
Abstract
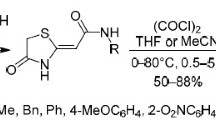
(Z)-3-(2-Aryl-2-oxoethylidene)morpholin-2-ones were synthesized by the reaction of aroylpyruvic acids with ethanolamine or 2-propanolamine. The products reacted with oxalyl chloride to form 8-aroyl-3,4-dihydropyrrolo[2,1-c][1,4]oxazin-1,6,7(1H)-triones.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Heterocyclic enaminoketones react with oxalyl chloride to form hetareno[e]pyrrole-2,3-diones [1–13] or 4-heterylfuran-2,3-diones [14] or their mixture [15]. To obtain more evidence which would make it possible to predict what of the two mentioned directions would be realized, we synthesized substituted 3-methylenemorpholin-2-ones, representatives of a new class of heterocyclic enaminoketones, and studied their reaction with oxalyl chloride. The structure of the substituted morpholinones seems to be borderline for the realization of one of the alternative directions of the reaction with oxalyl chloride
Heating aroylpyruvic acids 1a–1f with ethanolamine 2a or 2-propanolamine 2b in the presence of acetic acid in a 1 : 1 : 1 ratio in toluene with a Dean–Stark trap for 4–8 h (until water no longer evolved) gave (Z)-3-(2-aryl-2-oxoethylidene)morpholin-2-ones 3a–3k (Scheme 1). The structure of compounds 3a, 3e, and 3i was confirmed by X-ray diffraction (XRD) analysis. Compounds 3a–3c were described earlier [16], which compounds 3d–3k were synthesized for the first time. Under the conditions of synthesis of compounds 3a–3c described in [16], specifically heating under reflux in 1,4-dioxane for 1–1.5 h) [16], we faced strong tarring of the reaction mixture and lower yields of products.
Compounds 3a–3k are high-melting light yellow crystalline substances soluble in DMSO, DMF, acetone, ethyl acetate, chloroform, and 1,4-dioxane, sparingly soluble in aromatic hydrocarbons, and insoluble in alkanes and water.
The IR spectra of compounds 3a–3k contain absorption bands of the NH bond (3198–3247 cm–1, broad), lactam C1=O group (1730–1744 cm–1), and C=O group of the aroyl fragment (1615–1623 cm–1).
The 1H NMR spectra of compounds 3a–3k display, along with proton signals of the methylene groups of the morpholine ring and the aromatic rings and their substituents, a singlet of the methine proton (6.50–6.56 ppm) and a singlet of the NH proton (10.48–10.66 ppm).
The 13C NMR spectra of compounds 3a–3k show characteristic signals of the ketone carbonyl of the aroyl fragment (186.7–189.1 ppm) and lactam C1=O group (160.1–160.8 ppm).
The reaction of compounds 3a–3k with oxalyl chloride under the conditions usual for the synthesis of five-membered dioxaheterocycles (heating in anhydrous chloroform under reflux for 1–1.5 h) [17] afforded, instead of the expected 4-(2-oxo-5,6-dihydro-1,4-oxazin-3-yl)-5-arylfuran-2,3(2H)-diones 4, 8-aroyl-3,4-dihydropyrrolo[2,1-c][1,4]oxazine-1,6,7(1H)-triones 5a–5hFootnote 1 (Scheme 1).

1.
Compounds 5a–5h are high-melting red crystalline substances melting with decomposition, soluble in DMSO, DMF, acetone, acetonitrile, and 1,4-dioxane, sparingly soluble in aromatic hydrocarbons, ethyl acetate, and chloroform, and insoluble in alkanes and water.
The IR spectra of compounds 5a–5h display stretching absorption bands of the lactam C1=O and C6=O groups (1749–1761 cm–1), ketone C6=O group (1716–1732 cm–1), and the aroyl C=O group (1646–1673 cm–1).
The 1H NMR spectra of compounds 5a–5d display, along with the proton signals of the aromatic rings and their substituents, signals of the 4-CH2 (ppm) and 3-CH2 groups of the oxazine ring at 3.82–3.83 and 4.71–4.72 ppm, respectively.
The 13C NMR spectra of compounds 5a–5h show signals of the ketone carbonyls of the aroyl fragment (186.6–187.8 ppm) and the C7=O group (180.8–181.5 ppm), lactam C6=O (156.8–157.0 ppm) and C1=O groups (154.8–155.0 ppm), as well as C8a atom (138.8–148.5 ppm).
Apparently, the reaction of compounds 3 with oxalyl chloride occurs via closure of a pyrroledione ring because it is thermodynamically more stable that the alternative furandione ring. This reaction provides a new synthetic approach to the functionalized pyrrolo[2,1-c]oxazine-1,6,7-trione system.
In view of the poor understanding of the fine structure of 3-(2-aryl-2-oxoethylidene)morpholin-2-ones 3, we performed an XRD analysis of these compounds.
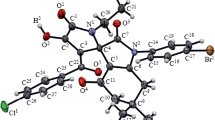
Compounds 3a, 3e, and 3i crystallize in centrosymmetricspace groups of the monoclinic or triclinic (compound 3i) crystal systems (Figs. 1–3). The C4C3C5 fragment of compound 3i is disordered over two positions [minor component occupancy 0.22(3)] because the two enantiomers are present in the same position (in Fig. 3, the atoms of the minor component are not shown).

General view of a molecule of (Z)-3-(2-oxo-2-phenylethylidene) morpholin-2-one (3e) by the XRD data (represented by 50% probability thermal ellipsoids).

General view of a molecule of (Z)-3-(2-(4-methoxyphenyl)-2-oxoethylidene)-6-methylmorpholin-2-one (3e) by the XRD data (represented by 50% probability thermal ellipsoids).

General view of a molecule of (Z)-3-(2-(4-bromo-phenyl)-2-oxoethylidene)-6-methylmorpholin-2-one (3i) by the XRD data (represented by 50% probability thermal ellipsoids).
The molecules of all the three compounds have in general similar geometries. The oxazine rings have a distorted boat conformation, with the C3 and O1 (3a) and C5 and O3 atoms (3e) deviating by 0.81 and 0.29 Å and the C3 and O2 atoms (3i), by 0.86 and 0.29 Å from the planes formed by the other ring planes. In all the compounds, the planar enaminoketone fragment formed by six-membered chelate rings by the intramolecular hydrogen bonds N1–H1∙∙∙O3 (3a) and N1–H1∙∙∙O1 (3e and 3i). Therewith, the amino groups, too, are always involved in the formation of intramolecular hydrogen bonds (IHB). Thus, the molecules of compounds 3a and 3i in the crystals form cetrosymmetric fimers due to the IHBs N1–H1∙∙∙O3 [1–x, 1–y, 1–z] (3a) and N1–H1∙∙∙O1 [1–x, 1–y, 1–z] (3i). The molecules of compound 3e in the crystal form infinite chains by the N1–H1∙∙∙O1 IHBs [x, y–1, z].
EXPERIMENTAL
The 1H and 13C NMR spectra were measured on a Bruker Avance III HD 400 spectrometer (400 and 100 MHz, respectively), internal reference HMDS. The IR spectra were recorded on a Perkin Elmer Spectrum Two spectrometer in mineral oil. The elemental analyses were obtained on a vario Micro cube analyzer. The purity of the synthesized compounds was confirmed by TLC on Merck Silica gel 60 F254 plates, eluents toluene, ethyl acetate, and toluene–ethyl acetate (5 : 1), vizualization by exposure to UV light (λmax 254 nm).
The XRD analysis of compounds 3a, 3e, and 3i was performed on an Xcalibur Ruby single-crystal diffractometer with a CCD detector by a standard procedure (MoKα-radiation, 295(2) K, ω scans, step 1°). Empirical absorption corrections were applied using SCALE3 ABSPACK algorithm [20]. The structures were decoded using SHELXS program [21] and refined by full-matrix least-squares on F2 with anisotropic thermal factors for nonhydrogen atoms using SHELXL program [22] with OLEX2 graphic interface [23]. Hydrogen atoms (except for the OH and NH hydrogen refined independently with isotropic thermal factors) were included riding on their carrier atoms.
(Z)-3-(2-Oxo-2-phenylethylidene)morpholin-2-one (3a). Acetic acid, 7.44 mL (130.1 mmol), and 7.87 mL (130.1 mmol) of monoethanolamine were added to a solution of 25.00 g (130.1 mmol) of benzoylpyruvic acid in 300 mL of toluene. The mixture was refluxed with Dean–Stark trap for 6 h (until water no longer evolved), the solvent was removed, and the residue was recrystallized from ethyl acetate. Yield 89%, mp 128–130°C (EtOAc), 127–128°C [16]. IR spectrum, ν, cm–1: 3236 br (NH), 1731 (C1=O), 1616 (COPh). 1H NMR spectrum, δ, ppm: 3.61 d.d (2H, C4H2, J 8.8, 5.0 Hz), 4.58 d.d (2H, C3H2, J 5.6, 4.8 Hz), 6.55 s (1H, CH=), 7.46–7.56 m (3Harom), 7.90 d (2Harom, J 6.9 Hz), 10.64 s (1H, NH). 13C NMR spectrum , δ, ppm: 38.2, 67.3, 91.8, 126.9, 128.6, 131.7 138.9, 146.2, 160.5 (C1=O), 189.1 (COAr). Found, %: C 66.09; H 5.29; N 6.66. C12H11NO3. Calculated, %: C 66.35; H 5.10; N 6.45; O 22.10.
X-ray diffraction study of compound 3a. Monoclinic crystal system, space group P21/n, C12H11NO3, M 217.22, a 6.7074(19), b 10.683(3), c 14.832(8) Å, β 92.75(3)°, V 1061.6(7) Å3, Z 4, dcalc 1.359 g/cm3; μ 0.099 mm–1. Final divergence factors: R1 0.0522 [for 1686 reflections with I > 2σ(I)], wR2 0.1543 (for all 2512 unique reflections), S 1.050.
Compounds 3b–3k were prepared similarly to compound 3a.
(Z)-3-[2-(4-Chlorophenyl)-2-oxoethylidene]morpholin-2-one (3b). Yield 87%, mp 152–154°C (EtOAc), 153–154°C [16]. IR spectrum, ν, cm–1: 3209 br (NH), 1731 (C1=O), 1612 (COAr). 1H NMR spectrum, δ, ppm: 3.62 d.d (2H, C4H2, J 8.6, 4.7 Hz), 4.58 t (2H, C3H2, J 5.1 Hz), 6.51 s (1H, CH=), 7.52 d (2Harom, J 8.6 Hz), 7.90 d (2Harom, J 8.5 Hz), 10.66 s (1H, NH). 13C NMR spectrum, δ, ppm: 38.2, 67.2, 91.5, 128.7, 128.8, 136.6 137.5, 146.6, 160.4 (C1=O), 187.6 (COAr). Found, %: C 57.47; H 4.25; N 5.30. C12H10ClNO3. Calculated, %: C 57.27; H 4.01; Cl 14.09; N 5.57; O 19.07.
(Z)-3-[2-(4-Bromophenyl)-2-oxoethylidene]morpholin-2-one (3c). Yield 88%, mp 156–158°C (EtOAc), 157–158°C [16]. IR spectrum, ν, cm–1: 3242 br (NH), 1737 (C1=O), 1620 (COAr). 1H NMR spectrum, δ, ppm: 3.61 d.d (2H, C4H2, J 9.0, 4.9 Hz), 4.57 d.d (2H, C3H2, J 5.6, 4.8 Hz), 6.50 s (1H, CH=), 7.68 d (2Harom, J 8.7 Hz), 7.83 d (2Harom, J 8.8 Hz), 10.59 s (1H, NH). 13C NMR spectrum, δ, ppm: 38.2, 67.2, 91.4, 125.5, 129.0, 131.6, 137.9, 146.4, 160.4 (C1=O), 187.7 (COAr). Found, %: C 48.99; H 3.58; N 4.40. C12H10BrNO3. Calculated, %: C 48.67; H 3.40; Br 26.98; N 4.73; O 16.21.
(Z)-3-[2-(4-Methylphenyl)-2-oxoethylidene]morpholin-2-one (3d). Yield 79%, mp 166–168°C (EtOAc). IR spectrum, ν, cm–1: 3230 br (NH), 1739 (C1=O), 1617 (COAr). 1H NMR spectrum, δ, ppm: 2.35 s (3H, CH3), 3.60 d.d (2H, C4H2, J 8.8, 4.9 Hz), 4.57 d.d (2H, C3H2, J 5.6, 4.7 Hz), 6.54 s (1H, CH=), 7.28 d (2Harom, J 7.6 Hz), 7.80 d (2Harom, J 8.2 Hz), 10.59 s (1H, NH). 13C NMR spectrum, δ, ppm: 21.0, 38.2, 67.3, 91.8, 127.0, 129.2, 136.3, 141.8, 146.0, 160.6 (C1=O), 188.9 (COAr). Found, %: C 67.73; H 5.86; N 5.85. C13H13NO3. Calculated, %: C 67.52; H 5.67; N 6.06; O 20.76.
(Z)-3-[2-(4-Methoxyphenyl)-2-oxoethylidene]morpholin-2-one (3e). Yield 75%, mp 175–177°C (EtOAc). IR spectrum, ν, cm–1: 3243 br (NH), 1744 (C1=O), 1620 (COAr). 1H NMR spectrum, δ, ppm: 3.59 d.d (2H, C4H2, J 8.9, 4.9 Hz), 3.82 s (3H, OCH3), 4.56 d.d (2H, C3H2, J 5.7, 4.9 Hz), 6.53 s (1H, CH=), 7.00 d (2Harom, J 8.9 Hz), 7.88 d (2Harom, J 8.9 Hz), 10.53 s (1H, NH). 13C NMR spectrum, δ, ppm: 38.1, 55.3, 67.2, 91.7, 113.8, 128.9, 131.6, 145.6, 160.6 (C1=O), 162.1, 188.1 (COAr). Found, %: C 63.53; H 4.98; N 5.88. C13H13NO4. Calculated, %: C 63.15; H 5.30; N 5.67; O 25.88.
X-ray diffraction study of compound 3e. Monoclinic crystal system, space group P21/n, C13H13NO4, M 247.24, a 14.043(6), b 6.4550(13), c 14.316(5) Å, β 113.73(4)°, V 1188.0(7) Å3, Z 4, dcalc 1.382 g/cm3; μ 0.103 mm–1. Final divergence factors: R1 0.0540 [for 1946 reflections with I > 2σ(I)], wR2 0.1597 (for all 2936 unique reflections), S 1.049.
(Z)-3-[2-(4-Nitrophenyl)-2-oxoethylidene]morpholin-2-one (3f). Yield 35%, mp 267–269°C (EtOAc). IR spectrum, ν, cm–1: 3202 br (NH), 3111, 3074 (NO2), 1730 (C1=O), 1623 (COAr). 1H NMR spectrum, δ, ppm: 3.65 d.d (2H, C4H2, J 8.3, 5.4 Hz), 4.60 d.d (2H, C3H2, J 5.8, 4.7 Hz), 6.56 s (1H, CH=), 8.13 d (2Harom, J 8.9 Hz), 8.31 d (2Harom, J 8.9 Hz), 10.53 s (1H, NH). 13C NMR spectrum, δ, ppm: 38.4, 67.1, 91.7, 123.8, 128.3, 144.0, 147.4, 149.1, 160.1 (C1=O), 186.7 (COAr). Found, %: C 55.46; H 3.98; N 10.36. C12H10N2O5. Calculated, %: C 54.97; H 3.84; N 10.68; O 30.51.
(Z)-6-Methyl-3-(2-oxo-2-phenylethylidene)morpholin-2-one (3g). Yield 83%, mp 136–138°C (EtOAc). IR spectrum, ν, cm–1: 3223 br (NH), 1737 (C1=O), 1621 (COPh). 1H NMR spectrum, δ, ppm: 1.35 d (3H, CH3, J 6.3 Hz), 3.33–3.39 m, 3.62–3.67 m, 4.81–4.89 m (3H, NCH2CHO), 6.55 s (1H, CH=), 7.49 t (2Harom, J 7.3 Hz), 7.55 t (1Harom, J 7.2 Hz), 7.90 d (2Harom, J 6.9 Hz), 10.60 s (1H, NH). 13C NMR spectrum, δ, ppm: 17.5, 43.5, 74.6, 91.6, 126.8, 128.5, 131.6, 138.8, 145.5, 160.6 (C1=O), 189.0 (COAr). Found, %: C 67.90; H 5.25; N 6.34. C13H13NO3. Calculated, %: C 67.52; H 5.67; N 6.06; O 20.76.
(Z)-3-[2-(4-Chlorophenyl)-2-oxoethylidene]-6-methylmorpholin-2-one (3h). Yield 84%, mp 156–158°C (EtOAc). IR spectrum, ν, cm–1: 3247 br (NH), 1737 (C1=O), 1615 (COAr). 1H NMR spectrum, δ, ppm: 1.35 d (3H, CH3, J 6.4 Hz), 3.33–3.40 m, 3.62–3.68 m, 4.81–4.89 m (3H, NCH2CHO), 6.51 s (1H, CH=), 7.53 d (2Harom, J 8.6 Hz), 7.91 d (2Harom, J 8.6 Hz), 10.61 s (1H, NH). 13C NMR spectrum, δ, ppm: 17.6, 43.6, 74.7, 91.4, 128.7, 128.8, 136.6, 137.5, 146.0, 160.5 (C1=O), 187.6 (COAr). Found, %: C 58.98; H 4.34; N 5.56. C13H12ClNO3. Calculated, %: C 58.77; H 4.55; Cl 13.34; N 5.27; O 18.06.
(Z)-3-[2-(4-Bromophenyl)-2-oxoethylidene]-6-methylmorpholin-2-one (3i). Yield 87%, mp 170–172°C (EtOAc). IR spectrum, ν, cm–1: 3232 br (NH), 1733 (C1=O), 1615 (COAr). 1H NMR spectrum, δ, ppm: 1.35 d (3H, CH3, J 6.4 Hz), 3.33–3.39 m, 3.62–3.68 m, 4.81–4.89 m (3H, NCH2CHO), 6.51 s (1H, CH=), 7.68 d (2Harom, J 8.7 Hz), 7.84 d (2Harom, J 8.8 Hz), 10.62 s (1H, NH). 13C NMR spectrum, δ, ppm: 17.5, 43.5, 74.6, 91.2, 125.4, 128.9, 131.5, 137.8, 145.9, 160.4 (C1=O), 187.6 (COAr). Found, %: C 50.73; H 3.61; N 4.71. C13H12BrNO3. Calculated, %: C 50.34; H 3.90; Br 25.76; N 4.52; O 15.48.
X-ray diffraction study of compound 3i. Triclinic crystal system, space group P–1, C13H12BrNO3, M 310.15, a 6.9751(18), b 8.513(2), c 11.587(2) Å, α 79.891(19), β 73.58(2), γ 84.83(2)°, V 649.1(3) Å3, Z 2, dcalc 1.587 g/cm3, μ 3.166 mm–1. Final divergence factors: R1 0.0687 [for 1703 reflections with I > 2σ(I)], wR2 0.1851 (for all 2997 unique reflections), S 1.037.
The results of the XRD analysis were deposited in the Cambridge Crystallographic Data Center under CCDC 1988255 (3a), 1988256 (3e), and 1988257 (3i) and are available by request at www.ccdc.cam.ac.uk/data_request/cif.
(Z)-6-Methyl-3-[2-(4-methylphenyl)-2-oxoethylidene]morpholin-2-one (3j). Yield 85%, mp 156–158°C (EtOAc). IR spectrum, ν, cm–1: 3198 br (NH), 1737 (C1=O), 1622 (COAr). 1H NMR spectrum, δ, ppm: 1.34 d (3H, CH3, J 6.4 Hz), 2.35 s (3H, CH3), 3.31–3.37 m, 3.60–3.66 m, 4.79–4.87 m (3H, NCH2CHO), 6.54 s (1H, CH=), 7.28 d (2Harom, J 8.6 Hz), 7.80 d (2Harom, J 8.1 Hz), 10.55 s (1H, NH). 13C NMR spectrum, δ, ppm: 17.5, 20.9, 43.5, 74.6, 91.6, 126.9, 129.0, 136.2, 141.7, 145.2, 160.6 (C1=O), 188.8 (COAr). Found, %: C 68.88; H 5.95; N 5.98. C14H15NO3. Calculated, %: C 68.56; H 6.16; N 5.71; O 19.57.
(Z)-3-[2-(4-Methoxyphenyl)-2-oxoethylidene]-6-methylmorpholin-2-one (3k). Yield 78%, mp 148–150°C (EtOAc). IR spectrum, ν, cm–1: 3228 br (NH), 1731 (C1=O), 1617 (COAr). 1H NMR spectrum, δ, ppm: 1.34 d (3H, CH3, J 6.4 Hz), 3.30–3.36 m, 3.59–3.65 m, 4.79–4.87 m (3H, NCH2CHO), 3.82 s (3H, OCH3), 6.52 s (1H, CH=), 7.01 d (2Harom, J 8.9 Hz), 7.88 d (2Harom, J 8.9 Hz), 10.48 s (1H, NH). 13C NMR spectrum, δ, ppm: 17.5, 43.5, 55.3, 74.6, 91.5, 113.8, 128.9, 131.5, 145.0, 160.8(C1=O), 162.1, 188.1 (COAr). Found, %: C 64.13; H 5.90; N 5.67. C14H15NO4. Calculated, %: C 64.36; H 5.79; N 5.36; O 24.49.
8-Benzoyl-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5a). A solution of 1.10 mL (12.7 mmol) oxalyl chloride in 5 mL of anhydrous chloroform was added in portions to a stirred solution of 2.297 g (10.6 mmol) of compound 3a in 30 mL of anhydrous chloroform. The mixture was refluxed for 100 min and cooled. The red crystals that formed were filtered off and sried in a vacuum. Yield 95%, mp 216–218°C (CHCl3). IR spectrum, ν, cm–1: 1750 (C6=O, C1=O), 1722 (C7=O), 1646 (COPh). 1H NMR spectrum, δ, ppm: 3.83 t (2H, C4H2, J 5.1 Hz), 4.72 t (2H, C3H2, J 5.0 Hz), 7.53 t (2Harom, J 5.0 Hz), 7.68 t (1Harom, J 7.4 Hz), 8.02 d (2Harom, J 7.1 Hz). 13C NMR spectrum, δ, ppm: 36.3 (C4), 67.2 (C3), 113.2, 128.6, 129.1, 134.0, 136.4, 148.1 (C8a), 154.9 (C1=O), 156.9 (C6=O), 181.3 (C7=O), 187.7 (COPh). Found, %: C 62.42; H 3.03; N 5.45. C14H9NO5. Calculated, %: C 62.00; H 3.34; N 5.16; O 29.49.
Compounds 6b–6h were prepared similarly to compound 6a.
8-(4-Chlorobenzoyl)-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5b). Yield 96%, mp 226–228°C (CHCl3). IR spectrum, ν, cm–1: 1761 (C6=O, C1=O), 1718 (C7=O), 1655 (COAr). 1H NMR spectrum, δ, ppm: 3.83 t (2H, C4H2, J 5.1 Hz), 4.72 t (2H, C3H2, J 5.1 Hz), 7.60 d (2Harom, J 8.8 Hz), 8.03 d (2Harom, J 8.7 Hz). 13C NMR spectrum, δ, ppm: 36.3 (C4), 67.2 (C3), 112.6, 128.8, 130.9, 135.3, 148.4 (C8a), 154.9 (C1=O), 156.9 (C6=O), 165.2, 181.0 (C7=O), 186.6 (COAr). Found, %: C 55.53; H 2.33; N 4.79. C14H8ClNO5. Calculated, %: C 55.01; H 2.64; Cl 11.60; N 4.58; O 26.17.
8-(4-Bromobenzoyl)-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5c). Yield 94%, mp 213–215°C (CHCl3). IR spectrum, ν, cm–1: 1749 (C6=O, C1=O), 1716 (C7=O), 1661 (COAr). 1H NMR spectrum, δ, ppm: 3.83 t (2H, C4H2, J 5.2 Hz), 4.72 t (2H, C3H2, J 5.1 Hz), 7.74 d (2Harom, J 8.5 Hz), 7.95 d (2Harom, J 8.6 Hz). 13C NMR spectrum, δ, ppm: 36.4 (C4), 67.3 (C3), 112.7, 128.4, 131.1, 131.8, 135.7, 148.5 (C8a), 155.0 (C1=O), 157.0 (C6=O), 181.1 (C7=O), 186.9 (COAr). Found, %: C 48.31; H 2.21; N 4.31. C14H8BrNO5. Calculated, %: C 48.03; H 2.30; Br 22.82; N 4.00; O 22.85.
8-(4-Methylbenzoyl)-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5d). Yield 90%, mp 210–212°C (CHCl3). IR spectrum, ν, cm–1: 1755 (C6=O, C1=O), 1732 (C7=O), 1655 (COAr). 1H NMR spectrum, δ, ppm: 2.38 s (3H, CH3), 3.82 t (2H, C4H2, J 5.1 Hz), 4.71 t (2H, C3H2, J 5.0 Hz), 7.29 d (2Harom, J 7.8 Hz), 7.67 d (2Harom, J 8.2 Hz). 13C NMR spectrum, δ, ppm: 21.2 (C6H4-Me), 36.3 (C4), 67.2 (C3), 113.4, 128.6, 129.1, 134.1, 147.9 (C8a), 154.9 (C1=O), 156.9 (C6=O), 164.9, 181.5 (C7=O), 187.2 (COAr). Found, %: C 63.38; H 3.68; N 5.32. C15H11NO5. Calculated, %: C 63.16; H 3.89; N 4.91; O 28.04.
8-Benzoyl-3-methyl-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5e). Yield 91%, mp 197–199°C (CHCl3). IR spectrum, ν, cm–1: 1753 (C6=O, C1=O), 1732 (C7=O), 1668 (COPh). 1H NMR spectrum, δ, ppm: 1.44 d (3H, CH3, J 6.4 Hz), 3.42 d.d (1H, C4H2, J 13.3, 9.6 Hz), 4.09 d.d (1H, C4H2, J 13.3, 9.6 Hz), 5.01 m (1H, C3H2), 7.53 t (2Harom, J 7.8 Hz), 7.68 t (1Harom, J 7.4 Hz), 8.01 d (2Harom, J 7.1 Hz). 13C NMR spectrum, δ, ppm: 17.4 (CH3), 41.2 (C4), 75.5 (C3), 113.2, 128.6, 129.2, 134.0, 136.5, 147.5 (C8a), 155.0 (C1=O), 157.0 (C6=O), 181.4 (C7=O), 187.8 (COPh). Found, %: C 63.49; H 3.67; N 5.14. C15H11NO5. Calculated, %: C 63.16; H 3.89; N 4.91; O 28.04.
3-Methyl-8-(4-chlorobenzoyl)-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5f). Yield 83%, mp 206–208°C (CHCl3). IR spectrum, ν, cm–1: 1750 (C6=O, C1=O), 1722 (C7=O), 1671 (COAr). 1H NMR spectrum, δ, ppm: 1.44 d (3H, CH3, J 6.4 Hz), 3.42 d.d (1H, C4H2, J 13.3, 9.6 Hz), 4.09 d.d (1H, C4H2, J 13.3, 3.1 Hz), 5.00 m (1H, C3H2), 7.60 d (2Harom, J 8.6 Hz), 8.03 d (2Harom, J 8.6 Hz). 13C NMR spectrum, δ, ppm: 17.2 (CH3), 41.0 (C4), 75.3 (C3), 112.4, 128.6, 130.8, 135.2, 138.8 (C8a), 154.8 (C1=O), 156.8 (C6=O), 180.8 (C7=O), 186.6 (COAr). Found, %: C 56.66; H 3.01; N 4.57. C15H10ClNO5. Calculated, %: C 56.35; H 3.15; Cl 11.09; N 4.38; O 25.02.
8-(4-Bromobenzoyl)-3-methyl-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5g). Yield 86%, mp 223–225°C (CHCl3). IR spectrum, ν, cm–1: 1754 (C6=O, C1=O), 1725 (C7=O), 1673 (COAr). 1H NMR spectrum, δ, ppm: 1.45 d (3H, CH3, J 6.4 Hz), 3.42 d.d (1H, C4H2, J 13.2, 9.5 Hz), 4.09 d.d (1H, C4H2, J 13.4, 3.0 Hz), 5.01 m (1H, C3H2), 7.74 d (2Harom, J 8.1 Hz), 7.95 d (2Harom, J 8.1 Hz). 13C NMR spectrum, δ, ppm: 17.3 (CH3), 41.2 (C4), 75.4 (C3), 112.6, 128.2, 130.9, 131.7, 135.7, 147.7 (C8a), 154.9 (C1=O), 156.9 (C6=O), 181.0 (C7=O), 186.9 (COAr). Found, %: C 49.79; H 2.56; N 4.08. C15H10BrNO5. Calculated, %: C 49.48; H 2.77; Br 21.94; N 3.85; O 21.97.
3-Methyl-8-(4-methylbenzoyl)-3,4-dihydro-1H-pyrrolo[2,1-c][1,4]oxazine-1,6,7-trione (5h). Yield 75%, mp 179–181°C (CHCl3). IR spectrum, ν, cm–1: 1753 (C6=O, C1=O), 1724 (C7=O), 1668 (COAr). 1H NMR spectrum, δ, ppm: 1.45 d (3H, CH3, J 6.4 Hz), 2.39 s (3H, CH3), 3.43 d.d (1H, C4H2, J 13.3, 9.5 Hz), 4.07 d.d (1H, C4H2, J 13.3, 3.1 Hz), 5.01 m (1H, C3H2), 7.33 d (2Harom, J 8.0 Hz), 7.91 d (2Harom, J 8.2 Hz). 13C NMR spectrum, δ, ppm: 17.4 (Me), 21.1 (C6H4–Me), 41.2 (C4), 75.3 (C3), 113.5, 129.2, 129.3, 134.1, 144.6, 147.2 (C8a), 154.9 (C1=O), 156.9 (C6=O), 181.5 (C7=O), 187.2 (COAr). Found, %: C 64.64; H 4.17; N 4.87. C16H13NO5. Calculated, %: C 64.21; H 4.38; N 4.68; O 26.73.
Notes
For preliminary communication, see [18].
REFERENCES
Maslivets, A.N., Mashevskaya, I.V., Krasnykh, O.P., Shurov, S.N., and Andreichikov, Y.S., Zh. Org. Khim., 1992, vol. 28, p. 2545.
Aliev, Z.G., Krasnykh, O.P., Maslivets, A.N., and Atovmyan, L.O., Izv. Akad. Nauk, Ser. Khim., 2000, vol. 12, p. 2080.
Maslivets, A.N., Golovnina, O.V., Krasnykh, O.P., and Aliev, Z.G., Chem. Heterocycl. Compd., 2000, vol. 36, p. 105. https://doi.org/10.1007/BF02256855
Tolmacheva, I.A., Mashevskaya, I.V., and Maslivets, A.N., Russ. J. Org. Chem., 2001, vol. 37, p. 596. https://doi.org/10.1023/A:1012458608681
Mashevskaya, I.V., Makhmudov, R.R., Aleksandrova, G.A., Golovnina, O.V., Duvalov, A.V., and Maslivets, A.N., Khim.-Farm. Zh., 2001, vol. 35, p. 20.
Tolmacheva, I.A., Mashevskaya, I.V., and Maslivets, A.N., Russ. J. Org. Chem., 2002, vol. 38, p. 281. https://doi.org/10.1023/A:1015590306099
Maslivets, A.N., Mashevskaya, I.V., Duvalov, A.V., Kol’tsova, S.V., and Feshin, V.P., Russ. J. Org. Chem., 2002, vol. 38, p. 738. https://doi.org/10.1023/A:1019679526434
Aliev, Z.G., Maslivets, A.N., Golovnina, O.V., Krasnykh, O.P., Atovmyan, L.O., Zh. Strukt. Khim., 2002, vol. 43, p. 576.
Kistanova, N.S., Mashevskaya, I.V., Bozdyreva, K.S., and Maslivets, A.N., Chem. Heterocycl. Compd., 2003, vol. 39, p. 673. https://doi.org/10.1023/A:1025170821406
Vostrov, E.S., Gilev, D.V., and Maslivets, A.N., Chem. Heterocycl. Compd., 2004, vol. 40, p. 532. https://doi.org/10.1023/B:COHC.0000033556.58356.5c
Bozdyreva, K.S., Smirnova, I.V., and Maslivets, A.N., Russ. J. Org. Chem., 2005, vol. 41, p. 1081. https://doi.org/10.1007/s11178-005-0296-6
Semenova, T.D. and Krasnykh, O.P., Russ. J. Org. Chem., 2005, vol. 41, p. 1222. https://doi.org/10.1007/s11178-005-0321-9
Chervyakov, A.V. and Maslivets, A.N., Russ. J. Org. Chem., 2013, vol. 49, p. 943. https://doi.org/10.1134/S1070428013060286
Maslivets, A.N., Lisovenko, N.Yu., Golovnina, O.V., Vostrov, E.S., and Tarasova, O.P., Chem. Heterocycl. Compd., 2000, vol. 36, p. 483. https://doi.org/10.1007/BF02269553
Silaichev, P.S., Kryuchkova, M.A., and Maslivets, A.N., Russ. J. Org. Chem., 2009, vol. 45, p. 1730. https://doi.org/10.1134/S1070428009110293
Andreichikov, Yu.S., Voronova, L.A., Astaf’eva, I.Yu., Tendryakova, S.V., and Belykh, Z.D., USSR Inventor’s Certificate no. 621676, 1978.
Maslivets, A.N. and Mashevskaya, I.V., 2,3-Digidro-2,3-pirroldiony (2,3-Dihydropyrrol-2,3-diones), Perm: Perm. Gos. Univ., 2005.
Tretyakov, N.A., Shavrina, T.V., and Maslivets, A.N., Russ. J. Org. Chem., 2019, vol. 55, p. 719. https://doi.org/10.1134/S1070428019050221
Allen, F.H., Kennard, O., Watson, D.G., Brammer, L., Orpen, A.G., and Taylor, R., J. Chem. Soc., Perkin Trans. 2, 1987, S1. https://doi.org/10.1039/P298700000S1
CrysAlisPro, Agilent Technologies, Version 1.171.37.33 (release 27-03-2014 CrysAlis171 .NET).
Sheldrick, G.M., Acta Crystallogr. Sect. A., 2008, vol. 64, p. 112. https://doi.org/10.1107/S0108767307043930
Sheldrick, G.M., Acta Crystallogr. Sect. C., 2015, vol. 71, p. 3. https://doi.org/10.1107/S2053229614024218
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J, Howard, J.A.K., and Puschmann, H., J. Appl. Cryst., 2009, vol. 42, p. 339. https://doi.org/10.1107/S0021889808042726
Funding
The work was financially supported by the Russian Foundation for Basic Research (project no. 19-33-90222) and the Government of the Perm Krai.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Tret’yakov, N.A., Dmitriev, M.V. & Maslivets, A.N. Synthesis of Pyrrolo[2,1-c][1,4]oxazine-1,6,7-triones by the Reaction of 3-Methylenemorpholin-2-ones with Oxalyl Chloride. Russ J Org Chem 56, 1367–1373 (2020). https://doi.org/10.1134/S1070428020080060
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070428020080060