
Abstract
The direct template reactions afford the cobalt(II) complexes (I–III) with the hydroxy- and acetyl-substituted 2,6-bis(pyrazol-3-yl)pyridine ligands containing the phenyl and 4-tert-butylphenyl group in position 1. All compounds are isolated in the individual state and characterized by elemental analysis and NMR spectroscopy. The X-ray diffraction data obtained for crystals of compound I (СIF file CCDC 1577238) and the data for solutions (using the proposed approach to analysis of paramagnetic NMR shifts on the basis of quantum chemical calculations) show that in the complexes the metal ion exists in the high-spin state (S = 3/2) and undergoes no temperature-induced spin transition in a temperature range of 120–300 K.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The development of new types of functional materials is the most important task of chemical science. Transition metal complexes often have unique properties, which makes them attractive for the preparation of various related materials. Nowadays one of the urgent and intensely developed trends of their application is the production of units for molecular electronics, primarily, for information storage and processing. Compounds appropriate for these purposes should be characterized by bistability: a capability of existing in two different electronic states under certain conditions.
The cobalt(II) complexes represent a typical example of these bistable molecular systems [1–7]. The cobalt(II) ion with the electronic configuration d7 can exist in the low-spin (S = 1/2) and high-spin (S = 3/2) states. The possibility of transition to occur between these two states in the cobalt(II) ion has first been found for the complexes with the coordination polyhedron CoN6 formed by the bidentate and tridentate ligands [8]. The intermediate values of magnetic moments obtained for these complexes assume an equilibrium mixture of molecules of the complex in the low- and high-spin states.
The spin state of the cobalt(II) ion is determined, to a significant extent, by its coordination environment primarily affecting the electronic structure of the complex. However, even slight changes at the “periphery” of the molecule can change the populations of the spin states [9, 10]. In some cases, the shift of the spin equilibrium is a consequence of steric effects of the substituents rather than the electronic ones [11, 12] and of the solvent effect as well [13, 14].
The earlier studied cobalt(II) complexes with the 2,6-bis(pyrazol-3-yl)pyridine ligands, as a rule, existed in the high-spin state and underwent no temperature-induced spin transition [15, 16]. In this work, we synthesized three new cobalt(II) complexes, namely, Co(LOHPh)2(ClO4)2 (I), Co(LOAcPh)2(ClO4)2 (II), and Co(LOHPht-Bu)2(ClO4)2 (III) (LOHPh is 2,6-bis(1-phenyl-5-hydroxypyrazol-3-yl)pyridine, LOAcPh is 2,6-bis(1-phenyl-5-acetylpyrazol-3-yl)pyridine, and LOHPht-Bu is 2,6-bis(1-(4-tert-butylphenyl)-5-hydroxypyrazol-3-yl)pyridine), with the 2,6-bis(pyrazol-3-yl)pyridine ligands containing various substituents (Scheme 1) and studied the influence of these substituents on the spin state of the cobalt(II) ion using NMR spectroscopy.

Scheme 1.
The structure and spin state of the cobalt(II) ion in complex I at 120 K in crystal and at ambient temperature in solution were confirmed by the X-ray diffraction data and using an original approach to analysis of paramagnetic NMR shifts based on quantum chemical calculations, respectively.
EXPERIMENTAL
All procedures related to the synthesis of new complexes were carried out in air using commercially available organic solvents. Compound Co(ClO4)2 ⋅ 6H2O [17], 4-tert-butylphenylhydrazine [18], diethyl-3,3'-(pyridine-2,6-diyl)bis(3-oxopropanoate) [19], and LOHPh [19] were synthesized using previously published procedures. Ligand LOAcPh was synthesized by the acylation of LOHPh [20]. Analyses to carbon, nitrogen, and hydrogen were conducted on a CarloErba, model 1106 microanalyzer.
Synthesis of LOHPht-Bu. A weighed sample of 4‑tert-butylphenylhydrazine hydrochloride (398 mg, 2 mmol) was added at ambient temperature to a solution of diethyl-3,3'-(pyridine-2,6-diyl)bis(3-oxopropanoate) (307 mg, 1 mmol) in acetic acid (50 mL). The reaction mixture was refluxed using a reflux condenser for 24 h under a nitrogen atmosphere. The cooled reaction mixture was mixed with ice (200 mL) due to which a white precipitate was formed. The precipitate was filtered and recrystallized from ethanol. The yield was 65%.
1H NMR (DMSO-d6), δ, ppm: 1.33 (s, 18H, Me), 6.19 (s, 2H, 4-Pyr), 7.52 (d, 3JHH = 7.3 Hz, 4H, o‑Ph(t-Bu)), 7.73 (d, 3JHH = 7.3 Hz, 4H, m-Ph(t-Bu)), 7.88 (m, 3H, Py), 11.78 (s, 2H, OH). 13C{1H} NMR (DMSO-d6), δ, ppm: 31.15 (s, Me), 34.29 (s, t-Bu), 86.02 (s, 4-Pr), 117.79 (s, 3-Py), 121.29 (s, m-Ph(t-Bu)), 125.67 (s, o-Ph(t-Bu)), 136.31 (s, iso-Ph), 137.13 (s, 4-Py), 148.56 (s, iso-Ph(t-Bu)), 150.25 (s, 5-Pr), 151.53 (s, 2-Py), 153.59 (s, 3-Pr). MS (ESI) m/z = 505.3 (M–H).
Synthesis of Co(LOHPh)2(ClO4)2 (I). Weighed samples of Co(ClO4)2 ⋅ 6H2O (8.8 mg, 0.024 mmol) and LOHPh (20 mg, 0.051 mmol) were dissolved or suspended in anhydrous ethanol (1.5 mL) under an argon atmosphere. The reaction mixture was stirred at ambient temperature for 1.5 h until the precipitate dissolved. Then a light orange-brown solution was evaporated to dryness, and a light yellow precipitate was washed with hexane (2 × 5 mL), dissolved in acetonitrile, and filtered. The filtrate was evaporated to dryness and dried in vacuo. The yield of compound I was 18.5 mg (74%).
For C46H34N10O12Cl2Co | |||
Found, % | C, 52.40 | H, 3.30 | N, 13.09 |
Anal. calcd., % | C, 52.69 | H, 3.27 | N, 13.36 |
1H NMR (CD3CN), δ, ppm: –1.52 (br.s, 2H, 4‑Py), 2.05 (br.s, 4H, п-Ph), 2.75 (br.s., 8H, m-Ph), 14.61 (br.s, 8H, o-Ph), 27.19 (br.s, 4H, 3-Py), 29.01 (br.s, 4H, OH), 55.27 (br.s, 4H, 4-Pyr). 13С{1H} NMR (CD3CN, δ, ppm): –442.13 (br.s, 2-Py), –35.71 (br.s, 3-Pyr), 65.55 (br.s, 4-Py), 109.72 (br.s, o-Ph), 123.41 (br.s, p-Ph), 125.34 (br.s, m-Ph), 179.55 (br.s, 4-Pyr), 209.03 (br.s, iso-Ph), 573.18 (br.s, 3-Py).
Synthesis of Co(LOAcPh)2(ClO4)2 (II). A weighed sample of LOAcPh (30 mg, 0.063 mmol) was dissolved or suspended in anhydrous ethanol (2 mL), Co(ClO4)2 ⋅ 6H2O (11 mg, 0.03 mmol) was added, and the mixture was refluxed with stirring under an argon atmosphere for 1.5 h. An orange precipitate was filtered off, washed with ethanol (2 × 5 mL) and diethyl ether (7 mL), dissolved in dichloromethane (10 mL), and filtered. The filtrate was evaporated to dryness and dried in vacuo. The yield of compound II was 26 mg (71%).
For C54H42N10O16Cl2Co | |||
Found, % | C, 52.09 | H, 3.43 | N, 11.21 |
Anal. calcd., % | C, 53.29 | H, 3.45 | N, 11.51 |
1H NMR (CD2Cl2), δ, ppm: 1.26 (br.s, 2H, 4-Py), 3.74 (br.s, 8H, m-Ph), 4.37 (br.s, 4H, p-Ph), 6.91 (br.s, 12H, CH3), 9.53 (br.s, 8H, o-Ph), 36.30 (br.s, 4H, 3‑Py), 57.96 (br.s, 4H, 4-Pyr). 13С{1H} NMR (CD2Cl2), δ, ppm: –387.89 (br.s, 2-Py), –46.43 (br.s, 3‑Pyr), 31.30 (br.s, Me), 99.25 (br.s, 4-Py), 108.95 (br.s, o-Ph), 126.87 (br.s, p-Ph), 127.73 (br.s, m-Ph), 177.28 (br.s, 4-Pyr), 199.06 (br.s, C=O), 207.28 (br.s, iso-Ph), 561.41 (br.s, 3-Py).
Synthesis of Co(LOHPht-Bu)2(ClO4)2 (III). A weighed sample of LOHPht-Bu (30 mg, 0.059 mmol) was dissolved or suspended in anhydrous ethanol (3 mL), Co(ClO4)2 ⋅ 6H2O (10 mg, 0.028 mmol) was added, and the mixture was stirred at ambient temperature for 2 h. The reaction mixture was filtered, and the filtrate was evaporated to dryness. A dark yellow precipitate was washed with diethyl ether (5 mL) and hexane (3 × 5 mL), dissolved in acetonitrile (8 mL), and filtered. The filtrate was evaporated to dryness and dried in vacuo. The yield was 25 mg (69%).
For C62H66N10O12Cl2Co | |||
Found, % | C, 58.10 | H, 5.17 | N, 10.61 |
Anal. calcd., % | C, 58.49 | H, 5.19 | N, 11.01 |
1H NMR (CD3CN), δ, ppm: –2.00 (br.s, 36H, Ме), –0.19 (br.s, 2H, 4-Py), 1.94 (br.s, 4H, p-Ph), 2.99 (br.s, 8H, m-Ph), 12.55 (br.s, 8H, o-Ph), 28.79 (br.s, 8H, 3-Py, OH), 54.38 (br.s, 4H, 4-Pyr). 13С{1H} NMR (CD3CN, δ, ppm): –439.49 (br.s, 2-Py), ‒38.92 (br.s, 3-Pyr), 26.65 (br.s, Me), 32.12 (br.s, t-Bu), 69.86 (br.s, 4-Py), 108.28 (br.s, o-Ph), 123.16 (br.s, p-Ph), 146.47 (br.s, m-Ph), 175.47 (br.s, 4-Pyr), 205.56 (br.s, iso-Ph), 576.24 (br.s, 3-Py), 1078.80 (br.s, 5-Pyr).
X-ray diffraction analysis of single crystals of complex I obtained by slow evaporation in air from an acetonitrile solution was carried out on a Bruker APEX2 CCD diffractometer (MoKα radiation, graphite monochromator, ω scan mode). The structure was solved by a direct method and refined by least squares in the full-matrix anisotropic approximation for \(F_{{hkl}}^{2}.\) Hydrogen atoms of the OH groups and solvate water molecules were localized from the difference electron density Fourier syntheses and refined in the isotropic approximation by the riding model. All calculations were performed using the SHELXTL PLUS program package [21]. The main crystallographic data and refinement parameters for complex I are presented in Table 1. The high values of the R factors and GOOF parameters are due to the fact that the crystals of complex I turned out to be accretions. We failed to describe their twinning as a superposition of individual components, but the quality of the obtained data was sufficient to reliably establish the molecular and crystal structures of complex I. The full data were deposited with the Cambridge Crystallographic Data Centre (CIF file (CCDC 1577238; http://www.ccdc. cam.ac.uk/).
1Н and 13С{1Н} NMR spectra were recorded for solutions of complexes I–III in CD3CN and CD2Cl2 on a Bruker Avance 600 spectrometer (600.22 and 150.96 MHz, respectively). The values of chemical shifts in the spectra were determined relative to the residual signal of the solvent (1H 1.94, 13C 118.26 ppm for CD3CN; 1H 5.32, 13C 54.00 ppm for CD2Cl2; 1H 2.50, 13C 39.52 ppm for DMSO-d6). The spectra were recorded using the following parameters: for 1Н NMR spectra, range 1000 ppm, recording time 0.1 s, relaxation delay 0.1 s, pulse duration 6.5 μs, scan number 1024; for 13С{1H} NMR spectra, range 3000 ppm, recording time 0.1 s, relaxation delay 0.1 s, pulse duration 9 μs, scan number more than 32 000. If the signal to noise ratio is necessary to enhance, the obtained free induction decays were processed by exponential weighing with the coefficient to 3 and 50 Hz in the case of 1H and 13C NMR spectra, respectively.
Quantum chemical calculations for cationic complex I were performed using the ORCA v. 4 program package in the framework of the density functional theory (DFT). The geometry of complex I was optimized without imposing symmetry restraints using the PBE non-hybrid functional [22] and def2-TZVP basis set [23]. The structure determined by X-ray diffraction analysis was used as the initial approximation. Solvation effects were took into account in terms of the conductor-like polarizable continuum model (CPCM) in the ORCA v. 4 program package. Acetonitrile was chosen as a solvent to record NMR spectra in solutions. The g tensor and hyperfine coupling tensors for protons and carbon nuclei were calculated on the basis of the geometry obtained for complex I using the PBE0 hybrid functional [24] and def2-TZVP basis set supplementing additional Gaussian primitives with a high exponential for a more precise description of the electron density in the nucleus region.
RESULTS AND DISCUSSION
Cobalt(II) complexes I–III with the 2,6-bis(pyrazol-3-yl)pyridine ligands were synthesized in high yields by the direct template reactions in ethanol via the scheme presented. Complexes I and III did not precipitate from the reaction mixture, and the reaction occurred at ambient temperature. Complex II turned out to be insoluble, and the starting ligand was poorly soluble under the reaction conditions and, hence, the synthesis was carried out under more drastic conditions (reflux).
Complexes I–III were isolated in the individual state and characterized by elemental analysis and NMR spectroscopy. The structure of complex I at 120 K was also confirmed by X-ray diffraction analysis.
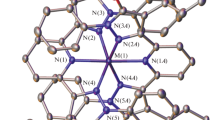
Complex I crystallizes as a hydrate with four water molecules (presumably captured by the crystal from air during crystallization). In crystal the complex occupies the partial position with symmetry 222 (Fig. 1). Each hydroxy group of the ligand is joint by strong hydrogen bonds with one solvate water molecule (O⋅⋅⋅O 2.577(9) Å, angle OHO 151(1)°), which is involved, in turn, in hydrogen bonding with the disordered perchlorate anions (O⋅⋅⋅O 2.88(1)/2.89(1) Å, angle OHO 138(1)°/172(1)°).

General view of complex I in the representation of atoms by thermal vibration ellipsoids (p = 50%). Disordered perchlorate anions and hydrogen atoms, except for those belonging to the OH groups and solvate water molecules, are omitted.
All three nitrogen atoms of the ligand (pyridine N(1) and pyrazole N(2)) are coordinated to the metal ion (coordination number of Со is 6) with the distances (Table 2) typical of cobalt(II) ions in the high-spin state (2.0–2.2 Å [25]). The latter also follows from the trigonal prismatic distortion [26] of its coordination polyhedron CoN6 (Fig. 2a), which is octahedron in the case of a low-spin transition metal ion with the configuration d7. In particular, the dihedral angle θ between the root-mean-square planes of two ligands bound to the metal is equal to 90° in the case of an ideal octahedron, whereas this angle is only 68.3(3)° in complex I. The observed trigonal prismatic geometry of the N6 environment of the cobalt(II) ion is a consequence of “rigidity” of the 2,6-bis(pyrazol-3-yl)pyridine ligand [26] with the angle of bite N(2)Co(1)N(2) equal to 150.2(3). The stabilization of the geometry can also be contributed by the phenyl substituents in position 1 of the pyrazole cycle. Two these substituents of the same ligand in the complex are arranged above and under the plane of the pyridine ring of another ligand (Fig. 2b) to form intramolecular stacks with a minimum C···C distance of ~3.5 Å and a dihedral angle between the planes of ~10° corresponding to the strong stacking interaction. It is most likely that the latter results in the unexpected turn of the phenyl group relative to the pyrazole cyclane plane at ~50° (Fig. 2a).

View of complex I illustrating (a) the angle between the planes of the ligands and (b) intramolecular stacking interaction. Parameters θ and b are the dihedral angle between the root-mean-square planes of the bis(pyrazol-3-yl)pyridine ligands and the shortest C⋅⋅⋅C distance between the planes of the phenyl and pyridine substituents, respectively.
Thus, the X-ray diffraction data unambiguously indicate that at 120 K the cobalt(II) ion in complex I exists in the high-spin state. It can be assumed that the presence of the phenyl and 4-tert-butylphenyl substituents in complexes II and III would result in a similar trigonal prismatic distortion of the coordination polyhedral cobalt(II) ion stabilizing its high-spin state.
The high-spin state of all the three complexes I–III at ambient temperature was confirmed by the data of NMR spectroscopy, including the use of an original approach to analysis of paramagnetic shifts on the basis of quantum chemical calculations.
Although NMR spectroscopy did not find wide use for paramagnetic compounds because of a number of difficulties in recording and data interpreting, the information obtained in this case can often be much more detailed than that for diamagnetic compounds commonly studied by the NMR method. For example, the values of paramagnetic shift and relaxation time of the nucleus are directly related to its coordinates relative to the paramagnetic ion, which makes it possible to obtain an information on the structure of the paramagnetic compound in solution [27].
One of the most popular approaches using NMR spectroscopy for studying paramagnetic compounds is the Evans method [28, 29], which allows one to measure the magnetic susceptibility (χ) in solution. The essence of the method is that the magnetic susceptibility of the whole solution changes upon the addition of a paramagnetic compound to the solution, and this change affects, in turn, the chemical shifts in the NMR spectra. The accuracy of the Evans method is fairly low and, as a rule, the inaccuracy of evaluation of the population of spin states exceeds 10% [30].
As an alternative to the Evans method we proposed to obtain the same information directly from chemical shifts in NMR spectra, because each spin state is characterized by a particular set of shifts. The chemical shift observed for a paramagnetic compound consists of two components: diamagnetic (δdia) and paramagnetic. The latter, in turn, is usually divided into the contact (δcont) and pseudo-contact (δpc) ones as follows:
The δcont contribution is caused by the spin density redistribution to the nucleus and is proportional to the spin density as follows:
where giso is the isotropic g tensor, and ρi is the spin density on nucleus i. As a rule, δcont weakens with an increase in the number of bonds separating the nucleus and paramagnetic center and becomes negligible in the case of five to six bonds between them.
The reason for the δpc contribution is the dipolar interaction between the magnetic moments of the nucleus and unpaired electron. As a consequence, its value strictly depends on the mutual arrangement of interacting particles in the space
where r, θ, and φ are the spherical coordinates of the nucleus in the system of coordinates of the χ tensor, and Δχax and Δχrh are the axial and orthorhombic anisotropies of the magnetic susceptibility tensor, respectively. The influence of the interaction of this type can be observed in the NMR spectra at the distances to 60 Å from the paramagnetic ion, which makes it possible to use the pseudo-contact contribution as a source of structural information. This principle formed a basis for the concept of “paramagnetic shift probes” [27]. For molecules with axial symmetry, Eq. (3) is simplified to the following equation:
If paramagnetic chemical shifts for the chosen compound are only pseudo-contact, then their interpretation is a trivial task under the condition that the coordinates of nuclei in the molecule are known (for example, from the X-ray diffraction data). In this case, the problem is solved by the approximation of the experimental chemical shifts at the varied parameter of magnetic susceptibility tensor anisotropy. However, the convergence of the experimental shifts with the calculated ones in terms of this approach is often unsatisfactory, indicating that the contact contribution should be taken into account. The latter can be calculated by the standard methods of quantum chemistry. In particular, as shown in practice, DFT combined with hybrid functionals (PBE0, B3LYP, and others) makes it possible to calculate the contact shifts for the 3d-metal complexes with a sufficient accuracy [31, 32].
For complexes I–III, the chemical shifts of the nuclei in the 1Н and 13С NMR spectra reach several hundreds of ppm (see Experimental), which unambiguously indicates the high-spin state of the cobalt(II) ion (S = 3/2) in these complexes in solution at ambient temperature.
We also compared the observed chemical shifts for complex I with the shifts calculated in the framework of the above described approach for a more reliable interpretation of the NMR data. In this case, the chemical shifts can be calculated using the following equation, which is valid for molecules with axial symmetry, such as complexes I–III:
The values of δcont were obtained from the quantum chemical calculation of complex I (see Experimental) taking into account the high-spin state of the cobalt(II) ion (S = 3/2) according to Eq. (2). The δdia contribution was determined as the corresponding chemical shift of the diamagnetic analog of complex I. Thus calculated chemical shifts were adjusted with the experimentally measured values for complex I (Fig. 3). This confirms the assumption about the high-spin state of the cobalt(II) ion based on an analysis of the chemical shifts in the 1Н and 13С NMR spectra. The value of magnetic susceptibility tensor anisotropy obtained (Δχax = 5.9 × 10–32 m3) is typical of the high-spin cobalt(II) complexes [27], additionally indicating that the obtained result is reliable. The closeness of the NMR chemical shifts of the corresponding nuclei for complexes I–III suggests the same (high-spin) state of the cobalt(II) ion in solution at ambient temperature. This, in turn, indicates the intramolecular nature of the effect, which can be attributed to the presence of the phenyl fragment in all the three complexes.

Experimental and theoretical paramagnetic chemical shifts in the (a) 1Н and (b) 13С NMR spectra of complex I.
Thus, we obtained and characterized the new cobalt(II) complexes with the 2,6-bis(pyrazol-3-yl)pyridine ligands containing the phenyl and 4-tert-butylphenyl group in position 1. The complexes differ in the nature of the substituent (and, correspondingly, its electronic characteristics) in position 5. The X-ray diffraction data (Co–N bond lengths and trigonal prismatic distortion of the coordination polyhedron CoN6) for one of the complexes unambiguously indicate that the cobalt(II) ion in crystal at 120 K exists in the high-spin state (S = 3/2). The absence of the temperature-induced spin transition for the whole series of the synthesized cobalt(II) complexes at 120–300 K was confirmed by the data of NMR spectroscopy at ambient temperature, including the original approach to analysis of paramagnetic shifts based on quantum chemical calculations. To conclude, the proposed modification of the ligand (replacement of the phenyl substituent by 4-tert-butylphenyl in position 1 and introduction of the acetoxy group to position 5) exerts no effect on the spin state of the cobalt(II) ion in the corresponding complexes, which remain high-spin in a range of 120–300 K.
ACKNOWLEDGMENTS
The structures of the synthesized compounds were studied using the equipment of the X-ray Structural Centre at the Nesmeyanov Institute of Organoelement Compounds (Russian Academy of Sciences).
This work was supported by the Russian Science Foundation, project no. 17-13-01456.
REFERENCES
Krivokapic, I., Zerara, M., Daku, M.L., et al., Coord. Chem. Rev., 2007, vol. 251, no. 3, p. 364.
Goodwin, H.A., Spin Crossover in Transition Metal Compounds II, Berlin: Springer, 2004.
Charpin, P., Nierlich, M., Vigner, D., et al., J. Chem. Crystallogr., 1988, vol. 18, no. 4, p. 429.
Figgis, B., Gerloch, M., Lewis, J., et al., J. Chem. Soc. A, 1968, p. 2086.
Onggo, D., Rae, A.D., and Goodwin, H.A., Inorg. Chim. Acta, 1990, vol. 178, no. 2, p. 151.
Sieber, R., Decurtins, S., and Stoeckli-Evans, H., Chem.-Eur. J., 2000, vol. 6, no. 2, p. 361.
Stoufer, R.C., Busch, D.H., and Hadley, W.B., J. Am. Chem. Soc., 1961, vol. 83, no. 17, p. 3732.
Figgins, P.E. and Busch, D.H., J. Am. Chem. Soc., 1960, vol. 82, no. 4, p. 820.
Judge, J.S. and Baker, W., Inorg. Chim. Acta, 1967, vol. 1, p. 68.
Harris, C., Lockyer, T., Martin, R., et al., Aust. J. Chem., 1969, vol. 22, no. 10, p. 2105.
Fisher, H.M. and Stoufer, R.C., Inorg. Chem., 1966, vol. 5, no. 7, p. 1172.
Simmons, M.G. and Wilson, L.J., Inorg. Chem., 1977, vol. 16, no. 1, p. 126.
Hathcock, D.J., Stone, K., Madden, J., and Slattery, S.J., Inorg. Chim. Acta, 1998, vol. 282, no. 2, p. 131.
Constable, E.C., Housecroft, C.E., Kulke, T., et al., Dalton Trans., 2001, no. 19, p. 2864.
Coronado, E., Giménez-López, M.C., Gimenez-Saiz, C., et al., Polyhedron, 2003, vol. 22, no. 14, p. 2375.
Clemente-León, M., Coronado, E., Giménez-López, M.C., and Romero, F.M., Inorg. Chem., 2007, vol. 46, no. 26, p. 11266.
Hynes, M.J. and O’Shea, M.T., Dalton Trans., 1983, no. 2, p. 331.
Jansen, M. and Dannhardt, G., Eur. J. Med. Chem., 2003, vol. 38, no. 10, p. 855.
Polezhaev, A.V., Chen, C.-H., Kinne, A.S., et al., Inorg. Chem., 2017, vol. 56, no. 16, p. 9505.
Nelyubina, Yu.V., The 11th Japanese-Russian Workshop on Open Shell Compounds and Moleuclar Spin Devices, 2017.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, no. 1, p. 112.
Perdew, J.P., Burke, K., and Ernzerhof, M., Phys. Rev. Lett., 1996, vol. 77, no. 18, p. 3865.
Weigend, F. and Ahlrichs, R., Phys. Chem. Chem. Phys., 2005, vol. 7, no. 18, p. 3297.
Adamo, C. and Barone, V., J. Chem. Phys., 1999, vol. 110, no. 13, p. 6158.
Spin-Crossover Materials: Properties and Applications, Halcrow, M.A., Ed., New York: Wiley, 2013.
Alvarez, S., J. Am. Chem. Soc., 2003, vol. 125, no. 22, p. 6795.
Bertini, I., Luchinat, C., Parigi, G., and Ravera, E., NMR of Paramagnetic Molecules: Applications to Metallobiomolecules and Models, Elsevier, 2016.
Evans, D., J. Chem. Soc., 1959, p. 2003.
Piguet, C., J. Chem. Educ., 1997, vol. 74, no. 7, p. 815.
Yatsunyk, L.A. and Walker, F.A., Inorg. Chem., 2004, vol. 43, no. 2, p. 757.
Novikov, V.V., Pavlov, A.A., Nelyubina, Y.V., et al., J. Am. Chem. Soc., 2015, vol. 137, no. 31, p. 9792.
Pavlov, A.A., Savkina, S.A., and Belov, A.S., Inorg. Chem., 2017, vol. 56, p. 6943.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Pavlov, A.A., Belov, A.S., Savkina, S.A. et al. Synthesis and Spin State of the Cobalt(II) Complexes with Substituted 2,6-Bis(pyrazol-3-yl)pyridine Ligands. Russ J Coord Chem 44, 489–495 (2018). https://doi.org/10.1134/S1070328418080067
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328418080067