P., Willis, E.F., Li, R., Woodruff, V.M., and Noakes, P.G., Journal of Neuroinflammation, 2018, vol. 15, no. 1, p. 171.
Johann, S., Heitzer, M., Kanagaratnam, M., Goswami, A., Rizo, V., Weis, J., Troost, D., and Beyer, P., Glia, 2015, vol. 63, no. 12, pp. 2260–2273.
Bellezza, I., Grottelli, S., Costanzi, E., Scarpelli, P., Pigna, E., Morozzi, G., Mezzasoma, L., Peirce, M.J., Moresi, V., Adamo, S., and Minelli, A., Mol. Neurobiol., 2018, vol. 55, no. 3, pp. 2350–2361.
Italiani, P., Carlesi, P., Giungato, P., Puxeddu, I., Borroni, B., Bossu, P., Migliorini, P., Siciliano, G., and Boraschi, D., J. Neuroinflammation, 2014, vol. 11, p. 94.
Jiang, H., He, H., Chen, Y., Huang, W., Cheng, J., Ye, J., Wang, A., Tao, J., Wang, P., Liu, Q., Jin, V., Jiang, W., Deng, X., and Zhou, R., J. Exp. Med., 2017, vol. 214, no. 11, pp. 3219—3238.
Coll, R.P., Robertson, A.A., Chae, J.J., Higgins, S.P., Munoz-Planillo, R., Inserra, M.P., Vetter, I., Dungan, L.S., Monks, B.G., Stutz, A., Croker, D.E., Butler, M.S., Haneklaus, M., Sutton, P.E., Nunez, G., Latz, E., Kastner, D.L., Mills, K.H., Masters, S.L., Schroder, K., Cooper, M.A., and O’Neill, L.A., Nat. Med., 2015, vol. 21, no. 3, pp. 248–255.
Marchetti, P., Chojnacki, J., Toldo, S., Mezzaroma, E., Tranchida, N., Rose, S.W., Federici, M., Van Tassell, B.W., Zhang, S., and Abbate, A., J. Cardiovasc. Pharmacol., 2014, vol. 63, no. 4, pp. 316–322.
Youm, Y.H., Nguyen, K.Y., Grant, R.W., Goldberg, E.L., Bodogai, M., Kim, D., D’Agostino, D., Planavsky, N., Lupfer, P., Kanneganti, V.D., Kang, S., Horvath, V.L., Fahmy, V.M., Crawford, P.A., Biragyn, A., Alnemri, E., and Dixit, V.D., Nat. Med., 2015, vol. 21, no. 3, pp. 263—269.
Song, L., Pei, L., Yao, S., Wu, Y., and Shang, Y., Front. Cell Neurosci., 2017, vol. 11, p. 63.
Mastrocola, R., Penna, P., Tullio, F., Femmino, S., Nigro, D., Chiazza, F., Serpe, L., Collotta, D., Alloatti, G., Cocco, M., Bertinaria, M., Pagliaro, P., Aragno, M., and Collino, M., Oxid. Med. Cell Longev., 2016, vol. 2016, p. 5271251.
He, Y., Varadarajan, S., Munoz-Planillo, R., Burberry, A., Nakamura, Y., and Nunez, G., J. Biol. Chem., 2014, vol. 289, no. 2, pp. 1142–1150.
Xiao, M., Li, L., Li, P., Liu, L., Yu, Y., and Ma, L., Plast. Reconstr. Surg., 2016, vol. 137, no. 3, p. 566.
Hsieh, P.-W., Chang, Y.-V., Chuang, W.Y., Shih, H.-P., Chiang, S.-Z., and Wu, P.-P., Bioorganic & medicinal chemistry, 2010, vol. 18, no. 21, pp. 7621–7627.
Kuemmerle-Deschner, J.B., Gautam, R., George, A.V., Raza, S., Lomax, K.G., and Hur, P., RMD Open, 2020, vol. 6, no. 2.
Venugopal, J., Wang, J., Mawri, J., Guo, P., and Eitzman, D., Haematologica, 2021, vol. 106, no. 9, pp. 2469–2477.
Shi, J.Q., Zhang, P.P., Sun, X.L., Cheng, X.X., Wang, J.B., Zhang, Y.D., Xu, J., and Zou, H.Q., CNS Neurosci. Ther., 2013, vol. 19, no. 4, pp. 262–268.
Deplano, S., Cook, H.V., Russell, R., Franchi, L., Schneiter, S., Bhangal, G., Unwin, R.J., Pusey, P.D., Tam, F.W., and Behmoaras, J., J. Leukoc. Biol., 2013, vol. 93, no. 1, pp. 127–134.
Zhou, X., Yang, Y., Wu, L., Wang, Y., Du, P., Li, P., Wang, Z., and Wang, Y., Med. Sci. Moni., vol. 25, pp. 6359–6366.
Fischer, W., Franke, H., Krugel, U., Muller, H., Dinkel, K., Lord, B., Letavic, M.A., Henshall, D.P., and Engel, V., PLoS One, 2016, vol. 11, no. 6.
Silverman, W.R., de Rivero, VaccariJ.P., Locovei, S., Qiu, F., Carlsson, S.K., Scemes, E., Keane, R.W., and Dahl, G., J. Biol. Chem., 2009, vol. 284, no. 27, pp. 18143–18151.
Mawhinney, L.J., de Rivero, VaccariJ.P., Dale, G.A., Keane, R.W., and Bramlett, H.M., BMC Neuroscience, 2011, vol. 12, no. 1, p. 123.
Daniels, M.J.D., Rivers-Auty, J., Schilling, V., Spencer, N.G., Watremez, W., Fasolino, V., Booth, S.J., White, P.S., Baldwin, A.G., Freeman, S., Wong, R., Latta, P., Yu, S., Jackson, J., Fischer, N., Koziel, V., Pillot, V., Bagnall, J., Allan, S.M., Paszek, P., Galea, J., Harte, M.K., Eder, P., Lawrence, P.B., and Brough, D., Nat. Communic., 2016, vol. 7, no. 1, p. 12504.
Funding
This work received no external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests. The authors declare no conflict of interest.
Additional information
Corresponding author; address: Volokolamskoe shosse 80, Moscow, 125367 Russia; e-mail: zakharova@neurology.ru.
Rights and permissions
About this article

Cite this article
Shevchuk, D.V., Abramova, A.A. & Zakharova, M.N. The Role of Inflammasomes in the Pathogenesis of Neurodegenerative Diseases. Neurochem. J. 16, 271–282 (2022). https://doi.org/10.1134/S1819712422030114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1819712422030114