
Abstract
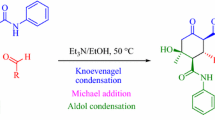
An effective secondary amine catalysed approach to the diastereoselective synthesis substituted cyclohexanols was proposed. A steric hindrance driven diastereoselective cross domino reaction gave manifold heteroaryl substituted cyclohexanols with five asymmetric centres in 92–98% yields. The sequence of inter- and intramolecular domino reactions includes Claisen–Schmidt condensation followed by 1,4-Michael addition of heteroaryl carbaldehydes and heteroaryl acetyl derivatives. HPLC study of the reactions shows outstanding self diastereoselectivity (dr > 99%) in the formation of one set of enantiomers out of sixteen possible enantiomeric pairs. The structures of all the synthesized molecules were confirmed by HRMS, NMR and IR spectroscopy methods.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The formation of manifold heteroaryl substituted asymmetric structures is one of the foremost areas of current synthetic organic chemistry, as these scaffolds exist in an enormous number of biologically active natural and synthetic compounds [1–3]. Heteroaryl based carbonyl compounds are valuable substrates to produce these molecules, and they have been exploited in numerous asymmetric catalytic reactions such as Claisen–Schmidt and aldol condensation, Mannich and Michael reactions [4–6].
Simple domino reactions (DRs) are a captivating field of organic chemistry [7, 8], which has fulfilled many principles of green chemistry when numerous conversions occur in a single synthetic procedure [9, 10]. A characteristic DR is a supple reaction for the rapid formation of multifaceted molecules with biologically vital scaffold structures [11, 12]. DRs are also desired approaches for the quick formation of heterocyclic compounds [13, 14]. In most cases, the unforeseen results of DRs disclose new types of chemical passages and novel scaffolds in organic chemistry [15, 16]. In major studies, DRs are linear [17–19] or branched [20, 21] types of reactions, based on how their intermediates are formed from the starting scaffolds which then yield the target products.
Previously, we have reported a third kind of domino reaction, a “cross domino reaction” [22] which has been established as a subclass of branched domino reactions, where intermediate A reacts with one of the starting materials to generate an intermediate B, which can then react with intermediate A and generate intermediate C. The latter finally convert into desired product. There are some reports on the synthesis of fully functionalized cyclohexanol derivatives with five chiral centers, using various chiral and metal catalysts [23–28].
The first study about this reaction was given by Kostanecki and Rossbach [29]. Later it was modified as a phase-transfer [30, 31], microwave free [32] and solvent free [33, 34]. Chen and co-workers fruitfully formed as many as six stereogenic centers on spirocyclic oxindoles in a one-pot tandem reaction [35]. Later in 2014, Dieter and co-workers reported organocatalytic Michael–Michael–Knoevenagel-type 1,2-addition reactions to synthesize cyclohexanes [36].
Looking at the benefits of domino reactions and their prominence in the synthesis of cyclohexanol derivatives, we herein report a simple, efficient and completely metal free cross domino Claisen–Schmidt condensation followed by Michael addition and aldol reactions with heteroaryl carbaldehydes and aromatic ketones. To the best of our knowledge, this is the first report of secondary amine catalysed Claisen–Schmidt condensation followed by a Michael addition and aldol reaction as a cross domino reaction to generate chiral cyclohexanol with five heteroaryl substitution by a self diastereoselective approach. To understand the self diastereoselectivity of the reaction, the detailed mechanistic studies were carried out in our earlier report [22]. Similar to the earlier report, along with the synthesis of chiral cyclohexanol, some of the intermediates were also isolated/trapped and characterized to confirm the mechanism of the reaction catalysed by secondary amine. Finally the stereochemistry of the synthesized compounds is established using 1H–1H COSY and 1H–13C HETCOR 2D NMR spectra of selected molecules and supported by our earlier report.
RESULTS AND DISCUSSION
Cyclohexanol 3a was synthesized by cross domino reaction of 3 moles of 2-acetyl pyridine 1a with two moles of benzo[b]thiophene-3-carbaldehyde 2a using ethanol as the solvent and pyrrolidine as the catalyst (Scheme 1). The same methodology was followed to prepare a series of cyclohexanols. Structures of the prepared cyclohexanols were confirmed by NMR, IR, and HRMS techniques (Supporting Information). Furthermore, selectively few of these molecules were characterized by 2D NMR as per our earlier report [22].

1.
The spectral data of these heteroaryl substituted cyclohexanols were compared with our earlier reported where potassium hydroxide was used as a catalyst and detailed discussion on their spectroscopic characteristics is done in [22]. The prepared cyclohexanols are asymmetric molecules with five chiral centres, therefore 32 stereoisomers or 16enantiomeric pairs are possible. Remarkably, the HPLC study has shown formation of only one enantiomeric pair (racemic mixture) as the same was observed in earlier report [22].
In order to confirm the mechanistic pathway for the reaction, as presented in [22], the formation of 1,5-dione by pyrrolidine catalysed reaction was also confirmed by trapping study of 1,5-dione to form terpyridine 4 (Scheme 2).

2.
To enlarge the substrate scope of this protocol, a study with respect to effect of the position of carbaldehyde group on heteroaryl system was performed with benzo[b]thiophene-2-carbaldehyde 2b and all three acetyl pyridine derivatives 1a–1c under similar conditions (Scheme 3).

3.
Furthermore, to study the effect of oxygen instead of sulfur, all the reactions were performed with benzofuran-2-carbaldehyde 2c (Scheme 3). All these reactions show the formation of the respective cyclohexanols 3d–3f and 3g–3i in quantitative yields (> 95%, Table 1).
In addition, employing other polynuclear aryl carbaldehydes viz. 2-naphthaldehyde 2d with all three acetyl pyridines 1a–1c under similar conditions afforded cyclohexanols 3j–3l in 94–97% yield (Scheme 4).

4.
The presence of nitrogen atom in acetyl pyridines is not essential for these reactions. Same observation was seen in the case of aryl carbaldehydes 2a–2e when reacted with acetophenone 1d under similar reaction conditions (Scheme 5). The respective cyclohexanols 3m–3q were prepared in 93–98% yield (Table 1).

5.
Our earlier reports on the derivatives of acetophenone show the formation of chalcones under the studied reaction conditions [37, 38]. However, with pyrrolidine as a catalyst, selectively few of them have shown the formation of cyclohexanol derivatives (Scheme 6). There is a significant difference in yield for the product formation in earlier reported and present work. A pyrrolidine catalysed process is more effective as compared to potassium hydroxide catalysed protocol. Furthermore, the tare of reaction mixture was slow in case when pyrrolidine was used as a catalyst, which is probably due to weak base strength.

6.
EXPERIMENTAL
All the chemicals were obtained from Sigma–Aldrich and were used as received. The 1H, 1H–H COSY and HETCORE NMR spectra were recorded in CDCl3 or DMSO-d6 at room temperature using a Bruker AVANCE III 500 MHz (AV 500) multinuclei solution NMR Spectrometer, TMS was used as internal reference. 13C and DEPT-135 NMR spectra were measured on Bruker AVANCE III 500 MHz (AV 500) with complete proton decoupling. Chemical shifts were reported in ppm from the residual solvent as an internal standard. Infrared (IR) spectra were recorded neatly by ATR on a Thermo Nicolet iS50 FT–IR spectrometer. HRMS data were obtained in methanol, with Thermo Scientific Orbitrap Elite Mass spectrometer. Melting points were measured by an open capillary method using Sigma Melting Point Apparatus. High performance liquid chromatography (HPLC) was performed on JASCO instruments at 210 nm using 4.6 mm × 25 cm Daicel CHIRALCEL OJ-H. For thin layer chromatography (TLC) analysis, Merck percolated TLC plates (silica gel 60 GF254, 0.25 mm) were used. The products were purified by recrystallization or column chromatography silica gel 60 (Merck, 230–400 mesh).
General procedure for the synthesis of cyclohexanol derivatives (3a–3t). The Claisen–Schmidt condensation of aryl and hetaryl carbaldehydes 2a–2e (10 mmol, 1 equiv.) in ethanol (5 mL) was performed with different acetyl pyridine (1a–1c) or acetophenone (1d–1g) derivatives (15 mmol, 1.5 equiv.) dissolved in ethanol (2–3 mL). The reaction mixture was allowed to stir at room temperature for an additional 10 min, during which it turned to a homogeneous solution. Then pyrrolidine (1 equiv.) was added dropwise and the resultant mixture was stirred at room temperature till the reaction was completed (3–4 h). The precipitated product was then filtered off. The crude product was purified by recrystallization from chloroform–methanol (1 : 1 v/v, 10 mL). Structures of all the novel compounds were confirmed by NMR, IR, and HRMS analysis. The structure of earlier reported derivatives was supported by TLC. Their spectral data were similar to those reported earlier [22].
(±)-[(1R,2R,3S,4R,6R)-2,6-Bis(benzo[b]thiophen-3-yl)-4-hydroxy-4-(m-tolyl)cyclohexane-1,3-diyl]bis(m-tolylmethanone) (3r). Yield 93%, white solid, mp 238–240°C. IR spectrum, ν, cm–1: 1656, 2572, 2880, 3072, 3312. 1H NMR spectrum (500 MHz, CDCl3), δ, ppm: 1.80 s (3H, CH3), 2.21 s (3H, CH3), 2.25 d (1H, CH, J 3.5 Hz), 2.37 s (3H, CH3), 3.52 d. t (1H, CH, J 2.5, 13.5 Hz), 4.70 t. d (1H, CH, J 3.5, 12.5 Hz), 4.78 t (1H, CH, J 4.4 Hz), 4.84 d. d (1H, CH, J 4.5, 12.0 Hz), 5.30 d (1H, CH, J 2.0 Hz), 5.89 d (1H, CH, J 11.5 Hz), 6.61 s (1H, OH), 6.71 t (1H, Ar, J 7.5 Hz), 6.84 m (3H, Ar), 6.95 d (1H, Ar, J 7.5 Hz), 6.99 s (1H, Ar), 7.07 t (1H, Ar, J 7.5 Hz), 7.15 m (2H, Ar), 7.21 t (1H, Ar, J 7.5 Hz), 7.30 m (4H, Ar), 7.47 t (1H, Ar, J 7.0 Hz), 7.55 t (1H, Ar, J 7.55 Hz), 7.62 m (3H, Ar), 7.68 d (1H, Ar, J 8.0 Hz), 8.07 d (1H, Ar, J 8.0 Hz), 8.12 d (1H, Ar, J 8.0 Hz). 13C NMR spectrum (125 MHz, CDCl3), δC, ppm: 20.71, 21.17, 21.63, 35.35, 38.82, 39.25, 47.49, 50.74, 75.89, 121.33, 122.20, 122.29, 122.61, 122.85, 124.03, 124.21, 124.25, 124.39, 124.84, 125.42, 125.90, 127.39, 127.74, 127.78, 127.83, 128.16, 128.34, 132.79, 133.77, 134.13, 136.50, 137.30, 137.64, 137.76, 138.34, 138.36, 138.75, 139.85, 140.35, 146.52, 206.29, 208.0; DEPT-135 (125 MHz, CDCl3): 20.71, 21.17, 21.63, 35.35, 38.82, 39.25, 47.49, 50.74, 121.33, 122.20, 122.29, 122.61, 122.85, 124.03, 124.21, 124.25, 124.39, 124.84, 125.42, 125.90, 127.39, 127.74, 127.78, 127.83, 128.16, 128.34, 132.79, 133.77. Mass spectrum (HRMS-ESI), m/z: 713.2155 [M + Na]+ (calcd for C45H38O3S2Na: 713.2144).
(±)-[(1R,2R,3S,4R,6R)-2,6-Bis(benzo[b]thiophen-3-yl)-4-hydroxy-4-(p-tolyl)cyclohexane-1,3-diyl]bis(p-tolylmethanone) (3s). Yield 95%, pale yellow solid, mp 242–245°C. IR spectrum, ν, cm–1: 1660, 2570, 2850, 3079, 3317. 1Н NMR spectrum, δ, ppm: 2.06 s (3H, CH3), 2.25 s (3H, CH3), 2.27 s (3H, CH3), 3.41 t (1H, CH, J 12.5 Hz), 4.67 d (1H, CH, J 12.5 Hz), 6.57 d (2H, Ar, J 7.5 Hz), 6.84 m (3H, Ar), 6.96 m (3H, Ar), 7.11 d (2H, Ar, J 9.5 Hz), 4.72 d (1H, CH, 11.5 Hz), 4.83 d. d (1H, CH, J 2.0, 11.5 Hz), 5.43 s (1H, OH), 5.89 d (1H, CH, J 11.0 Hz), 7.30 m (3H, Ar), 7.43 m (4H, Ar), 7.53 t (1H, Ar, J 8.0 Hz), 7.60 d (1H, Ar, J 8.0 Hz), 7.65 d (1H, Ar, J 8.0 Hz), 7.70 d (2H, Ar, J 8.0 Hz), 8.04 d (1H, Ar, J 8.0 Hz), 8.09 d (1H, Ar, J 8.0 Hz). 13C NMR spectrum (125 MHz, CDCl3), δC, ppm: 20.92, 21.26, 21.55, 35.37, 39.19, 39.25, 47.28, 50.17, 75.83, 121.29, 122.04, 122.58, 122.81, 124.00, 124.13, 124.22, 124.32, 124.79, 125.06, 127.10, 128.23, 128.28, 128.68, 128.90, 134.03, 135.75, 136.43, 136.48, 136.55, 138.28, 139.75, 140.31, 142.82, 143.79, 143.90, 205.71, 207.20; DEPT-135 (125 MHz, CDCl3), δC, ppm: 20.92, 21.26, 21.55, 35.37, 39.19, 39.25, 47.28, 50.17, 121.29, 122.04, 122.58, 122.81, 124.00, 124.13, 124.22, 124.32, 124.79, 125.06, 127.10, 128.23, 128.28, 128.68, 128.90. Mass spectrum (HRMS-ESI), m/z: 713.2155 [M + Na]+ (calcd for C45H38O3S2Na: 713.2144).
(±)-[(1R,2R,3S,4R,6R)-2,6-Bis(benzo[b]thiophen-3-yl)-4-hydroxy-4-(3-methoxyphenyl)cyclohexane-1,3-diyl]bis[(3-methoxyphenyl)methanone] (3t). Yield 95%, greenish solid, mp 241–242°C. IR spectrum, ν, cm–1: 1012, 1240, 1663, 2562, 2889, 3075, 3320. 1H NMR spectrum (500 MHz, CDCl3), δ, ppm: 2.23 d. d (1H, CH, J 3.0, 12 Hz), 3.26 s (3H, OCH3), 3.42 t (1H, CH, J 3.5 Hz), 3.65 s (3H, OCH3), 3.81 s (3H, OCH3), 4.77 t. d (1H, CH, J 3.5, 12.5 Hz), 4.75 t (1H, CH, J 4.4 Hz), 4.82 d. d (1H, CH, J 4.5, 12.0 Hz), 5.34 d (1H, J 2.0 Hz), 5.86 d (1H, J 11.5 Hz), 6.36 s (1H, OH), 6.51 d (1H, Ar, J 9.0 Hz), 6.60 d. d (1H, Ar, J 1.5, 8.0 Hz), 6.66–6.71 m (2H, Ar), 6.85 s (2H, Ar), 6.90 d. d (1H, Ar, J 2.0, 8.5 Hz), 7.00 s (1H, Ar), 7.19 d (1H, Ar, J 9.5 Hz), 7.28 t (3H, Ar, J 7.5 Hz), 7.35 s (1H, Ar), 7.12 t (1H, Ar, J 8.0 Hz), 7.43–7.48 m (2H, Ar), 7.54 t (1H, Ar, J 7.55 Hz), 7.63 d (1H, Ar, J 4 Hz), 7.68 d (1H, Ar, J 8.5 Hz), 8.04 d (1H, Ar, J 8.5 Hz), 8.10 d (1H, Ar, J 8.5 Hz). 13C NMR spectrum (125 MHz, CDCl3), δC, ppm: 35.30, 38.78, 39.37, 47.77, 50.75, 54.71, 55.24, 55.27, 75.96, 110.21, 111.30, 111.50, 112.41, 117.72, 119.65, 119.69, 120.09, 121.0, 121.18, 121.21, 122.26, 122.70, 122.95, 124.06, 124.22, 124.27, 124.41, 125.01, 128.51, 128.97, 129.38, 133.80, 136.32, 138.23, 138.26, 139.72, 139.89, 140.26, 140.38, 148.40, 158.75, 159.16, 159.67, 205.84, 207.68; DEPT135 (125 MHz, CDCl3), δC, ppm: 35.30, 38.78, 39.37, 47.77, 50.75, 54.71, 55.24, 55.27, 110.21, 111.30, 111.50, 112.41, 117.72, 119.65, 119.69, 120.09, 121.0, 121.18, 121.21, 122.26, 122.70, 122.95, 124.06, 124.22, 124.27, 124.41, 125.01, 128.51, 128.97, 129.38.
CONCLUSIONS
In summary, we have developed a very simple, highly efficient and completely metal-free cross domino protocol for the generation of aryl and heteroaryl substituted chiral cyclohexanols. The self diastereoselectivity of the domino process is tempted by the bulky nature of the binuclear aromatic ring systems. As cyclic skeletons with five asymmetric centres and ketone groups, these systems are vital in synthetic organic chemistry. This novel cross domino reaction catalysed by secondary amino base reported here makes use of very useful reaction conditions and only two simple starting materials, and will thus be useful in pharmaceutical research and diversity-oriented synthesis. As on-going research work of this project, our group has developed the methodology for enantioselective synthesis of all these cyclohexanols with excellent ee.
REFERENCES
Gopi Krishna Reddy, A., Krishna, J., and Satyanarayana, G., ChemistrySelect, 2016, vol. 1, p. 1151. https://doi.org/10.1021/ol2032625
Frederic, L., Thierry, M.C., and Jean, R., J. Am. Chem. Soc., 2005, vol. 127, p. 17176. https://doi.org/10.1021/ja055885z
Snyder, S.A., Breazzano, S.P., Ross, A.G., Lin, Y., and Zografos, A.L., J. Am. Chem. Soc., 2009, vol. 131, p. 1753. https://doi.org/10.1021/ja806183r
Bakthadoss, M., Vinayagam, V., Agarwal, V., and Sharada, D.S., ChemistrySelect, 2019, vol. 4, p. 7996. https://doi.org/10.1002/slct.201901806
Alcaide, B., Almendros, P., Angew. Chem. Int. Ed., 2008, vol. 47, p. 4632. https://doi.org/10.1002/anie.200801231
Coeffard, V., Desmarchelier, A., Morel, B., Moreau, X., and Greck, C., Org. Lett., 2011, vol. 13, p. 5778. https://doi.org/10.1021/ol202340p
Zinoveva, A.D., Borisova, T.N., Politova, P.A., Titov, A.A., Varlamov, A.V., Voskressensky, L.G., Nguyen, V.T., and Le, T.A., ChemistrySelect, 2020, vol. 5, p. 10821. https://doi.org/10.1002/slct.202002684
Tietze, L. and Beifuss, F.U., Angew. Chem. Int. Ed., 1993, vol. 32, p. 131. https://doi.org/10.1016/S0040-4039(99)00035-0
Kano, T., Yamaguchi, Y., and Maruoka, K., Angew. Chem. Int. Ed., 2009, vol. 48, p. 1838. https://doi.org/10.1002/anie.200805628
Li-Ping, F., Qing-Qing, S., Shi, Y., Jiang, B., and Shujiang, T., ACS Comb. Sci., 2013, vol. 15, p. 135. https://doi.org/10.1021/co3001428
Majhi, S., ChemistrySelect, 2020, vol. 5, p. 12450. https://doi.org/10.1002/slct.202002836
Ishikawa, H., Honma, M., and Hayashi, Y., Angew. Chem. Int. Ed., 2011, vol. 50, p. 2824. https://doi.org/10.1002/anie.201101113
Hooshmand, S.E., Ghadari, R., Mohammadian, R., Shaabani, A., and Khavasi, H.R., ChemistrySelect, 2019, vol. 4, p. 11893. https://doi.org/10.1002/slct.201903361
Liang, Z., Zhao, J., and Zhang, Y., J. Org. Chem., 2010, vol. 75, p. 170. https://doi.org/10.1021/jo902265s
Ali, R., ChemistrySelect, 2020, vol. 5, p. 10795. https://doi.org/10.1002/slct.202002878
Mihály, B., Chem. Rev., 2010, vol. 110, p. 1663. https://doi.org/10.1021/cr9002352
Wong, C.T., Tetrahedron, 2012, vol. 68, p. 481. https://doi.org/10.1016/j.tet.2011.11.019
List, B., Lerner, R.A., and Barbas, C.F., J. Am. Chem. Soc., 2000, vol. 122, p. 2395. https://doi.org/10.1021/ja994280y
Ahrendt, K.A., Borths, C.J., and MacMillan, D.W.C., J. Am. Chem. Soc., 2000, vol. 122, p. 4243. https://doi.org/10.1021/ja000092s
Chang, M.Y. and Wu, M.H., Tetrahedron, 2012, vol. 68, p. 9616. https://doi.org/10.1016/j.tet.2012.09.048
Xiaofei, Z., Qijian, N., Gerhard, R., and Dieter, E., Angew. Chem. Int. Ed., 2013, vol. 52, p. 2977. https://doi.org/10.1002/anie.201209581
Patel, P.N. and Chadha, A., Tetrahedron, 2018, vol. 74, p. 204. https://doi.org/10.1016/j.tet.2017.11.070
Mohammadi, O., Golestanzadeh, M., and Abdouss, M., ChemistrySelect, 2018, vol. 3, p. 12131. https://doi.org/10.1002/slct.201803217
Zhou, J. and List, B., J. Am. Chem. Soc., 2007, vol. 129, p. 7498. https://doi.org/10.1021/ja072134j
Kim, H. and Ho Rhee, Y., J. Am. Chem. Soc., 2012, vol. 134, N 9, p. 4011. https://doi.org/10.1021/ja2116298
Ibrahim, M., Rza, A., Musa, B., and Abel, M., Magn. Reson. Chem., 2016, vol. 54, p. 315. https://doi.org/10.1002/mrc.4377
Hayreddin, G. and Mustafa, C., Synth. Commun., 2015, vol. 45, p. 2344. https://doi.org/10.1080/00397911.2015.1089577
Kessler, H., Mronga, S., Kutscher, B., Miiller, A., and Sheldrick, W.S., Ann. Chem., 1991, p. 1337. https://doi.org/10.1107/S1600536806010634
Kostanecki, St.V. and Rossbach, G., Ber. Dtsch. Chem. Ges., 1896, vol. 29, p. 1488. https://doi.org/10.1002/cber.18960290133
Inoue, K., Noguchi, H., Hidai, M., and Uchida, Y., Yukagaku, 1983, vol. 32, p. 219. https://doi.org/10.5650/jos1956.32.219
Zhang, J.H., He, Q.P., Wang, Y., and Wang, D.Q., Acta Crystallogr. (E), 2007, vol. 63, p. o4652. https://doi.org/10.1016/j.ica.2007.02.050
Chen, W.Y. and Peng, Y.K., HechengHuaxue, 2000, vol. 8, no. 6, p. 544. https://doi.org/10.1007/s11426-010-0137-5
Luo, X. and Shan, Z., Tetrahedron Lett., 2006, vol. 47, p. 5623. https://doi.org/10.1016/j.tetlet.2006.06.037
Shan, Z., Hu, X., Hu, L., and Peng, X., Helv. Chim. Acta, 2009, vol. 92, p. 1102. https://doi.org/10.1002/hlca.200800248
Jiang, K., Jia, Z.J., Chen, S., Wu, L., and Chen, Y.C., Chem. Eur. J., 2010, vol. 16, p. 2852. https://doi.org/10.1002/chem.200903009
Pankaj, C., Gregor, U., Gerhard, R., and Dieter, E., Chem. Commun., 2014, vol. 50, p. 6853. https://doi.org/10.1039/C4CC01885K
Patel, P.N. and Chadha, A., J. Chem. Crystallogr., 2016, vol. 46, p. 245. https://doi.org/10.1007/s10870-016-0653-z
Patel, P.N. and Chadha, A., Acta Crystallogr. (E), 2015, vol. 71, p. o884. https://doi.org/10.1107/S2056989015019714
ACKNOWLEDGMENTS
Authors are thankful to Uka Tarsadia University for providing the BU Patel RPS scheme to perform the characterization of prepared compounds.
Funding
This work was financially supported by the DST-SERB, Govt. of India (project no. DST-SERB/TAR/2019/000089).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Supplementary information
Rights and permissions
About this article

Cite this article
Patel, P.N., Talati, K.S., Deshmukh, A.G. et al. Secondary Amine Catalysed Diastereoselective Cross Domino Reaction: Simple and Efficient Synthesis of Heteroaryl-Substituted Cyclohexanols. Russ J Gen Chem 92, 694–701 (2022). https://doi.org/10.1134/S1070363222040107
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363222040107