
Abstract
Sugar hydrazones were synthesized by condensation of pyrimidine-2-thione with aldopentoses, aldohexoses and ketohexose. The structures of the new pyrimidines were determined based on their spectral data and elemental analysis. The geometries of the E and Z isomers of the imino group were optimized at B3LYP/6-311G level of theory. DFT results indicated that E-isomer more stable than its Z-isomer in both gas phase and DMSO. The physicochemical properties of the pyrimidines were evaluated using Molsoft tools. The antimicrobial activity of the newly synthesized compounds was evaluated, and they showed good activity. Pyrimidine-hydrazone of D-mannose moiety showed the highest activity against Escherichia coli strain with MIC value 8 μg/mL. Additionally, molecular docking studies were performed on enoyl reductase from Escherichia coli active sites. The docking score of the ligands ranged between –6.8932 to –8.1090 kcal/mol. Moreover, molecular interaction studies revealed that enoyl reductase had strong hydrogen bonding interactions with pyrimidine ligands. Global chemical descriptors of pyrimidines were calculated to predict their reactivity and correlated with the docking score data.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Pyrimidine ring is of interest because it is present in the main skeleton of alkaloids and nucleic bases such as cytosine, thymine and uracil. Moreover, pyrimidine scaffolds possess a wide diversity of therapeutic properties including anticancer [1, 2], antibacterial [3, 4], antifungal [5], antiviral [2, 6], anti-inflammatory [7] and central nervous activities [8]. Furthermore, pyrimidine derivatives also showed different pharmacological activities like antitumor [9], analgesic [10] and antiallergic [11].
Hydrazones have attracted the intensive attentions of researchers in many fields of chemical, pharmaceutical, and life sciences in recent years [12–14]. This motif contains the toxophoric group (–HC=N–NH–) which could be formed by condensation of aldehydes/ketones with hydrazines in organic solvents such as methanol, ethanol, dichloromethane and others. Hydrazones derived from various heterocycles were another group of chemical agents used in many pharmaceuticals as well as biological activities and organic syntheses [15]. They also possess antimicrobial effects [16–18], anticonvulsant [19], antiproliferative [20], anti-inflammatory [21], antiviral [22], antimalarial [23] and antioxidant [24]. More importantly, hydrazones derivatives have been recorded to have anti-HIV [25] as well as anticancer properties [26]. Additionally, they have been widely used in DNA-binding agents, as diagnostic agents for medical purposes as well as genomic research [27].
The sugar moiety usually alters the pharmacological properties of the parent scaffold and also responsible for certain interactions with molecular targets [28–30]. Sugar-containing molecules are clinically useful as anti-infectives, anti-proliferative, and other forms of human diseases.
Both pyrimidines and sugar-hydrazones exhibit an array of antibacterial properties. More importantly, they both show promise for use in pharmaceutical and medicinal applications. In the following article, we attempt to design, synthesis and evaluation of a new library of antibacterial candidates, making it clear that the synthesis of these molecules can lead to new drug prototypes. Our design strategy based on the synthesis of pyrimidine-sugar-hydrazones. The substitution pattern was rationalized to mimic the structural features of antibacterial FDA approved drugs Trimethoprim, Furazolidone and Neomycin (Fig. 1).

Design strategy for the pyrimidine-sugar hydrazone as a new antibacterial agent.
All newly synthesized compounds were screened for the antibacterial and antifungal activity. Additionally, the attempts are also made to determine the mechanism of action of these molecules by finding the binding modes of synthesized ligands with enoyl reductase using docking studies. Finally, physicochemical properties of the tested compounds were computationally predicted as well as the optimized geometry to determine their stability.
RESULTS AND DISCUSSION
Chemistry
Owing to biological and pharmacological activities of pyrimidine compounds, we make multiple attempts for functionalization of pyrimidine at position 2. The synthetic pathway for the target pyrimidine-based compounds (III, IV) is simple, straight forward and outlined in Scheme 1. The structures of the synthesized pyrimidines were confirmed by their IR, 1H NMR, mass spectra, and elemental analysis.

Scheme 1 . Synthetic pathway of pyrimidine hydrazones (IIIa–f) and (IV).
The pyrimidine ring moiety was designed via multicomponent one pot Biginelli reaction by cyclocondensation of ethyl cyanoacetate, thiourea and benzaldehyde in the presence of ethanolic potassium carbonate followed by acidification using acetic acid to give compound (I). The electrophilic character of pyrimidine ring at position 2 due to presence of two adjacent electronegative nitrogen atoms promote a substitution reaction that allow the existence of hydrophilic parts required to bind via various electrostatic interactions and hydrogen bonding.
Treatment of (I) with hydrazine hydrate yielded 2‑hydrazinopyrimidine (II). Subsequent reaction of (II) with a variety of aldpentoses [namely, D-ribose, D-arabinose, D-xylose], aldohexoses [namely, D-galactose, D-glucose and D-mannose] and ketohexose like fructose yielded the corresponding hydrazones (IIIa–f) and (IV), respectively in a very good yields.
The FT-infrared spectra of compounds (IIIa–f) and (IV) showed broad absorption band in the region 3500–3395 cm–1 attributed to the overlapping between hydroxyl group of sugar moiety and NH groups. The carbonitrile group exhibited a medium absorption bands at 2235–2210 cm–1, however, the carbonyl of the lactam moiety showed strong intensity bands at 1669–1631 cm–1 [31]. Furthermore, compounds (IIIa–f) and (IV) exhibited an additional absorption bands at range 3131–3038 and 2974–2921 cm–1 corresponds to stretching of aromatic and aliphatic C–H bond, respectively [32, 33]. The 1H NMR spectral data of (IIIa–f) and (IV) were recorded in DMSO-d6. The 1H NMR spectra revealed the presence of two broad exchangeable singlets at range δ 12.41–11.24 and 11.12–9.81 ppm due to the presence of pyrimidine NH and hydrazone NH, respectively. The azomethine proton (CH=N) appeared either doublet signal at δ 7.66, 7.58 and 7.58 ppm as in compounds (IIIa), (IIId) and (IIIe) or overlapped with aromatic protons as in compounds (IIIb), (IIIc) and (IIIf). Additionally, 1H-NMR spectra exhibited two multiplets at δ 7.79–7.75 and 7.53–7.48 ppm were assigned to aromatic protons. Moreover, 1H-NMR spectra revealed the existence of broad exchangeable singlets at 6.05–3.51 ppm for OH protons whereas the alditolyl protons observed at the shielding area 6.05–2.90 ppm.
The mass spectrum of hydrazones (IIIa–c) showed molecular ion peak at m/z = 359 corresponding to [M]+, which confirms the proposed structure and their base peaks were observed at m/z 43 for compounds (IIIa) and (IIIb), and m/z 170 for compound (IIIc). Similarly, the molecular ion peak of hydrazones (IIId–f) showed molecular ion peak at 389 and their base peak was observed at m/z 43. The fragmentation pattern of (IIIa–f) is shown Scheme S1.
The mass spectra have one main fragmentation route which involves breaking of the sugar-carbonitrile moiety to yield the 2-amino-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile fragment (i) (13–89%). The radical cation fragment (i) can tautomerize to the pyrimidine ring (ii). Removal of cyanate radical from species (ii) led to the formation of the 2-amino-4-phenyl-pyrimidine cation (iii) as a strong intense peak (15–100%). Expulsion of cyanide radical and a hydrogen molecule from radical cation (ii) gives the cation (iv) as a moderate intense peak (4–30%). Elimination of cyanate radical from cation (iv) gives the 4-phenyl-(1H)-imidazole radical cation (v) as a moderate intense peak (10–46%). The removal of a CH radical from 4-phenyl-(1H)-imidazole radical cation (v) gives the 3-phenyl-1,2-diazete cation (vi) (7–25%). Expulsion of a nitrogen atom from species (vi) led to the formation of the radical cation species (vii) (3–30%). The radical cation species (viii) (51–58%) can be obtained by elimination of cyanide radical and a carbon atom from the radical cation (v). Removal of hydrogen cyanide from species (viii) affords the phenyl cation (ix) as a moderate intense peak (28–55%). Elimination of acetylene molecule from species (ix) affords the cyclobutadienyl cation (x) as a moderate intense peak (24–54%). In another route, species (ix) losses three carbon atoms and gain a hydrogen molecule at the same step affording propyl cation (xi) as a very strong intense peak (39–100%) which then gain a hydrogen atom affords the cation (xii) as a very strong intense peak too (54–100%). Finally, the repulsion of a hydrogen molecule from the cation (xii) gives the cation (xiii) as a strong intense peak (51–56%), Scheme S1 (see Supplementary Information).
DFT Studies
It is noticeable that the stereochemistry of hydrazones was investigated and reported to exist in a mixture of geometrical isomerism (E/Z) around the azomethine linkage (–C=N–) [34, 35], Scheme 2. The ratio of these isomers depends on several factors like nature of the solvent, their chelation ability, the position and the electronic effect of the attached substituents [36]. As predicted, the proposed structures of hydrazones (IIIa–f) and (IV) were consistent with the previously reported results for substituted hydrazones [37–39], which were proved to exist in the E-configuration of the C=N bond in polar solvent (DMSO solution) due to steric hindrance on the imine bond (Scheme 2).

Scheme 2 . E and Z isomers of hydrazones (IIIa–f).
The stability of hydrazones (IIIa–f) are confirmed by DFT calculations to estimate the optimized structures which carried out in both gas phase and DMSO at B3LYP 6–311G basis set. The calculations were performed for the two isomers (E/Z) of compound (IIIe), as a module, to predict the most stable isomer (Fig. 2). The results of the DFT calculations predicted that E form is the lower energy structure and hence more stable than its isomer by 14.74 and 3.71 kcal/mol in gas phase and DMSO, respectively. However, the small energy difference between the two conformations, either in gas phase or in DMSO, indicates their coexistence in equilibrium.

Optimized molecular structure of (E/Z) isomers of compound (IIIe).
Physicochemical Properties [40–42]
In silico computational methods were used to determine the physicochemical and biochemical properties of ligands (IIIa–f) and (IV). The Lipinski rule of five can be used to predicte the oral bioavailability of a drug candidate. This rule is based on the physicochemical parameters of the tested ligands, including: molecular weight (MW); a partition coefficient (clogP); number of hydrogen bond donors (HBD); and number of hydrogen bond acceptors (HBA). The synthesized ligands (IIIa–f) and (IV) were validated via Lipinski’s rule descriptors. Table 1 indicates that all the tested ligands had a single violation in terms of hydrogen donor. Furthermore, Veber descriptors like number of rotatable bonds (NROTB) and topological polar surface area (TPSA) were used as another predication for oral bioavailability. The NROTB is a measure for the molecular flexibility and for excellent oral bioavailability, NROTB shouldn’t more than ten. All ligands (IIIa–f) and (IV) have less than 10 rotatable bonds. Additionally, TPSA is a good descriptor characterizing drug absorption including bioavailability, blood–brain barrier penetration and intestinal absorption, predicted TPSA values of ligands (IIIa–c) were less than 140 Å2 which is consistent with the value known for most drugs that predict promising oral bioavailability.
Antimicrobial Activity
Hydrazones (IIIa–f) and (IV) are estimated their antimicrobial activity against gram-positive bacteria such as Bacillus subtilis and Staphylococcus aureus, gram-negative bacteria such as Escherichia coli and Proteus vulgaris, in addition to two fungal strains, Candida albicans and Aspergillus flavus. Gentamycin and Ketoconazole were used as antibacterial and antifungal reference drugs, respectively. The results (Table 2) indicated that the tested compounds showed significant antibacterial activity against gram-negative strains relative to gram-positive bacteria, whereas, showed weak antifungal activity. Compounds (IIIf) showed the highest activity against Escherichia coli and only compound (IIId) have considerable antifungal activity.
The minimum inhibitory concentration (MIC) is the lowest concentration causing full inhibition of the tested microorganism’s growth. MIC is a further test applied for selected compounds with good antimicrobial activity. The MIC were determined via the double dilution technique [43]. For each selected compound, six concentrations were prepared (8, 16, 32, 64, 128, and 256 μg/mL). Table 3 displays the MIC values of the tested compounds for different microorganisms. As shown in Table 3, compound (IIIf) exhibited the highest antibacterial activity against Escherichia coli with MIC 8 μg/mL. On the other hand, compound (IIId) had the highest antibacterial activity against Bacillus subtilis with MIC 16 μg/mL. Moreover, Compound (IIIb) presented the highest antibacterial activity against Proteus vulgaris with MIC 8 μg/mL.
Molecular Docking Studies
The significant experimental antibacterial activities of compounds (IIIa–e) provide a hint for conducting molecular docking studies to determine the protein–ligand interactions. The docking was performed via the MOE (2015) for the active sites of enoyl reductase from E. coli (PDB: 1C14 [44]) to predict the binding mode and support the biological results. Bacterial enoyl reductase catalyzes an essential step in biosynthesis of bacterial fatty acid. Enoyl reductase is an attractive target for antibacterial drug discovery due to its important role in the metabolism and its sequence conservation across many bacterial species. After reviewing the different chemical classes that docked into the enoyl reductase active site, such as amides, triazoles, pyridine and pyrimidine derivatives [45–48], we examined our compounds on the active site because some structural similarities exist.
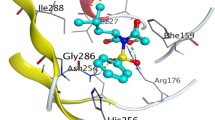
The hydrazones (IIIa–f) interact with enoyl reductase amino acids of its active site and give scores ranging from –6.8932 to –8.1090 kcal/mol (Table 4). Molecular interactions (Fig. 3) showed that the potential drug binding sites of enoyl reductase are GLY 13, SER 19, GLY 93, SER 91, VAL 65, ILE 92, TYR 146, ALA 95, MET 159 and GLN 40. The best score value was noticed for candidate (IIIf) (–8.1090 kcal/mol) which has the best biological activity and this supported by presence of five interactions with amino acids of enoyl reductase involving two hydrogen bonds donor between the two NH groups in compound (IIIf) and amino acid residue at the backbone of the enzyme namely GLY 93 as well as two hydrogen bonds acceptor, one between carbonitrile nitrogen with VAL 65 and the other between oxygen of hydroxyl group and SER 91. The last interaction is pi-H between phenyl moiety of (IIIf) and GLN 40 residue (Fig. 3), (IIIf). Moreover, ligand (IIIc) showed the lowest enoyl reductase inhibition among the nominated ligands, displayed three interaction including hydrogen bond donor with GLY 93, hydrogen bond acceptor with SER 19 and H-pi binding interaction with the active site at TYR 146 via its phenyl ring.

2D and 3D binding modes of (IIIa–f) (red tube) in enoyl reductase active sites (PDB: 1C14 [44]).

(Contd.)
Quantum Chemical Parameters
The DFT method were used to calculate the descriptor parameters [49–52] of hydrazones (IIIa–f) and (IV) to predict their reactivity and correlate with the docking score data. The molecular descriptors were calculated by using B3LYP/6-311G level and given in Table S1 at gas phase (see supplementary material).
The global chemical reactivity descriptors were calculated from HOMO and LUMO energies. They are namely [53–59] ionization potential (Ip), electron affinity (EA), electronegativity (χ), chemical potential (μ), hardness (η), softness (S) and the electrophilicity (ω). According to these parameters, the chemical reactivity varies with the structural configuration of molecules.
According to data in Table S1, we can deduce that hydrazone (IV) has the highest reactivity based on parameter ∆E, whereas (IIIc) is the highest depending on parameter S. The data points out that each parameter exhibits poor to good correlation with score of docking studies as indicated from correlation coefficient (0.2406–0.5926).
EXPERIMENTAL
Instruments and Apparatus
Melting points were determined by MEL-TEMP II melting point apparatus in open glass capillaries. The IR spectra were recorded as potassium bromide (KBr) discs on a Perkin-Elemer FT-IR, Faculty of Science, Alexandria University. The NMR spectra were carried out at ambient temperature (~25°C) on a (JEOL) 500 MHz spectrophotometer using tetramethylsilane (TMS) as an internal standard, NMR Unit, Faculty of Science, Mansoura University. Mass spectra and elemental analyses were analyzed at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt.
Agar Disk-Diffusion Method
The hydrazones were dissolved in DMSO (which has no inhibition activity) to obtain concentrations of 250 ppm and soaked in filter paper disks of 5 mm. The test was performed on medium potato dextrose agar (PDA) which contains an infusion of 200 g potatoes, 6 g dextrose, and 15 g agar. Uniform size filter paper disks (three disks per compound) were impregnated with equal volume (10 μL) from the specific concentration of dissolved tested compounds and then carefully placed on the incubated agar surface. After incubation for 36 h at 27°C in the case of bacteria and for 48 h at 24°C in the case of fungi, inhibition of the organisms (evidenced by a clear zone surrounding each disk) was measured and used to calculate the mean of inhibition zones [60].
Determination of MIC
All the bacteria were incubated and activated at 37°C for 24 h inoculation into nutrient broth and the fungi were incubated in malt extract broth for 48 h. The compounds were dissolved in DMSO and then diluted using cautiously adjusted Mueller–Hinton broth. Two-fold serial concentrations dilution method (8, 16, 32, 64, 128, and 256 µg/mL) of some compounds were employed to determine the MIC values. In each case, triplicate tests were performed, and the average was taken as the final reading. The tubes were then inoculated with the test organisms, grown in their suitable broth at 37°C for 24 h for tested microorganisms (1 × 108 CFU/mL for bacteria and 1 × 106 CFU/mL of fungi), each 5 mL received 0.1 mL of the above inoculum and was incubated at 37°C for 24 h.
Docking Program
Molecular docking simulations were performed to achieve the mode of interaction of prepared pyrimidines with the binding pocket of enoyl reductase. The newly released crystal structure of enoyl reductase as a receptor was retrieved from protein data bank (www.rcsb.org) with PDB ID: 1C14 [44]. Software version 2015.10 of Molecular Operating Environment (MOE) was used to prepare the input files and analyzing the result. All water molecules, ligands and ions were removed from pdb file for the preparation of protein input file. The active site was selected utilizing ‘Site Finder’ MOE 2015.10 feature. Prior to docking, the hydrazone structures were subjected to energy minimization and geometry optimization before docking. Docking simulations were conducted several times with various fitting protocols to observe the best molecular interactions and free binding energies. All docking results were sorted by scoring binding energy.
Synthesis of pyrimidines (IIIa–f) and (IV). 2-Thioxo-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (I). A mixture of ethylcyanoacetate (1.13 g, 0.01 mol), thiourea (0.76 g, 0.01 mol) and benzaldehyde (1.06 g, 0.01 mol) in ethanol (20 mL) containing potassium carbonate (1.38 g, 0.01 mol) was heated at reflux for 5 h. The precipitated potassium salt was collected by filtration and washed with ethanol. Dissolve the potassium salt in hot water (80°C) and stir until a clear solution is obtained. The solution is then acidified with acetic acid and continues stirring for 5 min. The deposited precipitate formed is collected and washed well with water, dried and crystalized from ethanol to give yellowish-white crystal; yield: 80%; mp: 311–314°C [lit. 300–302°C] [61]; TLC in 1 : 1 methanol-chloroform, Rf: 0.69; 1H NMR (500 MHz, DMSO-d6): δ 12.82 (brs, 2H, 2NH), 7.66 (d, J = 7.6 Hz, 2H, o-phenyl-H), 7.63 (t, 1H, J = 8.1 Hz, p-phenyl-H) and 7.56 ppm (t, 2H, J = 7.9 Hz, m-phenyl-H); 13C NMR (126 MHz, DMSO-d6): δ 176.30, 161.06, 158.58, 132.13, 129.44, 128.76, 128.48, 113.81 and 90.71 ppm.
2-Hydrazino-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (II). A mixture of 2-thioxo-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (I) (2.29 g, 0.01 mol) and hydrazine hydrate (2.5 g, 0.05 mol) in ethanol (50 mL) was heated at reflux for 10 h. The precipitate was filtrated and washed with ethanol without need for further purification to form yellow crystal; yield: 75%; mp: 227–230°C [lit. 237°C] [62]; TLC in 1 : 1 methanol–chloroform, Rf: 0.78; 1H NMR (500 MHz, DMSO-d6): δ 12.72 (brs, 1H, NH-pyrimidine), 7.77 (d, J = 7.6 Hz, 2H, phenyl-H), 7.49 (m, 3H, phenyl-H) and 7.01 ppm (brs, 3H, NH–NH2); 13C NMR (126 MHz, DMSO-d6): δ 170.17, 160.20, 137.31, 130.37, 128.19, 128.11 and 118.80 ppm.
Preparation of sugar-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIa–f) and (IV). A solution of 2-hydrazino-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile (II) (1.14 g, 0.005 mol) in ethanol (30 mL) was added to the appropriate solution of pentoses (namely, D-xylose, D-arabinose or D-ribose), D-hexoses [namely, D-fructose, D-galactose, D-glucose or D-mannose] in water (2 mL). The reaction mixtures were refluxed on water bath for 4–6 h (monitored by TLC) and then kept at room temperature for 24 h. The crystalline product which separated was filtered, washed with water and crystalized from ethanol.
D-Xylose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIa). Yellowish-white crystal; yield: 79%; mp: 188–190°C; TLC in 1 : 1 methanol–chloroform, Rf: 0.72; IR: 3438 (OH and NH), 2217 (C≡N) and 1631 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 11.24 (brs, 1H, NH), 10.12 (s, 1H, =N–NH–), 7.77 (m, 2H, phenyl-H), 7.66 (d,1H, J = 8.1 Hz, –CH=N–), 7.51 (m, 3H, phenyl-H), 6.07 (d, 1H, J = 5.1 Hz, alditolyl H), 5.42 (brs, 1H, OH), 5.03 (d, 1H, J = 6.2 Hz, alditolyl-H), 5.00 (m, 1H, OH), 3.75 (m, 2H, 2OH) and 3.65 ppm (m, 2H, alditolyl-H). The other alditolyl protons were associated with the solvent absorption in a large signal centered at δ 3.38 ppm; mass spectrum: molecular ion peak m/z 359, base peak m/z: 43.09. C16H17N5O5 requires: C: 53.46; H: 4.77; N: 19.49% found: C: 53.72; H: 4.39; N: 19.21%.
D-Arabinose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIb). Yellowish-white crystal; yield: 81%; mp: 223–225°C; TLC in 1 : 1 methanol–chloroform, Rf: 0.83; IR: 3461 (OH and NH), 2213 (C≡N) and 1669 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 12.37 (brs, 1H, ‒NH), 11.12 (s, 1H, =N–NH–), 7.79 (m, 2H, phenyl-H), 7.53 (m, 4H, –CH=N– and phenyl-H), 5.10 (m, 1H, OH), 3.64 (m, 2H, 2OH), 3.47 (s, 1H, alditolyl-H), 3.39 (s, 1H, alditolyl-H) and 3.57 ppm (s, 2H, alditolyl-H). The other alditolyl protons were associated with the solvent absorption in a large signal centered at δ 3.50 ppm; mass spectrum: molecular ion peak m/z 359, base peak m/z 43.10. C16H17N5O5 requires: C: 53.46; H: 4.77; N: 19.49% found: C: 53.22; H: 4.89; N: 19.77%.
D-Ribose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIc). Yellowish-brown crystal; yield: 63%; mp: 185°C; TLC in 1 : 1 methanol- chloroform, Rf: 0.83, IR: 3392 (OH and NH), 2210 (C≡N) and 1666 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 12.41 (brs, 1H, –NH), 9.81 (s, 1H, =N–NH–), 7.76 (m, 2H, phenyl-H), 7.49 (m, 4H, –CH=N– and phenyl 3H), 5.32 (s, 1H, OH), 3.95 (s, 1H, OH), 3.76 (s, 1H, OH), 3.44 (s, 1H, alditolyl-H) and 3.57 ppm (s, 2H, alditolyl-H). The other alditolyl protons were associated with the solvent absorption in a large signal centered at δ 3.50 ppm; mass spectrum: molecular ion peak m/z 359, base peak m/z 170.13. C16H17N5O5 requires: C: 53.46; H: 4.77; N: 19.49% found: C: 53.34; H: 4.62; N: 19.67%.
D-Galactose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIId). Yellowish-white crystal; yield: 94%; m.p.: 225–227°C; TLC in 1:1 methanol-chloroform, Rf: 0.78; IR: 3395 (OH and NH), 2218 (C≡N) and 1648 cm–1 (C=O), 1H NMR (500 MHz, DMSO-d6): δ 12.36 (brs, 1H, –NH), 10.08 (s, 1H, =N–NH–), 7.78 (m, 2H, phenyl-H), 7.58 (d, 1H, J = 7.9 Hz, –CH=N–), 7.51 (m, 3H, phenyl-H), 7.05 (m, H, alditolyl-H), 5.04 (m, 1H, OH), 3.56 (m, 1H, OH), 3.49 (m, 1H, alditolyl-H) 3.47 (m, 1H, OH), 3.15 (m, 1H, alditolyl-H) and 3.69 ppm (m, 2H, 2OH). The other alditolyl protons were associated with the solvent absorption in a large signal centered at δ 3.45 ppm; mass spectrum: molecular ion peak m/z 389, base peak m/z 43.07. C17H19N5O6 requires: C: 52.43; H: 4.92; N: 17.98% found: C: 52.34; H: 4.73; N: 17.85%.
D-Glucose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIe). Yellow powder; yield: 73%; mp: 148–150°C; TLC in 1 : 1 methanol-chloroform, Rf: 0.67; IR: 3295 (OH and NH), 2235 (C≡N) and 1669 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 11.51 (brs, 1H, –NH), 10.07 (s, 1H, =N–NH–), 7.75 (m, 2H, phenyl-H), 7.49 (m, 4H, ‒CH=N– and phenyl 3H), 6.05 (d, 1H, OH), 5.37 (d, 1H, OH), 5.02 (m, 2H, 2OH), 3.52 (m, 1H, OH) and 3.78 (m, 1H, alditolyl-H), 3.64 (m, 1H, alditolyl-H), 3.32 (m, 1H, alditolyl-H), 3.15 (m, 1H, alditolyl-H), 3.02 (m, 1H, alditolyl-H) and 2.91 (m, 1H, alditolyl-H) ppm; mass spectrum: molecular ion peak m/z 389, base peak m/z 43.13. C17H19N5O6 requires: C: 52.43; H: 4.92; N: 17.98% found: C: 52.65; H: 5.12; N: 18.15%.
D-Mannose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IIIf). Yellowish-brown powder; yield: 85%; mp: 180°C; TLC in 1 : 1 methanol-chloroform, Rf: 0.52; IR: 3315 (OH and NH), 2212 (C≡N) and 1661 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 12.17 (br, 1H, –NH), 9.88 (s, 1H, =N–NH–), 7.79 (m, 2H, phenyl-H), 7.49 (m, 4H, –CH=N– and phenyl-H), 5.26 (d, 1H, OH), 3.54 (m, 2H, 2OH), 3.45 (m, 1H, OH), 3.17 (m, 1H, alditolyl-H), 3.61 (m, 1H, alditolyl-H), 3.53 (m, 1H, alditolyl-H), 3.47 (m, 1H, alditolyl-H) and 3.36 (m, 1H, alditolyl-H) ppm. The other alditolyl protons were associated with the solvent absorption in a large signal centered at δ 3.46 ppm; mass spectrum: molecular ion peak m/z 389, base peak m/z 43.10. C17H19N5O6 requires: C: 52.43; H: 4.92; N: 17.98% found: C: 52.31; H: 5.07; N: 17.85%.
D-Fructose-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile-2-hydrazone (IV). Yellowish-white powder; yield: 58%; mp: 120–123°C; TLC in 1 : 1 methanol-chloroform, Rf: 0.69; IR: 3627 (OH and NH), 2234 (C≡N) and 1666 cm–1 (C=O); 1H NMR (500 MHz, DMSO-d6): δ 11.50 (brs, 1H, –NH), 10.05 (s, 1H, =N–NH–), 7.76 (m, 2H, phenyl-H), 7.48 (m, 3H, phenyl-H), 6.05 (s, 1H, OH), 5.00 (m, 2H, 2OH), 3.51 (m, 1H, OH), 3.78 (m, 1H, alditolyl-H), 3.64 (m, 1H, alditolyl-H), 3.32 (m, 1H, alditolyl-H), 3.15 (m, 2H, alditolyl-H), 3.02 (m, 2H, alditolyl-H) and 2.90 (m, 2H, alditolyl-H) ppm; mass spectrum: molecular ion peak m/z 389, base peak m/z 170.12. C17H19N5O6 requires: C: 52.43; H: 4.92; N: 17.98% found: C: 52.55; H: 4.78; N: 17.73%.
CONCLUSIONS
In the present study, hydrazones containing pyrimidine moiety (IIIa–f) and (IV) were synthesized and characterized. DFT studies indicated that E-isomer of imino group (CH=N) more stable than its Z-isomer in both gas phase and DMSO. The compounds were evaluated for their antimicrobial activity against gram-positive, gram-negative, and fungi by the agar well diffusion method, some of the synthesized compounds showed a significant antibacterial activity. Molecular docking studies were carried out on enoyl reductase from E. coli active sites. The MOE scores of hydrazones (IIIa–f) ranged between –6.8932 to ‒8.1090 kcal/mol in which the highest antibacterial effect have the highest score value.
REFERENCES
Abdel-Mohsen, H.T., Ragab, F.A., Ramla, M.M., and El Diwani, H.I., Eur. J. Med. Chem., 2010, vol. 45, pp. 2336–2344. https://doi.org/10.1016/j.ejmech.2010.02.011
El-Sayed, W.A., Ramiz, M.M., and Abdel-Rahman, A.A.-H., Z. Naturforsch. C, 2009, vol. 64, pp. 323–328. https://doi.org/10.1515/znc-2009-5-603
El-Sayed, W.A. and Abdel-Rehman, A.A.-H., Z. Naturforsch. B, 2010, vol. 65, pp. 57–66. https://doi.org/10.1515/znb-2010-0110
Ramiz, M.M.M., El-Sayed, W.A., El-Tantawy, A.I., and Abdel-Rehman, A.A.-H., Arch. Pharm. Res., 2010, vol. 33, pp. 647–654. https://doi.org/10.1007/s12272-010-0501-1
Gholap, A.R., Toti, K.S., Shirazi, F., Deshpande, M.V., and Srinivasan, K.V., Tetrahedron, 2008, vol. 64, pp. 10214–10223. https://doi.org/10.1016/j.tet.2008.08.033
El-Sayed, W.A., Rashad, A.E., Awad, S.M., and Ali, M.M., Nucleosides Nucleotides Nucleic Acids, 2009, vol. 28, pp. 261–274. https://doi.org/10.1080/15257770902946165
Falcão, E.P., de Melo, S.J., Srivastava, R.M., de Catanho, M.T.J., and Nascimento, S.C.D., Eur. J. Med. Chem., 2006, vol. 41, pp. 276–282. https://doi.org/10.1016/j.ejmech.2005.09.009
Gillespie, R.J., Bamford, S.J., Clay, A., Gaur, S., Haymes, T., Jackson, P.S., Jordan, A.M., Klenke, B., Leonardi, S., and Liu, J., Bioorg. Med. Chem., 2009, vol. 17, pp. 6590–6605. https://doi.org/10.1016/j.bmc.2009.07.078
Kreutzberger, A. and Schimmelpfennig, H., Arch. Pharm., 1981, vol. 314, pp. 34–41. https://doi.org/10.1002/ardp.19813140107
Ueda, T., Sakakibara, J., and Nakagami, J., Chem. Pharm. Bull., 1983, vol. 31, pp. 4263–4269. https://doi.org/10.1248/cpb.31.4263
Ozeki, K., Ichikawa, T., Takehara, H., Tanimura, K., Sato, M., and Yaginuma H., Chem. Pharm. Bull., 1989, vol. 37, pp. 1780–1787. https://doi.org/10.1248/cpb.37.1780
Emregül, K.C., Düzgün, E., and Atakol O., Corr. Sci., 2006, vol. 48, pp. 3243–3260. https://doi.org/10.1016/j.corsci.2005.11.016
Cozzi, P.G., Chem. Soc. Rev., 2004, vol. 33, pp. 410–421. https://doi.org/10.1039/B307853C
Fakhari, A.R., Khorrami, A.R., and Naeimi, H., Talanta, 2005, vol. 66, pp. 813–817. https://doi.org/10.1016/j.talanta.2004.12.043
Kaymakçıoğlu, B.K. and Rollas, S., Il Farmaco, 2002, vol. 57, pp. 595–599. https://doi.org/10.1016/S0014-827X(02)01255-7
Ma, C., Wang, Q., and Zhang, R., Heteroat. Chem., 2008, vol. 19, pp. 583–591. https://doi.org/10.1002/hc.20481
El-Atawy, M.A., Hamed, E.A., Alhadi, M., and Omar A.Z., Molecules, 2019, vol. 24, pp. 4198–4211. https://doi.org/10.3390/molecules24224198
Omar, A.Z., Mosa, T.M., El-Sadany, S.K., Hamed, E.A., and El-Atawy, M., J. Mol. Struct., 2021, vol. 1245, pp. 131020–131028. https://doi.org/10.1016/j.molstruc.2021.131020
Küçükgüzel, I., Küçükgüzel, Ş.G., Rollas, S., Ötük-Sanış, G., Özdemir, O., Bayrak, I., Altuğ, T., and Stables J.P., Il Farmaco, 2004, vol. 59, pp. 893–901. https://doi.org/10.1016/j.farmac.2004.07.005
Vicini, P., Geronikaki, A., Incerti, M., Busonera, B., Poni, G., Cabras, C.A., and La Colla P., Bioorg. Med. Chem., 2003, vol. 11, pp. 4785–4789. https://doi.org/10.1016/S0968-0896(03)00493-0
Salgın-Gökşen, U., Gökhan-Kelekçi, N., Göktaş, Ö., Köysal, Y., Kılıç, E., Işık, Ş., Aktay, G., and Özalp M., Bioorg. Med. Chem., 2007, vol. 15, pp. 5738–5751. https://doi.org/10.1016/j.bmc.2007.06.006
Kumar, K.S., Ganguly, S., Veerasamy, R., and De Clercq, E., Eur. J. Med. Chem., 2010, vol. 45, pp. 5474–5479. https://doi.org/10.1016/j.ejmech.2010.07.058
Aggarwal, S., Paliwal, D., Kaushik, D., Gupta, G.K., and Kumar, A., Comb. Chem. High Throughput Screen., 2018, vol. 21, pp. 194–203. https://doi.org/10.2174/1386207321666180213092911
Gacche, R., Gond, D., Dhole, N., and Dawane, B., J. Enzyme Inhib. Med. Chem., 2006, vol. 21, pp. 157–161. https://doi.org/10.1080/14756360500532671
Savini, L., Chiasserini, L., Travagli, V., Pellerano, C., Novellino, E., Cosentino, S., and Pisano, M.B., Eur. J. Med. Chem., 2004, vol. 39, pp. 113–122. https://doi.org/10.1016/j.ejmech.2003.09.012
Zhang, H.-Z., Drewe, J., Tseng, B., Kasibhatla, S., and Cai, S.X., Bioorg. Med. Chem., 2004, vol. 12, pp. 3649–3655. https://doi.org/10.1016/j.bmc.2004.04.017
Xavier, A. and Srividhya, N., J. Appl. Chem., 2014, vol. 7, pp. 6–15.
Olano, C., Méndez, C., and Salas, J.A., Nat. Prod. Rep., 2010, vol. 27, pp. 571–616. https://doi.org/10.1039/B911956F
Song, M.C., Kim, E., Ban, Y.H., Yoo, Y.J., Kim, E.J., Park, S.R., Pandey, R.P., Sohng, J.K., and Yoon, Y.J., Appl. Microbiol. Biotechnol., 2013, vol. 97, pp. 5691–5704. https://doi.org/10.1007/s00253-013-4978-7
Elshahawi, S.I., Shaaban, K.A., Kharel, M.K., and Thorson, J.S., Chem. Soc. Rev., 2015, vol. 44, pp. 7591–7697. https://doi.org/10.1039/C4CS00426D
Omar, A.Z., Mahmoud, M.N., El-Sadany, S.K., Hamed, E.A., and El-atawy, M.A., Dyes Pig., 2021, vol. 185, pp. 108887–108896. https://doi.org/10.1016/j.dyepig.2020.108887
Alamro, F.S., Ahmed, H.A., El-Atawy, M.A., Al-Zahrani, S.A., and Omar, A.Z., Molecules, 2021, vol. 26, pp. 4546–4557. https://doi.org/10.3390/molecules26154546
Al-Zahrani, S.A., Ahmed, H.A., El-Atawy, M.A., Abu Al-Ola, K.A., and Omar, A.Z., Materials, 2021, vol. 14, pp. 2587–2599. https://doi.org/10.3390/ma14102587
Almehmadi, M.A., Aljuhani, A., Alraqa, S.Y., Ali, I., Rezki, N., Aouad, M.R., and Hagar, M., J. Mol. Struct., 2021, vol. 1225, pp. 129148–129156. https://doi.org/10.1016/j.molstruc.2020.129148
El-Atawy, M.A., Omar, A.Z., Hagar, M., and Shashira, E.M., Green Chem. Lett. Rev., 2019, vol. 12, pp. 364–376. https://doi.org/10.1080/17518253.2019.1646813
D’yakonov, V.A., Finkelshtein, E.S., and Ibragimov A.G., Tetrahedron Lett., 2007, vol. 48, pp. 8583–8586. https://doi.org/10.1016/j.tetlet.2007.10.065
Kumar, P., Kadyan, K., Duhan, M., Sindhu, J., Singh, V., and Saharan, B.S., Chem. Cent. J., 2017, vol. 11, pp. 1–14. https://doi.org/10.1186/s13065-017-0344-7
Patorski, P., Wyrzykiewicz, E., and Bartkowiak G., J. Spectrosc., 2013, article no. 197475. https://doi.org/10.1155/2013/197475
Wyrzykiewicz, E. and Prukała, D., J. Heterocycl. Chem., 1998, vol. 35, pp. 381–387. https://doi.org/10.1002/jhet.5570350221
Fernandes, B.T., Segretti, C.F.M., Polli, C.M., and Parise-Filho, R., Lett. Drug Des. Discov., 2016, vol. 13, pp. 999–1006.
Boutard, N., Białas, A., Sabiniarz, A., Guzik, P., Banaszak, K., Biela, A., Bień, M., Buda, A., Bugaj, B., Cieluch, E., et al., Bioorg. Med. Chem. Lett., 2019, vol. 29, pp. 646–653. https://doi.org/10.1016/j.bmcl.2018.12.034
Chitti, S., Singireddi, S., Reddy, P.S.K., Trivedi, P., Bobde, Y., Kumar, C., Rangan, K., Ghosh, B., and Sekhar, K.V.G.C., Bioorg. Med. Chem. Lett., 2019, vol. 29, pp. 2551–2558. https://doi.org/10.1016/j.bmcl.2019.08.013
Ge, B., Bodeis, S., Walker, R.D., White, D.G., Zhao, S., McDermott, P.F., and Meng, J., J. Antimicrob. Chemother., 2002, vol. 50, pp. 487–494. https://doi.org/10.1093/jac/dkf162
Qiu, X., Abdel-Meguid, S.S., Janson, C.A., Court, R.I., Smyth, M.G., and Payne, D.J., Protein Sci., 1999, vol. 8, pp. 2529–2532. https://doi.org/10.1110/ps.8.11.2529
Takhi, M., Sreenivas, K., Reddy, C.K., Munikumar, M., Praveena, K., Sudheer, P., Rao, B.N., Ramakanth, G., Sivaranjani, J., Mulik, S., et al., Eur. J. Med. Chem., 2014, vol. 84, pp. 382–394. https://doi.org/10.1016/j.ejmech.2014.07.036
Panicker, C.Y., Varghese, H.T., Manjula, P., Sarojini, B., Narayana, B., War, J.A., Srivastava, S., Van Alsenoy, C., and Al-Saadi, A.A., Spectrochim. Acta A, 2015, vol. 151, pp. 198–207. https://doi.org/10.1016/j.saa.2015.06.076
Kini, S.G., Bhat, A.R., Bryant, B., Williamson, J.S., and Dayan, F.E., Eur. J. Med. Chem., 2009, vol. 44, pp. 492–500. https://doi.org/10.1016/j.ejmech.2008.04.013
Hirschbeck, M.W., Kuper, J., Lu, H., Liu, N., Neckles, C., Shah, S., Wagner, S., Sotriffer, C.A., Tonge, P.J., and Kisker, C., Structure, 2012, vol. 20, pp. 89–100. https://doi.org/10.1016/j.str.2011.07.019
Koopmans, T., Physica, 1933, vol. 1, pp. 104–113. https://doi.org/10.1016/S0031-8914(34)90011-2
Kaya S., Kariper S.E., Ungördü A., Kaya C., J. New Res. Sci., 2014, vol. 3, pp. 1–8.
Alexander, D. and Moccari, A., Corrosion, 1993, vol. 49, pp. 921–928.
Sastri,V. and Perumareddi, J., Corrosion, 1997, vol. 53, pp. 617–622.
Eren, B. and Ünal, A., Spectrochim. Acta A, 2013, vol. 103, pp. 222–231. https://doi.org/10.1016/j.saa.2012.10.055
Parr, R.G., Szentpaly, L.v., and Liu, S., J. Am. Chem. Soc., 1999, vol. 121, pp. 1922–1924. https://doi.org/10.1021/ja983494x
Chattaraj, P.K., Maiti, B., and Sarkar, U., J. Phys. Chem. A, 2003, vol. 107, pp. 4973–4975. https://doi.org/10.1021/jp034707u
Flippin, L.A., Gallagher, D.W., and Jalali-Araghi, K., J. Org. Chem., 1989, vol. 54, pp. 1430–1432. https://doi.org/10.1021/jo00267a035
Chattaraj, P.K., and Giri, S., J. Phys. Chem. A, 2007, vol. 111, pp. 11116–11121. https://doi.org/10.1021/jp0760758
Padmanabhan, J., Parthasarathi, R., Subramanian, V., and Chattaraj, P., J. Phys. Chem. A, 2007, vol. 111, pp. 1358–1361. https://doi.org/10.1021/jp0649549
Ayers, P.W. and Parr, R.G., J. Am. Chem. Soc., 2000, vol. 122, pp. 2010–2018. https://doi.org/10.1021/ja9924039
Balouiri, M., Sadiki, M., and Ibnsouda S.K., J. Pharm. Anal., 2016, vol. 6, pp. 71–79. https://doi.org/10.1016/j.jpha.2015.11.005
Kambe, S., Saito, K., Kishi, H., Sakurai, A., and Midorikawa H., Synthesis, 1979, vol. 1979, pp. 287–289.
Hussain, S., El-Barbary, A., and Mansour, S., J. Heterocycl. Chem., 1985, vol. 22, pp. 169–171. https://doi.org/10.1002/jhet.5570220141
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
This article doesn’t contain any studies involving human participants performed by any of the authors and doesn’t contain any studies involving animals performed by any of the authors.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Supplementary Information
Rights and permissions
About this article

Cite this article
Omar, A.Z., El-Aleem, N.G., Megid, S.M. et al. Design, Synthesis, Characterization, DFT Calculations, Molecular Docking Study, and Antimicrobial Activity of Hydrazones Bearing Pyrimidine and Sugar Moieties. Russ J Bioorg Chem 48, 1076–1088 (2022). https://doi.org/10.1134/S1068162022050156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162022050156