
Abstract
Inhibitors of aldose reductase provide a feasible mode of action against diabetic complications. Based on the marketed aldose reductase inhibitor epalrestat containing rhodanine nucleus, rhodanine-3-hippuric acid and its 5-arylidene derivatives were synthesized. The structure of newly synthesized compounds was confirmed by IR, 1H NMR, and 13C NMR spectrometry. In vitro aldose reductase inhibitory activity of the synthesized compounds was assayed and the results showed that most of the derivatives were potent against aldose reductase with IC50 values ranging from 0.2 to 2.36 µM. Two of the compounds with the highest efficacies among the tested compounds (IC50 values of 0.2 and 0.6 µM) were more potent than epalrestat. Molecular docking studies were undertaken to explore the binding modes of all compounds into the active site of aldose reductase in order to rationalize the inhibitory efficacy of these derivatives. Rhodanine-3-hippuric acid and all its arylidene derivatives fulfil Lipinski’s rule and show good drug-likeness property. In conclusion, novel compounds synthesized in the present study have proved to be potential drugs for diabetic complications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
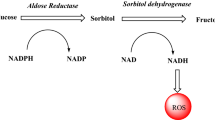
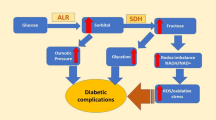
Diabetes mellitus (DM) is a band of metabolic disorders characterized by hyperglycaemia resulting from the defects in insulin discharge, insulin action, or both [1]. The unrelieved hyperglycaemia of diabetes is connected with long-term damage, functional disorder, and collapse of discrete organs especially the eyes, kidney, nerves, heart, and blood vessels [2]. Statistical data specified that over 400 million people worldwide were suffering from DM in the year 2015 and this quantity may rise to 642 million by 2040, which intimates that DM presents a great monetary and physical burden [3]. Several mechanisms describe the pathogenesis of diabetic complication. In this paper, we target the first mechanism, in which the hyperglycaemic condition is explained by increased flux of glucose in the polyol pathway leading to elevated accumulation of cellular sorbitol to cause osmotic stress in cells, implicating microvascular damages. The polyol pathway is commonly accepted to be the mechanism of most importance in the pathogenesis of diabetic complications [4]. The foremost enzyme present in the polyol pathway is aldose reductase (ALR2) [5], which converts glucose to sorbitol in the presence of NADPH as a cofactor. ALR2 with sorbitol dehydrogenase (SDH) convert sorbitol to fructose with NAD+ as cofactor, which presents the polyol pathway [6]. It is a part of the aldo-keto reductase superfamily [7]. ALR2 of the polyol metabolic pathway, inhibited by aldose reductase inhibitors (ARIs), has been a matter of interest, as it appears to be a hopeful pharmacotherapeutic target. It was primarily initiated to be implicated in the aetiology of chronic degenerative complications, such as retinopathy, neuropathy, and nephropathy [8] (Scheme 1 and Scheme 2).

Synthesis of rhodanine-3-hippuric acid (III).

Synthesis of substituted 5-benzylidene rhodanine-3-hippuric acid (V) and (Va–u).
All ARIs have the following two particular pharmacophore elements: (i) an acidic moiety capable of interacting with the anion-binding site of the catalytic site and (ii) a lipophilic scaffold that can bind to the highly flexible specificity pocket of the catalytic site. ARIs are classified into different groups according to their chemical structures: acetic acid derivatives, such as epalrestat and imirstat, and cyclic imides, especially spirohydantoins, like sorbinil and fidarestat [9]. Even though a series of structurally diverse ARIs has been developed up-to-date (Fig. 1) [10, 11], carboxylic acid derivatives play an important role and represent the major class of ARIs. They readily exhibit activity in the AR inhibition because of the structural importance of carboxylate anion head group, which may fit well in the anion-binding pocket of aldose reductase. A large number of aldose reductase inhibitors were developed based on their different chemical core structures [12–14]. Furthermore, all of them consist of a chemical group of acetic acid on the core. However, a lower tissue penetration has been found to be the major shortcoming for some individual potent carboxylic acid ARIs [15, 16]. Another chemical class of ARIs, which includes spirohydantoin analogues and their derivatives, has been developed. The key structures of spiro and hydantoins are responsible for the strongest inhibition against aldose reductase. In the binding interaction in the active site of AR, the hydantoin ring occupies the anion pocket as does the anion head group of carboxylate ARIs. Oxygen and nitrogen atoms are associated with carbonyl and amine groups in hydantoin produce a dynamic hydrogen bonding network with residues Tyr48, His110, and Trp111 of aldose reductase [17]. Sorbinil belongs to spirohydantoin group; it has structurally constrained spiro system, in whichring planes of the hydantoin and chroman are perpendicular with respect to each other, which may be the conformational shape of the inhibitors favored for the binding to aldose reductase. It has been proved as a good aldose reductase inhibitor both in vitro and in vivo, but in clinics 10% of the patients receiving sorbinil have some side reactions [18]. In another design, the orthogonal spirohydantoin moiety was changed into a more comfortable structure that was bridged by the sulphonyl group [19, 20]. In the present study, we blended the above two strategies and included amide and a carboxylic acid moiety with a rhodanine moiety and designed a novel series of compounds as ARIs. This may improve the cellular permeability and at the same time favour binding to AR active site via hydrogen bonds. Among all, Epalrestat is the well-known and first inhibitor for the treatment of diabetic neuropathy and highly marketed drug comprising the rhodanine nuclei; it is used to delay the progression of diabetic complications [21]. Attempts were made already to increase the efficacy of Epalrestat by modifying it in three domains: (i) phenyl ring was substituted with furyl, thienyl, or naphthyl, which resulted in reduced ARI activity; (ii) nitrile group introduced in place of methyl in the olefin chain did not make much difference in the activity; and (iii) replacement of acetic acid group with benzoic acid, phenylacetic acid, or H moieties keeping rhodanine moiety intact was studied [22]. None of the approaches resulted in desired improvement of the inhibitor activity.

Chemical structure of some known aldose reductase inhibitors.
In view of the fact that functionality of rhodanine is essential for ARI activity, we focused on the synthesis of new epalrestat analogues bearing other functional groups because epalrestat is the only ARI currently used for therapy in China, Japan, and India [23, 24]. Thus, as part of our initial efforts to discover new potentially active agents, we report herein the synthesis of rhodanine derivatives and their evaluation against aldose reductase enzyme. In addition, we performed molecular docking studies for all synthesized compounds and investigated the relationship with inhibition of aldose reductase.
RESULT AND DISCUSSION
Chemistry
A series of new aldose reductase inhibitors were synthesized as outlined in Table 1. Rhodanine-3-hippuric acid (III) was readily synthesized [25] by reacting p-amino hippuric acid (I) with carbon disulphide and chloroacetic acid (Scheme 1). Suitably substituted aryl aldehydes (IV) and (IVa–u) were reacted with compound (III) in the presence of sodium acetate and acetic acid [26] to yield the products (V) and (Va–u) (Scheme 2).
The newly synthesized rhodanine-3-hippuric acid (III) and derivatives (V) and (Va–u) were fully characterized by IR, 1H, and 13C NMR spectral data. The 1H NMR spectrum of rhodanine-3-hippuric acid (III) exhibited a doublet at δ 3.95 (2H, J 6.0, H9') for the N–CH2 protons and a sharp singlet at δ 4.40 due to the S–CH2 (2H, H5) protons of rhodanine moiety. Aromatic protons appeared as a pair of doublets, each integrating for two protons at δ 7.98 (2H, J 8.4, H2', H6') and 8.95 (2H, J 8.4, H3', H5'). A triplet at δ 8.95 (J1 5.8, J2 6.0, H8') was assignable to hippuric acid NH proton. The 13C NMR spectrum also reinforced the rhodanine-3-hippuric acid structure by the two resonating signals at δ 165.89 (C4) and 203.49 (C2) indicating the presence of carbonyl and thiocarbonyl groups of rhodamine, respectively. Signals at δ 37.24 due to S–CH2 (C5) of the rhodanine ring and at δ 41.25 (C9') due to N–CH2 of hippuric acid moiety were also observed. Signals resonating at δ 128.18–138.06 (C1', C2', C3', C4', C5', and C6') were attributed to the aromatic carbons of hippuric acid moiety. In 1H NMR spectrum of compound (Vf), all the signals are similar to that of compound (III) except the signal at δ 4.40 for S–CH2 (H5) protons disappeared, instead it showed a singlet at δ 7.86 (H6) which corresponds to the benzylic proton. The additional aromatic protons appeared between δ 7.55 to 8.03 ppm (H8, H9, H11, and H12). In the 13C NMR spectra of compound (Vf), a new signal at δ 124.00 was generated while the peak at δ 37.24 (C5) of compound (III) disappeared. Additionally, 6 more signals were observed in the aromatic region (C7, C8, C9, C10, C11, and C2). All other signals of compounds (V) and (Va–u) coincided with the signals of compound (III). These characteristic changes strongly confirmed the formation of 5-benzylidene rhodanine-3-hippuric acid derivatives. Identical results were observed with other derivatives of 5-benzylidene rhodanine-3-hippuric acid.
In Vitro Aldose Reductase Activity Assay
In the present study, newly synthesized compound (III) and its benzylidene derivatives (V) and (Va–u) were taken to recognize their effective inhibitory potential against ALR2 obtained from rat kidney tissue. For this performed assay we used DL-glyceraldehyde as the substrate which was reduced to glycerol in the presence of NADPH as the co-factor and the absorbance at 340 nm was measured using a spectrometer [27]. The exhibited results were expressed in 50% inhibition concentration method (IC50) and most of the synthesized analogues inhibited ALR2 in submicromolar concentrations as summarized in Table 1. In line with the previously reported work [28], the newly designed rhodanine-3-hippuric acid (III) and its derivatives (V) and (Va–u) carrying the carboxylic acid and amide functional group, behave as promisingly good AR inhibitors. Among the in vitro tested compounds, compound (Vi) was found to be the most potential ALR2 inhibitor with an IC50 value of 0.2 µM. Specifically, (Vi) is nearly four times more active than epalrestat. Furthermore, compound (Vn) also proved its efficacy against ALR2 and significantly higher than that of the reference drug (IC50 value of 0.6 µM). A slightly reduced inhibition potential was observed in compounds (Vg), (Vm), and (Vq), which displayed the activity at low submicromolar level with the IC50 values of 1.27, 1.3, and 1.2 µM, respectively. Among the 23 analogues, compounds (Vi) and (Vn) were found to be comparable with the standard epalrestat and the remaining compounds (III), (V), (Va–m), (Vo), and (Vq–Vu) were moderately active. Considering the basic rhodanine scaffold hippuric acid structure, introduction of a benzylidene moiety in the 4th position of the rhodanine ring did not make much difference in compound (V) in terms of its activity. Substituents on the benzylidene ring as in compounds (Vb), (Vg), (Vl), (Vm), (Vn), (Vi), and (Vq) were suspected to contribute greatly to the inhibitory activity. Among the monosubstituted compounds (Va–c) with a bromo substituent in the ortho, meta, and para positions, respectively, compound (Vb) with bromo substituent in the m-position showed an increase in the potency followed by the p- and o-derivatives. The same trend was followed with chloro substituted compounds (Vd–f), fluoro substituted compounds (Vh–j), methoxy substituted compounds (Vk–m), nitro substituted compounds (Vo–q), and methyl substituted compounds (Vs–u). Strikingly disubstituted derivative (Vg) and trisubstituted one (Vn) showed much higher potency when compared with their mono-substituted derivatives (Vd–f) and (Vk–m), respectively. No difference was observed between electron withdrawing or donating groups.
Molecular Docking
Aldose reductase (AR) is a potential target for drug design and is commonly accepted as an enzyme controlling the rate in the polyol pathway. On the basis of a number of AR crystal studies by crystallography and mutagenesis [29, 30], three distinct binding pockets in the active site of AR have already been suggested: the anion-binding pocket, lined by the residue Tyr48, His110, Trp20, and Trp111 side chains; hydrophobic specificity pocket formed by residues Leu300, Cys298, Cys303, Trp111, and Phe122; and hydrophobic pocket made of Trp20, Trp111, Phe122, and Trp219 [31].
Docking studies of new rhodanine-3-hippuric acid (III) and its benzylidene derivatives (V) and (Va–u) were performed to evaluate the binding interaction of the complex, which is an important parameter to describe the mechanistic details of the biological activity. Docking studies were performed on the aldose reductase receptor (PDB ID: 2FZD) using the AutoDock vina software. The observed binding energy is depicted in Table 1 and the results showed a better comprehension of the aldose reductase inhibitory potential. Herein, the putative binding mode of the most active compounds (Fig. 2), in particular compounds (Vi) and (Vn), was considered for the argument along with epalrestat, a reference compound. It was noted that compounds (Vi) and (Vn) fitted well with the active site of ALR2 and a better docking score (–9.4 and –9.1 kcal/mol, respectively) was achieved, which proved their high binding affinity. On the other hand, the epalrestat earned the docking score of –9.1 kcal/mol. The docking results support compounds (Vi) and (Vn) as more active ALR2 inhibitors.

2D and 3D structures of compounds (Vi) (a) and (Vn) (b) with 2FZD. Non-polar hydrogens bonding has been removed for clarity in the 3D structure. In 2D structure, the green colour circle indicates hydrogen bond interaction; pink circle, π–π stacked interaction; and yellow colour, π–S interaction.
The docking study of these 23 synthesized compounds (III), (V), and (Va–u) provides a better understanding of the inhibitory activity of aldose reductase at the molecular level and also sheds light on the interactions in the active site of aldose reductase. The results reveal that compounds (Vi) and (Vn) bound tightly in the active site of aldose reductase. All the compounds are well occupied in the receptor cavity and form hydrogen bonds and hydrophobic interactions. The amide carbonyl group (–CONH–) of all the compounds (III), (V), and (Va–u) is anchored into anion-binding site and forms hydrogen bonding with Trp111 and His110, which are the key residues in binding and catalysis. The exocyclic carbonyl or the thiocarbonyl group of the rhodanine moiety form hydrogen bonding with Trp20. The thiocarbonyl group also showed π–S interactions with Try79, Phe119 and Phe122. The ring sulphur atom of the rhodanine ring form π–sulfur and π–π stacked interactions with either Trp219 or Phe122. The benzylidine ring was trapped by the hydrophobic pocket formed by Val47, Pro218, Gln49, Phe122, and Cys303. The phenyl rings get tightly trapped in the hydrophobic pocket formed by Phe122, Leu300, Leu301, and Val30 and got anchored to the Cys298–Leu301 loop of the specificity pocket. It showed π–S interactions with Cys298 and π–π stacked interactions with Tyr20. The hydrogen bonded interactions with His110 and Trp111 in the anionic binding site is identical with the H-bonded interactions present in epalrestat (having carboxylate anion as a head group) and sorbinil (a spirohydantoin derivative).
Compounds (Vb), (Vl), (Vo), (Vq), (Vr), and (Vt) exhibited additional H-bonded interactions with Ser302. The binding poses of these compounds are the same as epalrestat in the anionic site of the enzyme. The common feature of these molecules is the presence of the carboxylic acid functional group at the terminal of rhodanine. It also exhibited other interactions with other enzyme residues due to the presence of a free amide group in between the acid functional group and rhodanine ring. Compound (III), (V), and (Va–u) fulfil Lipinski’s rule and show good drug-likeness properties: all the compounds have a molecular mass less than 500 Da and contain not more than 5 hydrogen bond donors and not more than 10 hydrogen bond acceptors. The octanol–water partition coefficient logP is also not greater than 5 (Table 2).
EXPERIMENTAL
Melting points were taken on a sigma melting point instrument and are uncorrected. Structures of all new compounds were elucidated using IR, 1H and 13C NMR spectroscopy, and elemental analysis. FT-IR spectra (ν, cm–1) of the synthesized compounds were recorded out with Shimadzu 360 FT-IR spectrophotometer using KBr pellets. 1H and 13C NMR spectra (δ, ppm) were measured at 400 and 100 MHz, respectively, on a Bruker-400 spectrometer using DMSO-d6 as a solvent and TMS as an internal standard. C, H, N, and S elemental analysis were determined using Perkin Elmer 2400 elemental analyser. All other chemicals were obtained from commercial sources and were of analytical grade.
General procedure for the synthesis of rhodanine-3-hippuric acid (III).p-Amino hippuric acid (I) (0.01mol) was dissolved in water. While stirring, the water solution of potassium hydroxide (0.02 mol) was added. The solution was cooled to 0°C, the solution of CS2 (0.01mol) was added. The mixture was kept for stirring for about 3 h and placed in a refrigerator for next 24 h. Then the water solution of chloroacetic acid (0.01 mol) was added and the mixture was stirred for 3 h. Subsequently, the solution of hydrochloric acid was added and refluxed for 2 h. While cooling, a precipitate appeared and then it was filtered and dried to afford the corresponding product (III).
2-(4-(4-Oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid(III). Yellow solid; yield 75%; mp 220°C; IR: 3384, 1740; 1H NMR: 8.95 (1H, t, J 5.8,6.0 Hz, H8′), 7.98 (2H, d, J 8.4, H3′ + H5′), 7.40 (2H, d, J 8.4 Hz, H2′ + 6′), 4.40 (2H, s, H5), 3.95 (2H, d, J 6.0 Hz, H9′); 13C NMR: 203.49 (C2), 173.89 (C10′), 171.13 (C7′), 165.89 (C4), 138.06 (C4′), 134.66 (C1′), 128.88 (C3′ + C5′), 128.18 (C2′ + C6′), 41.25 (C9′), 37.24 (C5); Anal. Calc. for C12H10N2O4S2: C, 46.44; H, 3.25; N, 9.03; O, 20.62; S, 20.66; Found: C, 46.42; H, 3.27; N, 9.08; O, 20.66; S, 20.68.
General procedure for the synthesis of 5-benzylidene rhodanine derivatives (V) and (Va–u). A mixture of compound (III) (0.003 mol) and substituted benzaldehyde (IV) and (IVa–u) (0.003 mol) were refluxed in glacial acetic and sodium acetate (0.003 mol) for 4–6 h. The reaction mixture was cooled and the resulted precipitate was filtered, washed with water, dried, and recrystallized (ethanol) to afford the corresponding product (V) and (Va–u).
2-(4-((Z)-5-Benzylidene-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (V). Yellow solid; yield 73%; mp 260°C; IR: 3400, 1727; 1H NMR: 8.96 (1H, s, H8′), 8.01 (2H, d, J 8.4, H3′ + H5′), 7.81 (1H, s, H6), 7.62 (2H, d, J 8.8, H2′ + H6′), 7.56 (1H, d, J 7.6, H10), 7.45 (2H, d, J 8.4 Hz, H8 + H12), 7.14 (2H, d, J 8.8 Hz, H9 + H11), 3.92 (2H, s, H9′); 13C NMR: 193.83 (C2), 171.08 (C10′), 166.46 (C7′), 164.12 (C4), 138.77 (C6), 134.66 (C7), 134.88 (C1′), 132.18 (C10), 128.38 (C11), 128.94 (C8), 128.88 (C4′), 127.87 (C3′ + C5′), 127.54 (C12), 127.26 (C2′ + C6′), 126.09 (C9), 124.53 (C5), 41.16 (C9′); Anal. Calc. for C19H14N2O4S2: C, 57.27; H, 3.54; N, 7.03; O, 16.06; S, 16.09; Found: C, 57.21; H, 3.59; N, 7.11; O, 16.12; S, 16.14.
2-(4-((Z)-5-(2-Bromobenzylidene)-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Va). Yellow solid; yield 70%; mp 255°C; IR: 3400, 1735; 1H NMR: 8.03 (2H, d, J 8.4 Hz, H3′ + H5′), 7.84 (1H, s, H6), 7.78 (2H, d, J 8.4, H2′ + H6′), 7.68 (1H, d, J 7.6, H9), 7.62 (1H, d, J 7.6, H12), 7.59 (1H, d, J 7.6, H11), 7.64 (1H, d, J 7.6, H10), 3.95 (2H, s, H9′); 13C NMR: 193.49 (C2), 171.25 (C10′), 166.29 (C7′), 165.72 (C4), 137.42 (C6), 135.98 (C7), 135.24 (C9), 134.72 (C1′), 134.27 (C10), 133.58 (C11), 132.39 (C4′), 131.86 (C3′ + C5′), 128.62 (C12), 128.19 (C2′ + C6′), 124.29 (C8), 124.26 (C5), 41.46 (C9′); Anal. Calc. for C19H13BrN2O4S2: C, 47.81; H, 2.74; Br, 16.74; N, 5.87; O, 13.41; S, 13.43; Found: C, 47.81; H, 2.79; Br, 16.72; N, 5.83; O, 13.41; S, 13.50.
2-(4-((Z)-5-(3-Bromobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vb). Yellow solid; yield 72%; mp 255°C; IR: 3405, 1725; 1H NMR: 8.02 (2H, d, J 8.4, H3′ + H5′), 7.92 (1H, s, H8), 7.85 (1H, s, H6), 7.74 (1H, d, J 7.6, H10), 7.67 (1H, d, J 7.6, H12), 7.56 (3H, m, H2′ + H6′ + H11), 3.97 (2H, s, H9′); 13C NMR: 193.44 (C2), 171.05 (C10′), 166.59 (C7′), 165.92 (C4), 137.57 (C6), 135.36 (C7), 134.92 (C1′), 133.38 (C10), 133.33 (C11), 131.59 (C8), 130.94 (C4′), 128.90 (C3′ + C5′), 128.54 (C12), 128.26 (C2′ + C6′), 125.09 (C9), 122.63 (C5), 41.26 (C9′); Anal. Calc. for C19H13BrN2O4S2: C, 47.81; H, 2.74; Br, 16.74; N, 5.87; O, 13.41; S, 13.43; Found: C, 47.85; H, 2.72; Br, 16.78; N, 5.83; O, 13.46; S, 13.45.
2-(4-((Z)-5-(4-Bromobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vc). Yellow solid; yield 69%; mp 260°C; IR: 3403, 1723; 1H NMR: 9.00 (1H, t, J 6, H8′), 8.02 (2H, d, J 8.4, H3′ + H5′), 7.84 (1H, s, H6), 7.79 (2H, d, J 8.4, H2′ + H6′), 7.64 (2H, d, J 8.8, H9 + H11), 7.56 (2H, d, J 8.4, H8 + H12), 3.97 (2H, d, J 6.0, H9′); 13C NMR: 193.5 (C2), 171.1(C10′), 166.7 (C7′), 165.9 (C4), 137.6 (C6), 134.9 (C1′), 132.5 (C9,11), 132.3 (C3′ + C5′), 132.2 (C7), 131.4 (C4′), 128.9 (C8 + C12), 128.2 (C2′ + C6′), 124.6 (C10), 124.1 (C5), 41.2 (C9′); Anal. Calc. for C19H13BrN2O4S2: C, 47.81; H, 2.74; Br, 16.74; N, 5.87; O, 13.41; S, 13.43; Found: C, 47.83; H, 2.75; Br, 16.76; N, 5.84; O, 13.44; S, 13.41.
2-(4-((Z)-5-(2-Chlorobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)aceticacid (Vd). Yellow solid; yield 65%; mp 255°C; IR: 3407, 1719; 1H NMR: 8.99 (1H, t, J 5.6, H8′), 8.02 (2H, d, J 8.2, H3′ + H5′), 7.95 (1H, s, H6), 7.70–7.65 (2H, m, H2′ + 6′), 7.58 (4H, q, J1 7.2, J2 8.0, H9 + H10 + H11 + H12), 3.97 (2H, d, J 6.0, H9′); 13C NMR: 193.6 (C2), 171.1 (C10′), 166.4 (C7′), 165.9 (C4), 137.5 (C6), 134.9 (C1′), 134.7 (C7), 132.3 (C8), 130.8 (C4′), 130.5 (C9), 129.4 (C10), 128.8 (C3′ + C5′), 128.4 (C12), 128.2 (C2′ + C6′), 127.34 (C11), 126.9 (C5), 41.2 (C9′); Anal. Calc. for C19H13ClN2O4S2: C, 52.71; H, 3.03; Cl, 8.19; N, 6.47; O, 14.78; S, 14.81; Found: C, 52.71; H, 3.04; Cl, 8.16; N, 6.48; O, 14.73; S, 14.88.
2-(4-((Z)-5-(3-Chlorobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Ve). Yellow solid; yield 65%; mp 255°C; IR: 3397, 1716; 1H NMR: 8.89 (1H, t, J 6, H8′), 8.14 (2H, d, J 8.8, H3′ + H5′), 7.82 (1H, s, H6), 7.75 (1H, s, H8), 7.69–7.63 (3H, m, H10 + H11 + H12), 7.54 (2H, d, J 8.8, H2′ + H6′), 3.94 (2H, d, J 5.6, H9′); 13C NMR: 193.6 (C2), 171.4 (C10′), 166.5 (C7′), 164.9 (C4), 137.5 (C6), 136.1 (C1′), 134.8 (C7), 134.3(C9), 131.8 (C11), 130.2 (C4′), 130.6 (C10), 128.5 (C3′ + C5′), 128.6 (C2′ + C6′), 128.2 (C8), 125.3 (C12), 123.5 (C5), 41.5 (C9′); Anal. Calc. for C19H13ClN2O4S2: C, 52.71; H, 3.03; Cl, 8.19; N, 6.47; O, 14.78; S, 14.81; Found: C, 52.73; H, 3.06; Cl, 8.15; N, 6.44; O, 14.75; S, 14.85.
2-(4-((Z)-5-(4-Chlorobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vf). Yellow solid; yield 65%; mp 260°C; IR: 3395, 1707; 1H NMR: 9.04 (1H, t, J 6.0, H8′), 8.02 (2H, d, J 8.4, H2′ + H6′), 7.86 (1H, s, H6), 7.71 (2H, d, J 8.8, H3′ + H5′), 7.64 (2H, d, J 8.8, H8 + H12), 7.56 (2H, d, J 8.4, H9 + H11), 3.97 (2H, d, J 5.6, H9′); 13C NMR: 193.4 (C2), 171.1 (C10′), 166.7 (C7′), 165.9 (C4), 137.6 (C6), 135.6 (C1′), 134.9 (C10), 132.2 (C9 + C11), 131.8 (C7), 131.3 (C4′), 129.6 (C3′ + C5′), 128.8 (C8 + C12), 128.2 (C2′ + C6′), 124.0 (C5), 41.2 (C9′); Anal. Calc. for C19H13ClN2O4S2: C, 52.71; H, 3.03; Cl, 8.19; N, 6.47; O, 14.78; S, 14.81; Found: C, 52.74; H, 3.06; Cl, 8.14; N, 6.45; O, 14.74; S, 14.83.
2-(4-((Z)-5-(2,3-Dichlorobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido) acetic acid (Vg). Yellow solid; yield 69%; mp 260°C; IR: 3283, 1722; 1H NMR: 8.99 (1H, t, J 6.0, H8′), 8.02 (2H, d, J 8.4, H3′ + H5′), 7.92 (1H, s, H6), 7.82 (1H, dd, J 2.0, H11), 7.65–7.57 (4H, m, H2′ + H6′ + H10 + H12), 3.97 (2H, d, J 6.0, H9′); 13C NMR: 193.5 (C2), 171.1 (C10′), 166.3 (C8′), 165.8 (C4), 137.4 (C6), 135.0 (C1′), 133.4 (C7), 133.2 (C9), 132.3 (C4′), 132.2 (C10), 129.2 (C8), 128.8 (C3′ + C5′), 128.4 (C11), 128.2 (C2′ + C6′), 127.9 (C12), 127.1 (C5), 41.2 (C9′); Anal. Calc. for C19H12Cl2N2O4S2: C, 48.83; H, 2.59; Cl, 15.17; N, 5.99; O, 13.69; S, 13.72; Found: C, 48.86; H, 2.55; Cl, 15.19; N, 5.94; O, 13.65; S, 13.75.
2-(4-((Z)-5-(2-Fluorobenzylidene)-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vh). Yellow solid; yield 63%; mp 250°C; IR: 3341, 1759; 1H NMR: 8.96 (1H, t, J 5.6, H8′), 8.01 (2H, d, J 8.4, H3′ + H5′), 7.86 (2H, m, H2′ + H6′), 7.84 (1H, s, H6), 7.68 (1H, d, J 7.6, H12), 7.59 (1H, d, J 7.6, H10), 7.48 (1H, d, J 7.6, H11), 7.42 (1H, d, J 8.2, H9), 3.91 (2H, d, J 6.0, H9′); 13C NMR: 193.68 (C2), 171.10 (C10′), 166.72 (C7′), 165.57 (C4), 161.81 (C8), 137.52 (C6), 134.76 (C1′), 133.42 (C12), 131.86 (C7), 129.62 (C4′), 128.94 (C10), 128.86 (C3′ + C5′), 128.52 (C2′ + C6′), 121.25 (C5), 118.97 (C11), 116.06 (C9), 41.12 (C9′); Anal. Calc. for C19H13FN2O4S2: C, 54.80; H, 3.15; F, 4.56; N, 6.73; O, 15.37; S, 15.40; Found: C, 54.89; H, 3.17; F, 4.56; N, 6.76; O, 15.36; S, 15.48.
2-(4-((Z)-5-(3-Fluorobenzylidene)-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vi). Yellow solid; yield 70%; mp 255°C; IR: 3297, 1706; 1H NMR: 8.99 (1H, t, J 6.0, H8′), 8.02 (2H, d, J 8.8, H3′ + H5′), 7.86 (1H, s, H6), 7.79 (1H, s, H8), 7.64–7.61 (3H, m, H10 + H11 + H12), 7.57 (2H, d, J 8.8, H2′ + H6′), 3.97 (2H, d, J 5.6, H9′); 13C NMR: 193.4 (C2), 171.1 (C10′), 166.6 (C7′), 165.8 (C4), 137.5 (C6), 135.1 (C1′), 134.9 (C7), 134.1(C9), 131.3 (C11), 130.9 (C4′), 130.4 (C10), 128.9 (C3′ + C5′), 128.26 (C2′ + C6′), 128.2 (C8), 125.0 (C12), 123.3 (C5), 41.3 (C9′); Anal. Calc. for C19H13FN2O4S2: C, 54.80; H, 3.15; F, 4.56; N, 6.73; O, 15.37; S, 15.40; Found: C, 54.82; H, 3.17; F, 4.54; N, 6.78; O, 15.35; S, 15.45.
2-(4-((Z)-5-(4-Fluorobenzylidene)-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vj). Yellow solid; yield 61%; mp 245°C; IR: 3311, 1739; 1H NMR: 9.00 (1H, t, J 5.6, H8′), 8.02 (2H, d, J 8.4, H3′ + H5′), 7.88 (1H, s, H6), 7.78 (2H, m, H8 + H12), 7.56 (2H, d, J 8.4, H2′ + H6′), 7.44 (2H, t, J 8.8, H9 + H11), 3.97 (2H, d, J 6.0, H9′); 13C NMR: 193.6 (C2), 171.1 (C10′), 166.7 (C7′), 165.9 (C4), 161.9 (C10), 137.6 (C6), 134.8 (C1), 133.2 (C12), 133.1 (C8), 131.7 (C7), 129.7 (C4′), 128.9 (C3′ + C5′), 128.2 (C2′ + C6′), 122.9 (C5), 116.8 (C11), 116.6 (C9), 41.2 (C9′); Anal. Calc. for C19H13FN2O4S2: C, 54.80; H, 3.15; F, 4.56; N, 6.73; O, 15.37; S, 15.40; Found: C, 54.84; H, 3.13; F, 4.58; N, 6.76; O, 15.35; S, 15.44.
2-(4-((Z)-5-(2-Methoxybenzylidene)-4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vk). Yellow solid; yield 67%; mp 240°C; IR: 3431, 1741; 1H NMR: 8.02 (2H, m, H3′ + H5′), 7.72 (1H, s, H6), 7.64 (2H, d, J 8.8, H2′ + H6′), 7.55 (1H, d, J 7.6, H12), 7.42 (1H, d, J 7.6, H10), 7.14 (2H, d, J 8.8, H9 + H12), 3.94 (2H, s, H9′), 3.84 (3H, s, H1″); 13C NMR: 193.39 (C2), 171.14 (C10′), 166.78 (C7′), 165.77 (C4), 161.43 (C8), 142.15 (C6), 136.73 (C1′), 134.36 (C4′), 133.25 (C3′ + C5′), 129.26 (C10), 128.32 (C2′ + C6′), 126.15 (C12), 124.28 (C11), 119.36 (C5), 115.21 (C7 + C9), 55.69 (C1″), 41.27 (C9′); Anal. Calc. for C20H16N2O5S2: C, 56.06; H, 3.76; N, 6.54; O, 18.67; S, 14.97; Found: C, 56.02; H, 3.79; N, 6.55; O, 18.64; S, 14.99.
2-(4-((Z)-5-(3-Methoxybenzylidene)-4-oxo-2-thioxothiazolidin-l]3yl)benzamido)acetic acid (Vl). Yellow solid; yield 64%; mp 245°C; IR: 3441, 1731; 1H NMR: 8.01 (2H, d, J 8.4, H3′ + H5′), 7.84 (1H, s, H6), 7.72 (2H, d, J 8.8, H2′ + H6′), 7.56 (1H, d, J 7.6, H11), 7.14 (2H, d, J 8.8, H8 + H12), 7.09 (1H, d, J 7.6, H10), 3.96 (2H, s, H9′), 3.81 (3H, s, H1″); 13C NMR: 193.16 (C2), 171.32 (C10′), 166.89 (C7′), 165.74 (C4), 160.76 (C9), 139.42 (C6), 136.72 (C1′), 134.25 (C4′), 133.02 (C3′ + C5′), 129.15 (C11), 128.21 (C2′ + C6′), 128.21 (C7), 119.64 (C5), 115.26 (C10,12), 112.16 (C8), 55.43 (C1″), 41.36 (C9′); Anal. Calc. for C20H16N2O5S2: C, 56.06; H, 3.76; N, 6.54; O, 18.67; S, 14.97; Found: C, 56.08; H, 3.74; N, 6.59; O, 18.68; S, 14.92.
2-(4-((Z)-5-(4-Methoxybenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vm). Yellow solid; yield 65%; mp 242°C; IR: 3421, 1711; 1H NMR: 8.01 (2H, m, H3′ + H5′), 7.83 (1H, s, H6), 7.67 (2H, d, J 8.8, H2′ + H6′), 7.55 (2H, d, J 8.4, H8 + H12), 7.16 (2H, d, J 8.8, H9 + H11), 3.97 (2H, s, H9′), 3.86 (3H, s, H1″); 13C NMR: 193.59 (C2), 171.06 (C10′), 166.86 (C7′), 165.87 (C4), 161.57 (C10), 137.28 (C6), 134.76 (C1′), 133.16 (C4′), 132.93 (C3′ + C5′), 128.94 (C8 + C12), 128.22 (C2′ + C6′), 125.54 (C7), 119.84 (C5), 115.23 (C9 + C11), 55.59 (C1″), 41.14 (C9′); Anal. Calc. for C20H16N2O5S2: C, 56.06; H, 3.76; N, 6.54; O, 18.67; S, 14.97; Found: C, 56.08; H, 3.72; N, 6.58; O, 18.63; S, 14.94.
2-(4-((Z)-5-(3,4,5-Trimethoxybenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido) acetic acid (Vn). Yellow solid; yield 69%; mp 239°C; IR: 3334, 1709; 1H NMR: 8.98 (1H, t, J1 6.0, J2 5.6, H8′), 8.02 (2H, d, J 8.4, H2′ + H6′), 7.80 (1H, s, H6), 7.55 (2H, d, J 8.4, H3′ + H5′), 6.99 (2H, s, H8 + H12), 3.98 (2H, d, J 5.6, H9′), 3.87 (6H, s, H1″ + H3″), 3.77 (3H, s, H2″); 13C NMR: 193.4 (C2), 171.1 (C10′), 166.6 (C7′), 165.9 (C4), 153.3 (C9,11), 140.0 (C6), 137.6 (C10), 134.9 (C1′), 133.28 (C4′), 128.90 (C3′ + C5′), 128.4 (C7), 128.2 (C2′ + C6′), 121.9 (C5), 108.1 (C8 + C12), 60.2 (C2″), 56.1 (C1″ + C3″), 41.3 (C9′); Anal. Calc. for C22H20N2O7S2: C, 54.09; H, 4.13; N, 5.73; O, 22.92; S, 13.13; Found: C, 54.04; H, 4.15; N, 5.76; O, 22.91; S, 13.15.
2-(4-((Z)-5-(2-Nitrobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vo). Yellow solid; yield 62%; mp 218°C; IR: 3397, 1724; 1H NMR: 9.01 (1H, t, J 6.0, H8′), 8.25 (1H, d, J 8, H9), 8.09 (1H, s, H6), 8.03 (2H, d, J 8.4, H3′ + H5′), 7.95 (1H, t, J 8.4, H11), 7.80–7.76 (2H, m, H10 + H12), 7.59 (2H, d, J 8.4, H-2′,6′), 3.98 (2H, d, J 6.0, H9′); 13C NMR: 193.8 (C2), 171.1 (C10′), 166.0 (C7′), 165.9 (C4), 147.9 (C8), 137.4 (C6), 134.9 (C1′), 134.7 (C11), 131.4 (C4′),129.3 (C10), 129.1 (C7), 128.8 (C3′ + C5′), 128.6 (C12), 128.2 (C2′ + C6′), 127.9 (C9), 125.6 (C5), 41.2 (C9′); Anal. Calc. for C19H13N3O6S2: C, 51.46; H, 2.95; N, 9.48; O, 21.65; S, 14.46; Found: C, 51.48; H, 2.92; N, 9.43; O, 21.69; S, 14.42.
2-(4-((Z)-5-(3-Nitrobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vp). Yellow solid; yield 59%; mp 290°C; IR: 3338, 1726; 1H NMR: 8.84 (1H, t, J 5.6, H8′), 8.53 (1H, m, H8), 8.34 (1H, dd, J 1.6, H10), 8.10 (1H, t, J 7.6, H12), 8.02 (3H, t, J 8.0, H11), 7.87 (1H, t, J 8.0, H11), 7.56 (2H, d, J 8.4, H2′ + H6′), 3.91(2H, d, J 5.6, H9′); 13C NMR: 193.2 (C2), 171.1 (C10′), 166.5 (C7′), 165.6 (C4), 148.3 (C9), 137.4 (C6), 135.7 (C1′), 135.1 (C7), 134.6 (C12), 131.1 (C4′), 130.1 (C11), 128.8 (C3′ + C5′), 128.2 (C2′ + C6′), 126.2 (C8), 124.9 (C10), 124.8 (C5), 41.8 (C9′); Anal. Calc. for C19H13N3O6S2: C, 51.46; H, 2.95; N, 9.48; O, 21.65; S, 14.46; Found: C, 51.44; H, 2.93; N, 9.47; O, 21.62; S, 14.44.
2-(4-((Z)-5-(4-Nitrobenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vq). Yellow solid; yield 55%; mp 230°C; IR: 3268, 1712; 1H NMR: 8.98 (1H, t, J1 6.0, J2 5.6, H8′), 8.37 (2H, d, J 8.8, H9 + H11), 8.03 (2H, d, J 8.8, H3′ + H5′), 7.94 (3H, J 8.8, singlet of H-6 merged with doublet of H2′ and H6′), 7.57 (2H, d, J 8.2, H8 + H12), 3.97 (2H, d, J 6.0, H9′); 13C NMR: 193.3 (C2), 172.0 (C10′), 171.1 (C7′), 166.5 (C4), 147.6 (C10), 139.08 (C6), 137.4 (C7), 135.0 (C1′), 131.4 (C3′ + C5′), 129.6 (C4′), 128.8 (C8 + C12), 128.2 (C2′ + C6′), 127.6 (C5), 124.4 (C9 + C11), 41.3 (C9′); Anal. Calc. for C19H13N3O6S2: C, 51.46; H, 2.95; N, 9.48; O, 21.65; S, 14.46; Found: C, 51.48; H, 2.94; N, 9.42; O, 21.64; S, 14.43.
2-(4-((Z)-5-(4-Formylbenzylidene) 4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vr). Yellow solid; yield 67%; mp 280°C; IR: 3326, 1722; 1H NMR: 10.08 (1H, s, H-1″), 8.98 (1H, t, J1 5.6, J2 6.0, H8′), 8.08 (2H, d, J 8.0, H3′ + H5′), 8.02 (2H, d, J 8.4, H2′ + H6′), 7.92 (1H, d, J 2.8, H6), 7.89 (2H, t, J1 2.8, J2 4.8, H9 + H11) 7.57 (2H, d, J 8.4, H8 + H12), 3.97 (3H, d, J 5.6, H9′); 13C NMR: 193.5 (C2), 192.6 (C1″), 171.1 (C10′), 166.6 (C7′), 165.8 (C4), 138.2 (C6), 137.5 (C7), 136.6 (C1′), 134.9 (C10), 131.0 (C9,11), 130.9 (C4), 130.2 (C3′+ C5′), 128.8 (C8 + C12), 128.2 (C2′ + C6′), 126.4 (C5), 41.34 (C9′); Anal. Calc. for C20H14N2O5S2: C, 56.33; H, 3.31; N, 6.57; O, 18.76; S, 15.04; Found: C, 56.37; H, 3.34; N, 6.54; O, 18.74; S, 15.02.
2-(4-((Z)-5-(2-Methylbenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vs). Yellow solid; yield 69%; mp 230°C; IR: 3423, 1717; 1H NMR: 8.02 (2H, m, H3′ + H5′), 7.86 (1H, s, H6), 7.79 (2H, d, J 8.8, H2′ + H6′), 7.54 (2H, d, J 7.6, H12), 7.47 (2H, d, J 8.8, H10), 7.39 (1H, d, J 7.6, H10), 4.06 (2H, d, J 4.0, H9′), 2.42 (3H, s, H-1″); 13C NMR: 193.56 (C2), 171.26 (C10′), 166.67 (C7′), 165.54 (C4), 141.96 (C6), 138.28 (C8), 137.98 (C1′), 134.25 (C7), 131.86 (C9), 130.78 (C10), 130.29 (C4′), 130.15 (C3′ + C5′), 128.96 (C12), 128.17 (C2′ + C6′), 127.17 (C11), 120.09 (C5), 41.38 (C9′), 21.16 (C1″); Anal. Calc. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; O, 15.52; S, 15.55; Found: C, 58.28; H, 3.99; N, 6.73; O, 15.59; S, 15.52.
2-(4-((Z)-5-(3-Methylbenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid (Vt). Yellow solid; yield 65%; mp 240°C; IR: 3432, 1711; 1H NMR: 8.01 (2H, m, H-3′,5′), 7.81 (1H, s, H6), 7.76 (2H, d, J 8.8, H2′ + H6′), 7.56 (2H, d, J 8.6, H8 + H12), 7.42 (1H, d, J 7.6, H11), 7.12 (1H, d, J 7.6, H10), 4.71 (2H, d, J 4.0, H-9′), 2.24 (3H, s, H1″); 13C NMR: 193.25 (C2), 171.14 (C10′), 166.78 (C7′), 165.48 (C4), 142.74 (C6), 138.81 (C9), 137.31 (C1′), 133.52 (C7), 130.76 (C10), 310.52 (C11), 130.28 (C4′), 130.14 (C3′ + C5′), 128.96 (C7), 128.57 (C12), 128.42 (C2′ + C6′), 121.08 (C5), 41.43 (C9′), 21.61 (C1″); Anal. Calc. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; O, 15.52; S, 15.55; Found: C, 58.22; H, 3.91; N, 6.74; O, 15.56; S, 15.50.
2-(4-((Z)-5-(4-Methylbenzylidene)4-oxo-2-thioxothiazolidin-3-yl)benzamido)acetic acid(Vu). Yellow solid; yield 63%; mp 235°C; IR: 3411, 1712; 1H NMR: 8.05 (2H, m, H3′ + H5′), 7.88 (1H, s, H6), 7.62 (4H, m, H2′ + H6′ + H8 + H12), 7.46 (2H, d, J 7.6, H9 + H11), 4.01 (2H, d, J 4.0, H9′), 2.44 (3H, s, H1″); 13C NMR: 193.74 (C2), 171.04 (C10′), 166.82 (C7′), 165.88 (C4), 141.57 (C6), 138.07 (C10), 137.73 (C1′), 133.05 (C7), 130.72 (C9,11), 130.26 (C4′), 130.22 (C3′,5′), 128.89 (C8 + C12), 128.18 (C2′ + C6′), 121.95 (C5), 41.24 (C9′), 21.14 (C1″); Anal. Calc. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; O, 15.52; S, 15.55; Found: C, 58.26; H, 3.95; N, 6.74; O, 15.56; S, 15.59.
Aldose reductase inhibitory assay. Rat kidney tissue has been received from the Slaughterhouse and used as an enzyme source of aldose reductase. Rat kidney tissue was homogenized in ice-cold potassium phosphate buffer (100 mM, pH 7.4). The homogenate was centrifuged at 12000 rpm at 4°C for 30 min. The resulting supernatant was stored at –80°C and used as the source of aldose reductase for further assay. The in vitro assay of the inhibition of aldose reductase was carried out according to the method described by Hayman and Kinoshita [32]. Assay mixture contained 0.2 mL of enzyme source, 0.1 M potassium phosphate buffer pH 6.2, 10 mM DL-glyceraldehyde (Sigma Aldrich, USA), distilled water and 0.104 mM NADPH (Sigma Aldrich, USA). The test compound was incubated with the enzyme for 20 min at 37°C. The reaction mixture devoid of NADPH was used as a respective blank and the reaction was initiated by addition of NADPH. The change in absorbance at 340 nm due to the oxidation of NADPH was measured using the Lambda 25 UV-Visible spectrophotometer. The inhibitory activity of test compounds was estimated on the basis of the decrease in the absorbance of NADPH at 340 nm and quantified using the formula:
where T is absorbance of test reaction and C, absorbance of control.
The 50% inhibitory concentration (IC50) was calculated by non-linear regression. The dose–response curve was obtained by plotting the percentage inhibition versus the concentrations.
Molecular docking method. To examine the molecular interaction behind the mechanism of inhibition bycompounds (III), (V), and (Va–n), molecular docking study was carried out on Auto Dock Vina software. Lamarckian Genetic Algorithm (LGA was employed for docking. To perform molecular docking, the protein structure of aldose reductase (PDB id 2FZD) was obtained from the Protein Data Bank and the docking protocol was tested by removing co-crystallized ligand and water molecules from the protein; then the ligand structures (III), (V), and (Va–n) were drawn using Chem3D ultra. Further, 3D structures were set for applying partial charges and energy minimization. The active site of the enzyme was well-defined in a way so as to include residues of the active site within the grid size of 40 × 40 × 40 Å. Molecules (III), (V), and (Va–n) showed binding energy of >–8.3 kcal/mol and formed hydrophobic and hydrophilic bonds. Prediction of the binding affinity by molecular docking was found to be consistent with the experimental study. The interactions were observed using Chimera software packages.
REFERENCES
Soni, L.K., Gupta, A.K., and Kaskhedikar, S.G., Med. Chem. Res., 2008, vol. 17, pp. 258–266.
Tang, W.H., Martin, K.A., and Hwa, J., Front. Pharmacol., 2008, vol. 3, pp. 1–8.
Han, Z., Hao, X., Ma, B., and Zhu, C., Eur. J. Med. Chem., 2016, vol. 121, pp. 308–317.
Zhu, C., Diabetes Mellitus: Insights Perspect., 2013, vol. 2, pp. 17–46.
Bozdag-Dundar, O., Das Evcimen, N., Ceylan-Unlusoy, M., Ertan, R., and Sarikaya, M., Med. Chem. Res., 2007, vol. 16, pp. 39–47.
Carper, D.A., Wistow, G., Nishimura, C., Graham, C., Watanabe, K., Fujii, Y., Hayashi, H., and Ayaishi, O.A., Exp. Eye Res.,1989, vol. 49, pp. 377–388.
Jez, J.M., Bennett, M.J., Schlegel, B.P., Lewis, M., and Penning, T.M., Biochem. J., 1997, vol. 326, pp. 625–636.
Demopoulos, V.J., Zaher, N., Zika, C., Anagnostou, C., Mamadou, E., Alexiou, P., and Nicolaou, I., Drug Design Rev. Online, 2005, vol. 2, pp. 293–304.
Ramunno, A., Cosconatid, S., Sartini, S., Maglio, V., Angiuoli, S., Pietra, V.L., Maro, S.D., Giustiniano, M., Motta, C.L., Settimo, F.D., Marinelli, L., and Novellino, E., Eur. J. Med. Chem., 2012, vol. 51, pp. 216–226.
Kador, P.F., Kinoshita, J.H., and Sharpless, N.E., J. Med. Chem., 1985, vol. 28, pp. 841–849.
Suzen, S. and Buyukbingol, E., Curr. Med. Chem., 2003, vol. 10, pp. 1329–1352.
La Motta, C., Sartini, S., Simorini, F., Taliani, S., Marini, A.M., Da Settimo, F., Marinelli, L., Limongelli, V., and Novellino, E., J. Med. Chem., 2008, vol. 51, pp. 3182–3193.
Chen, X., Zhu, C., Guo, F., Qiu, X., Zhang, S., He, M., Praveen, S., Jing, C., Liu, Y., and Ma, B., J. Med. Chem., 2010, vol. 53, pp. 8330–8344.
Chen, X., Yang, Y., Zhu, C., Gui, D., Hussain, S., Qiu, X., Zhang, S., He, M., Jing, C., Liu, Y., and Ma, B., Eur. J. Med. Chem., 2011, vol. 46, pp.1536–1544.
Hamada, Y. and Nakamura, J., Treat. Endocrinol., 2004, vol. 3, pp. 245–255.
Costantino, L., Vianello, P., Rastelli, G.V., Cignarella, G., and Barlocco, D., Med. Res. Rev., 1999, vol. 19, pp. 3–23.
Urzhumtsev, A., Tete-Favier, F., Mitschler, A., Barbanton, J., Barth, P., Urzhumtseva, L., Biellmann, J.F., Podjarny, A.D., and Moras, D., Structure, 1997, vol. 5, pp. 601–612.
Jaspan, J.B., Herold, K., and Bartkus, C., Am. J. Med., 1985, vol. 79, pp. 24–37.
Miwa, I., Hirano, M., Inagaki, K., Belbeoch, C., and Okuda, J., Biochem. Pharmacol., 1987, vol.36, pp. 2789–2794.
Miwa, I., Hirano, M., Kanbara, M., and Okuda, J., Biochem. Pharmacol., 1990, vol. 40, pp. 303–307.
Barakat, A., Al-Majid, A.M., AL-Najjar, H.J., Mabkhot, Y.N., Ghabbourc, H.A., and Fun, H., RSC Adv., 2014, vol. 4, pp. 4909–4916.
Thatikonda, N.R., Ravinder, M., Pankaj, B., Ravikanti, K., Chandrakant, B., Jagadeesh, B.N., Kolupula, S., Sanjay, K.B., and Rao, V.J., Eur. J. Med. Chem., 2014, vol. 71, pp. 53–66.
Rosanna, M., Antonello del, C., Marco, G., Roberta, M., Umberto, M., and Rosaria, O., Bioorg. Med. Chem. Lett., 2011, vol. 21, pp. 200–203.
Alexiou, P., Pegklidou, K., and Chatzopoulou, M., Curr. Med. Chem., 2009, vol.16, pp. 734–752.
Stawoska I., Tejchman W., Mazuryk O., Lycka A., Nowak-Sliwinska P., Zeslawska E., and Nitek W., J. Heterocycl. Chem., 2017, vol. 54, pp. 2889–2897.
Sundaram, K. and Ravi, S., Res. Chem. Intermed., 2015, vol. 41, pp.1011–1021.
Aliya, I., Yildiz, T., Khan, I., Abdul, H., Saeed, A., Norbert, F., Bajorath, J., and Iqbal, J., Bioorg. Chem., 2016, vol. 68, pp. 177–186.
Xin, H., Zhongfei, H., Li, Y., Li, C., Wang, X., Zhang, X., Qin, Y., Ma, B., and Changjin, Z., Bioorg. Med. Chem. Lett., 2017, vol. 27, pp. 887–892.
El-Kabbani, O., Ramsland, P., Darmanin, C., Chung, R.P.T., and Podjarny, A., Proteins: Struct., Funct., Bioinf., 2003, vol. 50, pp. 230–238.
Cosconati, S., Marinelli, L., La Motta, C., Sartini, S., Da Settimo, F., Olson, A.J., and Novellino, E., J. Med. Chem., 2009, vol. 52, pp. 5578–5581.
El-Kabbani, O., Darmanin, C., Schneide, T.R., Hazemann, I., Ruiz, F., Oka, M., Joachimiak, A., Schulze-Briese, C., Tomizaki, T., Mitschler, A., and Podjarny, A., Proteins: Struct., Funct., Bioinf., 2004, vol. 55, pp. 805–813.
Kinoshita, J.H., Invest. Ophthalmol., 1965, vol. 4, pp. 786–799.
Haraguchi, H., Hayashi, R., Ishizu, T., and Yagl, A., Planta Med., 2003, vol. 69, pp. 853–855.
ACKNOWLEDGMENTS
We thank Karpagam Academy of Higher Education for providing instrumentation specialities to do the present research work. The authors are thankful to SAIF-STIC, Cochin University of Science and Technology, for their instrumentation support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
This article does not contain any studies involving human participants performed by any of the authors and does not contain any studies involving animals performed by any of the authors.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Corresponding author: e-mail: sundarg2010@gmail.com
Rights and permissions
About this article

Cite this article
Stephen Kumar Celestina, Sundaram, K. & Ravi, S. Novel Derivatives of Rhodanine-3-Hippuric Acid as Active Inhibitors of Aldose Reductase: Synthesis, Biological Evaluation, and Molecular Docking Analysis. Russ J Bioorg Chem 45, 405–415 (2019). https://doi.org/10.1134/S1068162019050066
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162019050066