
Abstract
The reaction of AgNO3/Ni(NO3)2 · 6H2O with 4-nitroimidazolate (HNim) gave two new coordination poymers of [Ag(Nim)]n (I) and [Ni(Nim)(H2O)4]n (II). X-ray single-crystal analyses (СIF files CCDC nos. 1508955 (I) and 1821871 (II)) reveal that I and II show 2D grid layer and mononuclear network, respectively. The photoluminescence investigation indicates that I could be a prospective candidate for developing luminescence sensors for the highly sensing of Fe3+ analyte. Furthermore, the photocatalysis properties of II for degradation of the methyl violet and Rhodamine B have been examined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In recent years, as a newly emerged type of functional inorganic–organic hybrid material, coordination polymers (CPs) have attracted increasing research attention for various advanced applications [1–4]. They are also motivated largely by a demand for solving pollution problems in view of their possible applications in the green degradation of organic pollutants [5, 6]. However, these polymers have been controlled by the self-assembly of metal ions and organic ligands, the influencing factors of the ultimate Therefore, significant interest has arisen in the structural tuning of coordination polymers through rational design and selection of organic building blocks [7, 8].
Rigid 4-nitroimidazolate (HNim) and its derivates have been extensively used in the synthesis of multidimensional CPs containing a large variety of metal centers [9, 10]. Moreover, the nitryl group (NO2) can not only act as a hydrogen bond acceptor, but it can also take on some spatial effects in the formation of polymeric networks. As an effective combination of the abovementioned aspects and our previous work [11], we report here on two new coordination polymers, namely [Ag(Nim)]n (I) and [Ni(Nim)(H2O)4]n (II) have been synthesized. The photoluminescent sensing of I are potential candidates for detecting Fe3+ specie. Compound II is found to be able to photocatalytically degrade methyl violet (MV) in a relatively efficient way.
EXPERIMENTAL
Materials and methods. All chemicals were purchased without further purification. IR spectra were recorded with a Perkin-Elmer Spectrum One spectrometer in the region 4000–400 cm–1 using KBr pellets. Thermogravimetric (TG) analysis was carried out with a Metter–Toledo TA 50 under dry dinitrogen flux (60 mL min–1) at a heating rate of 5°C min–1. X-ray powder diffraction (XRPD) data were recorded on a Rigaku RU200 diffractometer at 60 kV, 300 mA for CuKα radiation (λ = 1.5406 Å) with a scan speed of 2°C/min and a step size of 0.02° in 2θ.
Synthesis of I. A mixture of HNim (0.2 mmol, 0.027 g), AgNO3 (0.4 mmol, 0.068 g), ammonia (25%, 10.0 mL) was stirred for 30 min in air and then transferred and sealed in a 25-mL Teflon-lined reactor and heated at 120°C for 60 h, and then cooled to room temperature at a rate of 5°C/h. Yellow block crystals of I were obtained in 67% yield based on silver.
Synthesis of II was analogous to that of I, except that AgNO3 was replaced by Ni(NO3)2 · 6H2O (0.116 g, 0.4 mmol). Green block crystals of II were obtained in 56% yield based on nickel.
Photoluminescence (PL) sensing method. The PL properties of I were investigated in H2O emulsions at room temperature using a RF-5301PC spectrofluorophotometer. The I–H2O emulsions were prepared by adding 3 mg of I powder into 3.00 mL of H2O and then ultrasonic agitation the mixture for 30 min before testing.
Photocatalytic properties. 50 mg of I, II were dispersed in 50 mL aqueous solution of MV or Rhodamine B (RhB) (10 mg/L) under stirring in the dark for 30 min to ensure the establishment of an adsorption–desorption equilibrium. Then the mixed solution was exposed to UV irradiation from an Hg lamp (250 W) and kept under continuous stirring during irradiation for 100 min. Samples of 5 mL were taken out every 10 min and collected by centrifugation for analysis by UV-Vis spectrometer. By contrast, the simple control experiment was also performed under the same condition without adding any catalysts.
X-ray crystallography. X-ray diffraction measurements were carried out on a Bruker SMART APEX diffractometer that was equipped with a graphite monochromated MoΚα radiation (λ = 0.71073 Å) by using a ω-scan technique. The intensities were corrected absorption effects by using SADABS. The structures were solved by using SHELXS-97 and refined by using SHELX-97 [12]. Absorption corrections applied by using multi-non-hydrogen atoms were refined anisotropically. Crystallographic details are given in Table 1. Selected bond dimensions are listed in Table 2.
Supplementary material for structure, I, II has been deposited with the Cambridge Crystallographic Data Centre (CCDC nos. 1508955 (I) and 1821871 (II); deposit@ccdc.cam. ac.uk or http://www.ccdc. cam.ac.uk).
RESULTS AND DISCUSSION
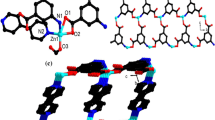
In the asymmetric unit of complex I, one Ag+ ion, one Nim ligand were observed. As shown in Fig. 1a, Ag(1) is nearly linear, linked to two N atoms from two symmetric Nim ligands. The lengths of Ag–N bond are in the expected range of 2.112(14)–2.129(13) (Table 2). Ag(I) coordinated with Nim ligands to generate a one-dimensional infinite chain. The distance of Ag–Ag is ~3.147(3) Å. As shown in Fig. 1b, the silver atom links Ag–Ag interactions into a 2D layer.

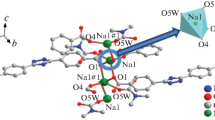
Coordination environment of Ag(I) center with 30% thermal ellipsoids (a); view of the 2D network in I (b); coordination environment of Ni(I) center with 30% thermal ellipsoids (c); view of the 1D supramolecular chain along the b axis in II (d).
Asymmetric unit of II is built by one Ni(I) center, one Nim ligand, and four coordinated lattice water ligands. As shown in Fig. 1c, each Ni(I) center is in distorted octahedral coordination geometry and is six-coordinated by two nitrogen atoms (N(1), N(1)#1 (#1 1 – x, 2 – y, –z) from two symmetric Nim ligands, four oxygen atom (O(3), O(3w)#1, O(4w), O(4w)#1 (#1 1 – x, 2 – y, –z) from four water molecules. The value of Ni–N bond distance is 2.104(3) Å, and the Ni–O bond separations are 2.149(3) and 2.156(4) Å, which are similar to those of other related Ni(II)-based [13]. The 1D supramolecular network is also stabilized by hydrogen-bonding interaction (C(1)–H(1)···O(1) (C···O 3.472(3) Å).
The IR spectra of CP II show a broad band at about 3381 cm–1, which can be attributed to the O–H stretching vibrations of coordination water molecules. The band at ~1520 cm–1 could be ascribed to the ν(C=N) absorption of imidazole rings of the Nim ligands [14]. The main characteristic absorption bands in the range of 1340–1540 cm–1 mainly attributed to the asymmetric and symmetric stretching vibrations of the nitrate groups.
TG experiments were carried out to measure the thermal stability of I, II, and the results are depicted in Fig. 2. Complex I shows a weight loss until 350°C and the organic components decomposed from 360–650°C. The remaining residue is 28.7% corresponding to the formation of AgO (obsd. 55.9%, calcd. 56.2%). For II, the first weight loss occurred in a temperature range from 35 to 195°C, corresponding to the loss of the coordinated water molecules (obsd. 19.8%, calcd. 20.3%). The remaining residue is attributed to the generation of CdO (obsd. 22.6%, calcd. 21.3%). The second weight loss occurred in a temperature range from 355 to 675°C, corresponding to the decomposition of the organic ligands.

TG curves of complexes I and II.
In order to check the phase purity of I, II, PXRD patterns of I, II were checked at room temperature. The peak positions of the simulated and experimental PXRD patterns are in agreement with each other, demonstrating the good phase purity of the bulk I, II.
The luminescent MOFs with different d10 metal ions were potential candidates due to their excellent photoactive materials [15]. The solid-state luminescent properties of pure HNim ligand and I were investigated carefully at room temperature. The maximum emission peaks of the HNim ligand and I are observed at ∼433 nm (λex = 280 nm), ∼437 nm (λex = 300 nm), respectively. Thus, the emission of I is very similar to that of the HNim ligand, which may arise from the ligand centered π–π* electronic transitions compared to the reported luminescent d10 MOFs [16].
We have tried to study the luminescent responses of I to different metal ions, the products of I were ground into powder and suspended in H2O solution containing the same concentration of M(NO3)n (M = Li+, Na+, K+, Ag+, Ca2+, Mg2+, Pb2+, Hg2+, Zn2+, Cd2+, Co2+, Ni2+, Al3+, and Fe3+) for 24 h. As depicted in Fig. 3a, the luminescent intensities of I are decreased sharply by Ag+, Hg2+, Pb2+, and Fe3+ ions, particularly for the Fe3+ ion, which has an excellent quenching effect for the system as a highly selective sensor. The quantitative sensitizations of quenching behavior were also examined via changing the concentration of Fe3+ cations in solution. The titration experiments indicated that the luminescent intensities were gradually weakened by increasing the amount of Fe3+ ions (Fig. 3b). The reason may be that the outer layers of Fe3+ ions comprised the unsaturated electronic state, which make the ions retain empty orbits and thus become the ideal acceptors for electrons. Thus, the unique quenching behavior herein may be largely ascribed to interactions of Fe3+ cations and oxygen atoms of nitrate from Nim ligands, which caused the electrons of Nim ligands to transfer from I to Fe3+ ions as electron acceptors, thus inducing the abovementioned luminescent decay.

Comparisons of luminescent intensities of I in different metal ions (a); different strength intensities of luminescent spectra of I with different amounts of Fe3+ ions (b); the relationship of luminescent intensities and Fe3+ concentration in I (c).
These MV and RhB were selected as model organic contaminant to study the photocatalytic activities of I, II. As shown in Fig. 4, the absorption peaks of MV and RhB decrease obviously with the increasing of reaction time in the presence of I, II. The calculation results show that the degradation rate of MV is 69.09% for I, 75.28% for II, while for RhB, the degradation rate for I and II is 38.58 and 40.24%, respectively. In addition, the control experiments on the degradation of organic pollutants were examined in the same reaction condition just without catalyst. The degradation rate of MV and RhB is just 33.39 and 34.20% within 100 min under UV irradiation without catalyst (Figs. 4c, 4f). As the above photocatalytic results shown, the photocatalytic performance of II is the better one due to metal nature. So, the metal center may influence their photocatalytic activities. The possible photocatalytic mechanism for the above degradation process is that when absorbed energy equal to or higher than its band gap energy (LUMO and the HOMO) of the materials I, II. When the HOMO seized one electron from one water molecule and then go back to stable state. The water molecule was oxidized into a •OH. Also, the electron of the LUMO reduced O2 to \({\text{O}}_{2}^{ - }\) by the combination of electrons (e–), which also turned to •OH. Finally, full •OH radical active works as an oxidizing agent to decompose MV/RhB [16–21].

UV-Vis absorption spectra of the MV and RhB solution during the decomposition reaction under 500 W Hg lamp irradiation in the presence of I (a) and (b); UV-Vis absorption spectra of the MV and RhB solution during the decomposition reaction under 500 W Hg lamp irradiation in the presence of II (d) and (e); curves of absorbance of the MV and RhB solution degraded by I or without any catalysis under UV light irradiation (c); curves of absorbance of the MV and RhB solution degraded by II or without any catalysis under UV light irradiation (f).
REFERENCES
Wang, D.E., Deng, K.J., Lv, K.L., et al., CrystEngComm, 2009, vol. 11, p. 1442.
Wang, S.B. and Wang, X.C., Small, 2015, vol. 11, p. 3097.
Liu, J., Liu, G.L., Gu, C.Y., et al., J. Mater. Chem., A, 2016, vol. 4, p. 11630.
Liu, J.Q., Wang, W.J., Luo, Z.D., et al., Inorg. Chem., 2017, vol. 56, p. 10215.
Tian, Y.-Q., Yao, S.-Y., and Gu, D., Chem. Eur. J., 2010, vol. 16, p. 1137.
Liao, Z.L., Li, G.D., Bi, M.H., and Chen, J.S., Inorg. Chem., 2008, vol. 47, p. 4844.
Wang, X.L., Luan, J., Lin, H.Y., et al., Dalton Trans., 2013, vol. 42, p. 8375.
Liu, J.Q., Wang, Y.Y., Wu, T., and Wu, J., CrystEngComm, 2012, vol. 14, p. 2906.
Li, Y.T., Yao, S.Y., Wang, Y., et al., CrystEngComm, 2011, vol. 13, p. 3470.
Lobban, C., Finney, J.L., and Kuhs, W.F., Nature, 1998, vol. 391, p. 268.
Wang, J., Li, C.T., Bai, C., et al., J. Mol. Struct., 2017, vol. 1141, p. 107.
Sheldrick, G.M., Acta. Crystallogr., Sect. A: Found. Adv., 2015, vol. 7, p. 3.
Liu, J.Q., J. Coord. Chem., 2011, vol. 64, p. 1807.
Chang, H.N., Hou, S.X., Hao, Z.C., and Cui, G.H., Polyhedron, 2018, vol. 141, p. 276.
Wu, Y.L., Yang, G.P., Zhao, Y.Q., et al., Dalton Trans., 2015, vol. 44, p. 3271.
Li, F.F., Ma, J., Song, S.Y., et al., Inorg. Chem., 2005, vol. 44, p. 9374.
Wu, Y., Wu, J., Luo, Z.D., et al., RSC. Adv., 2017, vol. 7, p. 10415.
Wu, Y., Lu, L., Feng, J.S., et al., J. Solid. State. Chem., 2017, vol. 245, p. 213.
Wu, Y., Wu, J., Xie, B., et al., J. Lumin., 2017, vol. 192, p. 775.
Wu, Y., Feng, J.S., Xie, B., et al., J. Inorg. Organomet., 2017, vol. 27, p. 1243.
Wang, K.X. and Chen, J.S., Acc. Chem. Res., 2011, vol. 44, p. 531.
ACKNOWLEDGMENTS
The authors acknowledge financial assistance from Sichuan University of Science and Engineering (nos. 2015RC26, 2015RC29 and 2017RCL02), the Education Committee of Sichuan Province (no. 17ZA0264), the Project of Zigong Science and Technology (no. 2016HG06) and the Opening Project of Key Laboratory of Green Catalysis of Sichuan Institutes of High Education (no. LYJ1705).
Author information
Authors and Affiliations
Corresponding author
Additional information
The article is published in the original.
Rights and permissions
About this article

Cite this article
Zhou, D., Lu, L., Luo, Y. et al. Structures, Photoluminescence, and Photocatalytic Properties of Two New Coordination Polymers based on 4-nitroimidazolate. Russ J Coord Chem 44, 812–817 (2018). https://doi.org/10.1134/S1070328418120126
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328418120126