
Abstract
A cooligomer has been synthesized with a yield of 93% by triple condensation of 2-allylphenol, formaldehyde, and ethylenediamine (0.5 : 4.0 : 1.0). The molecular weight and molecular weight distribution of the product were determined (Mw = 860 and Mn = 470), and it was revealed that cooligomer has sufficiently high thermal stability (a significant loss of cooligomer mass was observed at ~400°C). Thermal self-crosslinking (up to 280°C) of the cooligomer and its crosslinking with acrylonitrile in the presence of the initiator benzoyl peroxide (1%) followed by hydrolysis of the resulting polymer in the presence of KOH were carried out. Their structure was studied by IR spectroscopy. The sorption properties of the crosslinked hydrolyzed polymer were examined for the extraction of uranyl ions from model aqueous systems under batch conditions at various pH values, concentrations, and sorption times. It has been found that the maximum recovery of uranyl ions by the hydrolysis product of the crosslinked polymer at pH 7 is 90.8% and the sorption capacity is 203.5 mg/g. The dependence of the batch (“static”) sorption capacity of the crosslinked polymer on the equilibrium and initial concentration of uranyl ions was considered. It has been shown that the sorption capacity stabilizes at ~300 mg/g. The sorption properties of the crosslinked polymer were confirmed by both IR spectroscopy data and by the results of energy dispersive X-ray spectroscopy and scanning electron microscopy.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Oligomers and cooligomers prepared from phenols, aldehydes, amines, and other compounds containing various functional groups, such as phenolic hydroxyl and C=C and C≡C bonds, as well as nitrogen, phosphorus, or sulfur atoms, are the object of numerous studies, which is due to the wide possibilities of their use [1–8].
Unlike macromolecular compounds, structuring (curing) of oligomers can be carried out under “mild” conditions with little reaction heat. Shrinkage is low during the process, and the resulting crosslinked materials are characterized by solidity and thermal stability, which are important parameters that determine to a large extent their performance properties [9–15].
It should be noted that to solve many issues related to the removal of reaction heat and obtaining materials with desired properties by crosslinking of high-molecular-weight compounds, technologies are used that include the design of their structures at the synthesis stage. For example, hypercrosslinked terpolymers of styrene, vinylbenzyl chloride, and divinylbenzene (crosslinking agent) were synthesized under conditions that facilitate the regulation of pore volume, as was confirmed by Kupgan et al. [16] using molecular modeling.
Phenol–formaldehyde cooligomers remain in demand as precursors. Modified cooligomers of the phenol–formaldehyde type are of particular interest, which is largely due to their relative availability and easy carrying out of various chemical transformations either in thermal mode or in the presence of special crosslinking agents (for example, acrylonitrile and hexamethylenediamine) [17‒25]. Network products can be used as structural materials; adhesives; sorbents; and matrices for impregnating metal nanoparticles and metal sulfides, carbides, etc.
To obtain novolac resins used as raw materials for plastics, the condensation reaction of formaldehyde with a mixture of phenol, phenolphthalein, and m-cresol in the presence of an acid catalyst was studied [26]. Instead of m-cresol, bisphenol containing phthalimidine can also be used in this synthesis [27]. The materials obtained in these ways were recommended for use as plastics. Phenol–formaldehyde resins modified with Mannich bases and possessing biological activity are described in [28].
In recent years, it has been a practice to incorporate nanoparticles of metals and their oxides or sulfides into crosslinked materials. Thus, by incorporating silicon carbide and cork powder into a composite based on phenol–formaldehyde resin, a new material was developed as a matrix intended for aerospace engineering [29].
Phenol–formaldehyde resins, in particular sulfonated ones, are also used as ion exchange materials [30, 31]. Kargov et al. [32] studied the selectivity of phenol–formaldehyde resin-based ion exchangers for сesium and rubidium ions. The cited authors explain the selectivity of phenol–formaldehyde resins by the predominance of the dehydration stage on transfer from an aqueous solution to the phenolic phase. The higher selectivity of phenolic ion exchangers for rubidium and cesium ions in comparison with sulfonated polystyrene cation exchangers is explained by significantly lower moisture content in the polymer and stronger dehydration of ions in them.
Thus, interest in phenol–formaldehyde cooligomers as accessible raw materials is very high and new opportunities are opening up for targeted modeling of the structure of these cooligomers in order to carry out numerous transformations on their basis.
The purpose of this work is to perform the triple condensation of 2-allylphenol, formaldehyde, and ethylenediamine and, based on the resulting unsaturated product, to develop a sorbent material for extraction of uranyl ions from model aqueous systems. Despite the fact that numerous adsorption, precipitation, and other methods have already been proposed for the extraction of hazardous heavy metals and radioactive substances from environmental objects, this problem remains very relevant.
EXPERIMENTAL
The chemicals used for the synthesis were 2-allylphenol from Sigma-Aldrich (98%, Tbp = 220–221°C, \(d_{4}^{{20}}\) = 1.028 g/cm3, \(n_{D}^{{20}}\) = 1.578); formaldehyde (37% aqueous solution, KarmaLab); and ethylenediamine (99%, Tbp = 116°C, \(d_{4}^{{20}}\) = 0.899 g/cm3, \(n_{D}^{{20}}\) = 1.4565), acrylonitrile, and KOH purchased from Vekton. The solvents were benzene (Vekton) and acetone (analytical grade; Chemicals Base No. 1, Russia).
IR spectra of the obtained compounds were recorded on a Varian Fourier-transform IR spectrophotometer in the range of 4000–400 cm−1 at room temperature. Thermogravimetric and differential thermal analyses of the cooligomer were performed using an STA 449F3 instrument with the NETZSCH Proteus software.
Triple condensation of 2-allylphenol, formaldehyde, and ethylenediamine was carried out at a molar ratio of 0.5 : 4.0 : 1.0, respectively, according to Scheme 1.
A three-neck reaction flask equipped with a reflux condenser, a stirrer, a thermometer, and a dropping funnel was charged with 20 g (0.15 mol) of 2-allylphenol and 24 mL of formaldehyde (37% aqueous solution, 0.3 mol CH2O) with stirring, and the reaction was run at a temperature of 45‒50°C for 30‒40 min. Next, 18 g (0.3 mol) of ethylenediamine and the remaining amount of formaldehyde (48.4 mL or 0.9 mol) were introduced into the reaction mixture. The temperature was raised to 90°C while stirring, and the reaction was continued for 2 h. At the end of the reaction, the contents of the flask were dissolved in benzene and dried over Na2SO4, after which they were precipitated in heptane. The product yield was 93%.
The molecular weight and parameters of the molecular weight distribution of the cooligomer were determined by size exclusion liquid chromatography on a Kovo chromatograph (Czech Republic) with a refractometric detector. We used two 3.3 × 150 mm columns packed with the Separon-SGX stationary phase having a particle size of 7 μm and a porosity of 100 Å. The eluent was DMF used at a flow rate 0.3 mL/min and T = 20–25°C. Calibration was performed using PEG standards. Interpretation of MWD chromatograms was performed according to the method reported in [33].
Radical polymerization of cooligomer 1 (1a) with acrylonitrile was carried out as shown in Scheme 2.
A preliminarily prepared glass ampoule with a capacity of 20–30 mL was loaded with 10 g of 2-allylphenol–formaldehyde–ethylenediamine cooligomer, 5.2 g of acrylonitrile, and 2% (of the taken total amount of cooligomer and acrylonitrile) of the initiator benzoyl peroxide (BPO). The ampoule was cooled with liquid nitrogen, evacuated, sealed in a nitrogen stream and transferred to an ultrathermostat at a temperature of 80°C, where it was kept for 10 h. Upon completion of the process, the contents of the ampoule were treated with boiling acetone in a Soxhlet apparatus (to separate unreacted cooligomers). The crosslinked polymer was dried in a vacuum of 3–5 mmHg at 60–70°C. The yield of products 1b was 94%. They are insoluble and infusible substances of yellow color.
Hydrolysis of cooligomer (1b) crosslinked with acrylonitrile was carried out in the presence of a 10% sodium hydroxide aqueous solution by boiling for 10 h. The resulting product was dried under vacuum (3–4 mmHg). The yield of 1c was 84%.
The sorption properties of crosslinked polymer 1c were studied using a model system obtained by dissolving 134.5 mg/L uranyl nitrate of analytical grade (99.95%, Sigma-Aldrich, 2018) in distilled water.
To conduct the experiment, 30 mg of crosslinked polymer and a test aqueous solution containing uranyl nitrate were loaded into a 100 mL Teflon beaker. A buffer solution containing CH3COOH (99.9%, reagent grade, glacial; VitaReaktiv) and NH4OH (25%, analytical grade; Ruskhim) was introduced in an amount of 10 mL into the system; the amount was taken depending on the pH of the medium. After this, the volume of the mixture was brought to 50 mL by diluting with distilled water (EKROS-2205 (PE-2205) (A) aquadistiller). The system was kept at room temperature under batch conditions for 24 h. Upon completion of holding, the solution was filtered and the activity of the isotopes 235U and 238U (234mPa and 234Th) in the mother liquor was determined at 1500 Bq/L (Figs. 1, 2) using an HPGe γ-spectrometer (with a Canberra germanium photon detector).

Determination of uranium isotopes by HPGe spectrometry (gamma-ray spectra): (1) gamma-ray spectrum of a uranium standard, (2) gamma-ray spectrum of a test uranyl nitrate solution, and (3) background gamma-ray spectrum. Color drawings can be viewed in an electronic version.

Determination of uranium U-235 by HPGe spectrometry (gamma-ray spectra in the low energy range): (1) gamma-ray spectrum of a uranium standard, (2) gamma-ray spectrum of the test solution of uranyl nitrate, and (3) background gamma-ray spectrum.
The gamma-ray spectra of the sample were measured with a planar gamma detector, and the isotopic composition of the salts (the percentage of isotopes Uр-235, U-234, and U-238) was determined using the MGAU software (Fig. 3). The amount of radioisotope U-235 in the salt was 0.72 ± 0.05%.

Determination of the percentage of isotopes (1) Uр-235, (2) U-234, and (3) U-238 in a uranyl nitrate solution (program MGAU).
The efficiency of extraction of uranyl ions by the copolymers was judged by the decrease in the concentration of these isotopes in the solution before and after sorption, and the recovery (R) of uranyl ions and the sorption capacity (q) of the sorbent were calculated on this basis [18]:
Here R is the recovery (%), q is the sorption capacity of the sorbent (mg/g), C0 and C are respectively the initial and equilibrium concentrations of uranyl ions (mg/L), Vsorb is the volume of the sorption medium (mL), and msorb is the sorbent mass (mg).
Since the behavior of uranyl ions and the sorbent efficiency largely depend on acidity, the pH of the medium was adjusted using an ammonia solution and acetic acid (in certain proportions).
Images of polymer samples after sorption of uranyl ions were obtained with a Jeol JSM-6610LV scanning electron microscope.
RESULTS AND DISCUSSION
An IR study of the structure of the synthesized ternary cooligomer showed that at a 2-allylphenol: formaldehyde: ethylenediamine molar ratio of 0.5 : 4.0 : 1.0, cooligomer 1 is mainly formed (Scheme 1).

Scheme 1.
Cooligomer 1, IR spectrum, cm−1: 2965, 1462 (CH2); 2848, 928 (C‒H multiple bond of the allyl moiety); 1637 (C=C-allyl); 1592 (C=C-arom); 1218 (C‒O); 1138 (C‒N); 749 (trisubstituted aromatic ring); 3075 (O‒H) (Fig. 4).

IR spectra of cooligomer 1, self-crosslinked cooligomer 1a, polymer 1b based on cooligomer 1 and acrylonitrile, and its hydrolysis product 1c.
The results of analysis of the IR spectra demonstrated that despite the presence of formaldehyde taken in a sufficient amount (4.0 mol), cooligomer 1 formed has a linear structure; i.e., electrophilic substitution occurs in this case mainly in the ortho-position of 2-allylphenol, as shown by the presence of IR absorption bands of C–H bending vibrations of the trisubstituted aromatic ring (749 cm−1).
The molecular weight of cooligomer 1 was determined by the chromatographic method: Mw = 860, Mn = 470, Ð = 1.82, MW at the peak maximum \(M_{{\text{p}}}^{*}\) = 995 (Fig. 5). It can be seen that the product of 2-allylphenol condensation with formaldehyde and ethylenediamine (cooligomer 1) is characterized by a wide distribution and consists of di-, tri-, and tetramer, and fractions with a higher molecular weight of 995 can be traced at the peak maximums.

Chromatographic curve on cooligomer 1.
Figure 6 shows that in the temperature range of ~255–300°C, there is a primary slight (~19%) change in the cooligomer mass. This change may be due to the partial curing of cooligomer 1 as a result of both the subsequent cooligomer polycondensation reaction with the release of water and the cleavage of the allylic double bond (it is known that the thermal cleavage of the double bond of the allyl group occurs above 190°C) in the aforementioned temperature range. With a further change in temperature in the range of ~400–610°C, a 48% weight loss of the cooligomer is observed. From the TGA and DTA data, it follows that cooligomer 1 has sufficiently high thermal stability up to 400°C.

TGA and DTA results for cooligomer 1.
The self-crosslinking reaction was carried out at a temperature of 280°C. As can be seen from Scheme 2, resulting product 1a does not contain a multiple bond as confirmed by the IR data in Fig. 4. Thus, the IR spectrum of self-crosslinked product 1a has absorption bands intensities changed in comparison with cooligomer 1. For example, the absorption band of the allylic double bond of 1a is clearly changed in comparison with that of the allyl group of parent cooligomer 1; i.e., it is almost absent. The absence of multiple bond absorption bands in the IR spectrum indicates the occurrence of a thermal reaction with multiple bond opening in cooligomer 1.

Scheme 2.
IR spectrum of 1b, cm−1: 2936, 1464 (CH2); 1593 (C=C-arom); 754 (trisubstituted aromatic ring); 1140 (C‒N); 2239 (С≡N); 1218 (C‒O); 3597, 3617 (OH, NH) (Fig. 4). Also, in the case of crosslinking of cooligomer 1 with acrylonitrile (Scheme 2), the nitrile group band (2239 cm−1) is distinctly seen (1b). In addition, there is no absorption band due to the double bond of the allyl moiety (1637 cm−1) in this case as well, a finding that confirms the occurrence of the structuring reaction with double bond opening.
IR spectrum of 1c, cm−1: 2937, 1467, 1364 (CH2); 758 (trisubstituted aromatic ring); 1593 (C=C-arom); 1122 (C‒N); 1707 (COOH); 1211 (C‒O); 3621, 3695 (OH) (Fig. 4). It is noteworthy that the IR spectra in Fig. 4 exhibit broad bands due to the OH group. Their presence indicates the formation of an intramolecular hydrogen bond between the OH hydrogen and the amine nitrogen with the formation of a quasi-aromatic ring [6, 34].
Figure 7 presents results of a study on the effect of pH on the recovery of uranyl ions and the sorption capacity of sorbent 1c under batch conditions (temperature 25°C, initial concentration of uranyl ions in water 134.5 mg/L, holding time 24 h). It can be seen that a high recovery of uranyl ions (R) from the aqueous medium (90.8%) is achieved at pH 7. The sorption capacity is q ≈ 204 mg/g in this case.

Dependence of (1) the recovery of uranyl ions and (2) the sorption capacity of sorbent 1c on the pH of the solution.
It should be noted that the study of the sorption properties of copolymer 1b showed the recovery of uranyl ions at pH 7 to be 66.4% and q ≈ 156.8 mg/g. The relatively low sorption properties can be explained by the absence of a carboxyl group in copolymer 1b; thus, the hydrolysis of the nitrile group appears to be an important process. The sorption properties of the crosslinked polymer are also manifested due to the intramolecular hydrogen bond N…H, by which attraction or binding of metal ions from salts occurs [34]. In addition, many synthetic sorbents require high concentrations (100 mg/L or more) to trap uranyl ions, with the recovery not exceeding 90% in many cases [35]. However, the copolymers obtained in this work are active in much lower amounts (30 mg).
A method for extracting uranyl ions using interpolymer systems based on polymethacrylic acid and polyvinyl chloride was also described in [36]. It was shown that the maximum recovery of uranyl ions was 82.8% within 56 h. Meanwhile, the recovery within 24 h was ~76%, which shows the superiority of resulting sorbent 1c over many synthesized sorbents. Table 1 shows the results of studying the dependence of the recovery of \({\text{UO}}_{2}^{{2 + }}\) ions and the sorption capacity of sorbent 1b on the sorption time under batch conditions at pH 7 (temperature, 25°C; initial concentration of uranyl ions in water, 134.5 mg/L).
To analyze the kinetic curves of uranium sorption by crosslinked copolymer 1c, first-, pseudosecond-, and second-order kinetic models were used (Fig. 8) [37, 38].

Kinetics of uranium sorption by copolymer 1c: (1) diffusion, (2) chemisorption.
Below are the rate equations for second- (3) and pseudosecond-order (4) reactions:
Here k and k2 are the rate constants of second- and pseudosecond-order reactions, respectively (g/(mg min)), and qe is the amount of sorbed substance in the equilibrium state (mg/g).
Thus, as can be seen in Fig. 8, the pseudosecond-order kinetic model is more appropriate for uranium sorption.
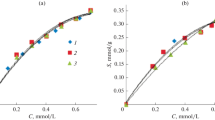
The dependence of the sorption capacity of crosslinked copolymer 1c on the initial and equilibrium concentration of uranyl ions is shown in Fig. 9. It can be seen that the sorption capacity of the sorbent is more than 200 mg/g in a solution of uranyl ion with an initial sorbent 1c concentration of 1 g/L. In this case, the equilibrium concentration of uranyl ions in the solution changes to 15 + 5 mg/L. However, when the equilibrium concentration of uranyl ions is more than 20 mg/L, the sorption capacity of 1c stabilizes at a value of ~300 mg/g and sorbent 1c exhibits maximum sorption capacity due to its saturation with uranyl ions.

Dependence of the static sorption capacity (q) of crosslinked polymer 1c on the (1) equilibrium and (2) initial concentration of uranyl ion.
After the sorption of uranyl ions by copolymer 1c, the IR spectrum shows a shift and change in the absorption bands due to COOH (1707 cm−1) and OH groups (Fig. 10). It is obvious that the process of sorption of uranyl ions by the crosslinked polymer is determined mainly by the COOH group and, additionally, by the coordination of the lone electron pair on nitrogen atoms and due to the phenolic OH group [34].

IR spectrum of copolymer 1c after sorption.
The sorption properties of sorbent 1c were also confirmed by the results of energy-dispersive X-ray spectroscopy (EDX) (Fig. 11) and scanning electron microscopy (SEM) (Fig. 12). Thus, the EDX spectrum and SEM images in Fig. 12 clearly reveal both the elemental composition of polymer 1c and sorbed uranyl ions. The mass concentration of the C atom in polymer 1c is ~62%, that of oxygen is ~23%, and the amount of uranium sorbed in 1c is on average ~9%. The data obtained is another piece of evidence that polymer 1c does indeed adsorb uranium. In addition, a uniform distribution of granules of sorbent 1c is shown after the sorption of uranyl ions, where uranium signals are clearly seen (Fig. 12d). Based on the X-ray fluorescence map of uranium (Lα, 13.8 keV), we conclude that the sorption process occurs both on the surface and in the bulk (in granules) of sorbent 1c; so, the relatively high amount of uranium in the sorbent is explained.

EDX spectrum of sorbent 1c.

SEM images of a sample of copolymer 1c after sorption: (a) sample of polymer 1c under a microscope, (b) presence of carbon in 1c, (c) presence of oxygen in 1c, and (d) presence of uranium in 1c.
Note that many known sorbents effectively extract uranyl ions mainly from concentrated solutions. The crosslinked polymers considered as a sorbent in this study are functional even in dilute solutions, which is very important from the point of view of their concentration and practical use.
To explore the possibility of regeneration of sorbent 1c, we studied the desorption of bound uranyl ions (100 mg/g) with nitric and hydrochloric acid solutions [18]. It was revealed that as the concentration of both acids increases, the desorption of uranium from the sorbent increases. Maximum desorption was achieved at a mineral acid concentration of 0.5 mol/L, being 90.3% in the case of desorption with nitric acid or 91.2% in the case of desorption with hydrochloric acid.
CONCLUSIONS
Ternary cooligomers based on 2-allylphenol, formaldehyde, and ethylenediamine with a molecular weight of up to 995 have sufficiently high thermal stability (up to ~400°C). The hydrolysis products of acrylonitrile-crosslinked copolymers based on ternary cooligomers of 2-allylphenol, formaldehyde, and ethylenediamine exhibit sorption ability towards uranyl ions.
It was found that the hydrolysis product of the crosslinked copolymer has the highest sorption activity at pH 7 and 24 h (recovery ~90.8%, q ~ 203.5 mg/g). The sorption properties of the copolymer were also confirmed by IR, EDX and SEM data.
REFERENCES
Y. Cui, X. Hou, W. Wang, and J. Chang, Materials (Basel) 10, 668 (2017).
K. N. Kornilov, Phosph. Sulfur, Silicon Relat. Elem. 192, 896 (2017).
E. V. Kolyakina and D. F. Grishin, Russ. Chem. Rev. 80, 683 (2011).
M. R. Bayramov, A. M. Magerramov, G. M. Mehdiyeva, Sh. J. Kuliyeva, and M. A. Agayeva, Processes Petrochem. Oil Refin. (PPOR), No. 4, 476 (2021).
A. M. Maharramov, M. R. Bayramov, M. A. Agayeva, G. M. Mehdiyeva, and I. G. Mamedov, Russ. Chem. Rev. 84, 1258 (2015).
A. M. Magerramov, M. R. Bairamov, G. M. Mehdiyeva, and M. A. Agaeva, Polym. Sci., Ser. B 54, 399 (2012).
M. R. Bairamov, N. Yu. Zeinalov, G. M. Mekhtieva, M. A. Agaeva, Sh. Dzh. Kulieva, M. A. Dzhavadov, and G. M. Gasanova, Neftepererab. Neftekhim., No. 11, 38 (2021).
A. M. Magerramov and M. R. Bairamov, Chemistry of Alkenylphenols (Tekhnosfera, Moscow, 2018) [in Russian].
F. Kalinina, D. Mognonov, L. R. Radnaeva, and V. Vasnev, Polym. Sci., Ser. A 44, 401 (2002).
T. R. Deberdeev, A. I. Akhmetshina, L. K. Karimova, E. K. Ignat’eva, R. Ya. Deberdeev, and A. A. Berlin, Polym. Sci., Ser. C 62, 145 (2020).
S. G. Dmitrienko, V. V. Irkha, T. B. Duisebaeva, Yu. V. Mikhailik, and Yu. A. Zolotov, J. Anal. Chem. 61, 14 (2006).
V. Z. Masloi, O. V. Masloi, N. N. Taranenko, and E. B. Loshkova, Vostoch.-Evrop. Zh. Peredovykh Tekhnol. 65, 32 (2013).
R Mehta and M. D. Dadmun, Macromolecules 39, 8799 (2006).
C.-Y. Yin and W.-C. Chen, Polymer 47, 3436 (2006).
A. L. Buchachenko, Russ. Chem. Rev. 72, 375 (2003).
G. Kupgan, Th. P. Liyana-Arachchi, and C. M. Colina, Polymer 99, 173 (2016).
G. Foyer, B.-H. Chanfi, B. Boutevin, S. Caillol, and G. David, Eur. Polym. J. 74, 296 (2016).
G. M. Mehdiyeva, M. R. Bairamov, Dzh. A. Nagiev, M. A. Agaeva, and Sh. Dzh. Kulieva, Russ. J. Phys. Chem. A 95, 769 (2021).
M. R. Bairamov, Sh. J. Guliyeva, G. M. Mehdiyeva, and M. A. Agayeva, Azerb. Chem. J., No. 1, 155 (2023).
M. R. Bayramov, A. M. Maharramov, G. M. Mehdiyeva, Sh. J. Guliyeva, and M. A. Aghayeva, Chem. Probl. 21, 85 (2023).
M. R. Bairamov, G. M. Mekhtieva, Dzh. A. Nagiev, Sh. Dzh. Gulieva, M. A. Agaeva, and A. M. Magerramov, Zh. Prikl. Khim. 96, 382 (2023).
A. M. Maharramov, M. R. Bayramov, Sh. J. Guliyeva, G. M. Mehdiyeva, N. M. Sadikhov, M. A. Agayeva, and B. A. Babayeva, Azerb. J. Chem. News 5, 59 (2023).
M. R. Bayramov, A. M. Magerramov, G. M. Mehdiyeva, Sh. J. Guliyeva, and M. A. Agayeva, Processes Petrochem. Oil Refin. (PPOR) 23, 198 (2022).
Y. Shi, L.-f. Wang, Y. Han, and C.-Y. Liao, China Foundry 13, 205 (2016).
M. N. Amiraslanova, N. R. Abdullaeva, L. I. Alieva, R. A. Rustamov, S. F. Akhmedbekova, E. I. Azizbeili, Sh. R. Alieva, F. A. Mamedzade, and A. P. Alieva, Plast. Massy, Nos. 5–6, 16 (2019).
L. N. Machulenko, A. I. Nechaev, S. A. Donetskaya, and S. N. Salazkin, Plast. Massy, Nos. 9–10, 36 (2015).
L. N. Machulenko, S. A. Donetskaya, and A. R. Potapova, Plast. Massy, Nos. 3–4, 28 (2016).
Sh. Yang, J.-Q. Wu, Y. Zhang, T.-Q. Yuan, and R.-C. Sun, J. Biobased Mater. Bioenergy 9, 266 (2015).
G. Pelin, C. E. Pelin, A. Stefan, Y. Dincay, E. Andronescu, O. Oprea, D. Ficai, and R. Trusca, Bull. Mater. Sci. 41, 281 (2018).
D. Ragavan, A. Girija, B. Kathereen, and R. K. Seenivasan, Int. J. Innovative Res. Dev. 3, 17 (2014).
I. Elodie Melro, E. A. Filipe, J. M. Artur, I. Valente, H. Duarte, A. Romano, and B. Medronho, Molecules 27, 2825 (2022).
S. I. Kargov, L. A. Shelkovnikova, and V. A. Ivanov, Russ. J. Phys. Chem. A 86, 860 (2012).
N. R. Bektashi, A. M. Mustafaev, and I. A. Huseynov, Russ. J. Appl. Chem. 84, 1211 (2011).
S. Akay, B. Kayan, D. Kalderis, M. Arslan, Y. Yagci, and B. Kiskan, J. Appl. Polym. Sci. 134, 38 (2017).
S. M. Badawy, Rad. Phys. Chem. 66, 67 (2003).
T. K. Dzhumadilov, A. A. Utesheva, R. G. Kondaurov, and Yu. V. Grazhulyavichyus, Khim. Zh. Kaz. 1, 176 (2021).
Y. S. Ho and G. McKay, Water Res. 33, 578 (1999).
Y. S. Ho, D. A. J. Wase, and C. F. Forster, Environ. Technol 17, 71 (1996).
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest.
Additional information
Translated by S. Zatonsky
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mehdiyeva, G.M., Bayramov, M.R. & Nagiev, J.A. Synthesis of Cooligomer Based on 2-Allylphenol, Formaldehyde, and Ethylenediamine and Study of Its Structured Product as a Sorbent for Extraction of Uranyl Ions from Aqueous Systems. Polym. Sci. Ser. B (2024). https://doi.org/10.1134/S1560090424600864
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S1560090424600864