
Abstract
Two isostructural Co/Ni(II)-based coordination polymers, namely, {[Co1/2(bpba)(H2O)]⋅2H2O}n (1) and {[Ni1/2(bpba)(H2O)]⋅2H2O}n (2), where Hbpba = 3,5-bis (pyridin-4-ylmethoxy) is benzoic acid, were synthesized hydrothermally and characterized. The Hbpba ligand contains two flexible pyridines and one rigid benzoic acid group. Due to the free rotation of methoxy groups, pyridine groups in Hbpba can participate in the coordination of metal ions. Photocatalysis of compounds 1 and 2 on six dyes was studied and the results shown that compounds 1 and 2 had the best degradability to rhodamine B and methyl blue, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Coordination polymers (CPs) constructed from metal ions or clusters and organic ligands [1–3] are one of the most promising functional crystalline materials owing to their high porosity and stability, controlled structures, and large specific surface area [4–10]. Some tiny changes, such as reaction time and temperature [11–13], solvent polarity [14, 15], or pH value [16], can lead to a great difference in structures and performances.
Currently, organic dyestuffs are widely applied into textiles, cosmetics, pharmaceutical, and solar cells [17, 18]. Although dyes brought diverse colors in daily life, they also cause many pollution that pose a serious threat to the river ecosystem and even human health due to their high chemical stability and carcinogenic effect. Therefore, the problem of dye pollution needs to be solved urgently by extensive scientists. Among various methods such as absorption [19, 20], photocatalysis [21–23], membrane separation [24, 25], and sedimentation [26], UV-light driven photocatalysis is considered an effective and environmentally friendly way to handle dye pollution. Nowadays, more and more research results show that MOFs have outstanding photocatalytic performance [27–30]. When exposed to light, UV light can induce the photocatalyst to generate LMCT, which can be deactivated by oxygenating H2O2 to OH radicals. Subsequently, one electron can be captured from H2O2 and generate OH radicals. OH radicals are highly active in oxidizing and decomposing the organic dyes to complete the photodegradation process [31, 32].
Herein, two isostructural Co/Ni(II)-based coordination polymers, {[Co1/2(bpba)(H2O)]⋅2H2O}n (1) and {[Ni1/2(bpba)(H2O)]⋅2H2O}n (2), were synthesized hydrothermally and characterized (Hbpba = 3,5-bis(pyridin-4-ylmethoxy) is benzoic acid) [33, 34]. The catalytic oxidation activity was tested against rhodamine B (RB), methyl blue (MB), methylene blue (MEB), neutral red (NR), methyl orange (MO), and a safranine T (ST) aqueous solution in the presence of H2O2. The results indicate that compound 1 can photodegrade RB with high-efficiency and selectivity, while compound 2 can photodegrade MB effectively and selectively.
EXPERIMENTAL
Materials and Measurements
All reagents were analytical grade and used without further purification. X-ray powder diffraction (XRD) measurements were carried out on a Bruker D8 Advance X-ray diffractometer using CuKα radiation (1.5418 Å), and the X-ray tube was operated at 40 kV and 40 mA. UV–Vis measurements were conducted on a TU-1900 UV–Vis spectrometer. Solid state diffuse reflectance UV–Vis spectra were obtained at room temperature using a Shimadzu UV-3600 double monochromator spectrophotometer, and BaSO4 was used as the 100% reflectance standard for all materials. The as-synthesized samples were characterized by thermogravimetric analysis (TGA) on a Perkin Elmer thermogravimetric analyzer Pyris 1 TGA up to 973 K using a heating rate of 10 K min–1 in an N2 atmosphere.
{[Co1/2(bpba)(H2O)]⋅2H2O}n (1) and {[Ni1/2(bpba)(H2O)]⋅2H2O}n (2)
A mixture of Co(NO3)2⋅6H2O (20.0 mg) or Ni(NO3)2⋅6H2O (20.0 mg) and Hbpba (4.0 mg), DMF (N,N-dimethyl formamide) and H2O (1 : 3 mL for compound 1; 1.5 : 3 mL for compound 2) was blended into a transparent glass bottle. After the reagents were adequately dissolved in the ultrasonic instrument, the glass bottle was placed into a Teflon-lined stainless steel vessel in an autoclave. The vessel was sealed and heated at 85°C for 48 h and then cooled to room temperature. Finally, a large number of red or green crystals were obtained (61% yield for compound 1 and 69% for compound 2 based on Hbpba).
Crystallographic Studies
Single crystal of 1 was prepared by the methods described in the synthetic procedures. X-ray crystallographic data of the complex were collected on a Bruker Apex Smart CCD diffractometer (graphite monochromator, Mo Kα radiation, λ = 0.71073 Å). The structure was solved by direct methods and all atoms except hydrogen were taken from the trial structure and then refined anisotropically using SHELXTL and full-matrix least-squares procedures based on F 2 values [35]. A semiempirical absorption correction was applied using SADABS [36]. The positions of hydrogen atoms were fixed geometrically at the calculated distances and allowed to ride on the parent atoms. The distributions of peaks in the channels of the title compound were chemically featureless to refine using conventional discrete-atom models. The relevant crystallographic data are presented in Table 1, while the selected bond angles are given in Table 2. CCDC numbers are 1983508 and 1983509 for compounds 1 and 2, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
RESULTS AND DISCUSSION
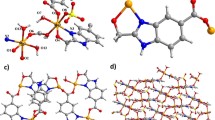
Complexes 1 and 2 are isostructural and crystallize in the monoclinic crystal system and space group C2/c. Only structure 1 is detailed here. As shown in Fig. 1a, the asymmetric unit of 1 contains one independent Co(II) cation, one bpba– anion, two coordinated and two lattice H2O molecules. The Co1 atoms are six-coordinated by two O atoms from the H2O molecules and two N atoms from the bpba– ligand in the equatorial position, and two O atoms from the bpba– ligand in the axial position. Only one flexible pyridyl part of the bpba– ligand is involved in coordination. The bpba– ligand is connected with Co(II) cations to form a 1D chain (Fig. 1b). The uncoordinated O or N bpba– atoms accept hydrogen bonding from coordinated water molecules (Table 3). The 1D chain is further stabilized by hydrogen bonds.

Strucrure of 1 (hydrogen atoms are omitted for clarity), symmetry codes: (#1) –x, y, –1/2 – z; (#2) –x, 1 – y, – z; (#3) x, 1 – y, –1/2 + z (a). A perspective of 1D chain along the a axis (b).
The powder diffraction of compounds 1 and 2 was measured; the results confirm the purities of 1 and 2, the experimental diffractograms of which almost matched their simulated diffractograms (Fig. 2). To estimate the stability of the coordination architectures, their thermal behavior was studied by TGA (Fig. 3). A gradual weight loss of 4.41% (4.80% for compound 2) is observed from 40 to 75°C (from 40 to 60°C for compound 2), which is attributed to the loss of one lattice water molecule (calculated 4.30% for compounds 1 and 2). The second weight loss of 8.50% (8.70% for compound 2, calculated as 8.60% for compounds 1 and 2) corresponds to the loss of two coordinated water molecules at 180°C for compound 1 (190°C for compound 2). The structure then decomposes starting at 317°C (330°C for compound 2).

Experimental (1) and simulates (2) powder diffraction patterns of compounds (a) 1 and (b) 2.

Thermal analysis plots of compounds 1 (1) and 2 (2) at room temperature.
The photocatalytic performances of compounds 1 and 2 were measured at room temperature using six dyes: RB, MB, MEB, NR, MO, and ST aqueous solution. 5 mg of compound 1 or 2 were added to 6 mL of an 8 ppm dye aqueous solution with 30% H2O2 in glass bottles under 365 nm UV light (WFH-203B) for 8 h. For compound 1, the photocatalytic efficiencies with respect to RB, MB, MEB, NR, MO, and ST were found to be 80.5, 64.3, 54.6, 23.5, 35.6, and 65.6%. For compound 2, the photocatalytic efficiencies with respect to RB, MB, MEB, NR, MO, and ST were found to be 90.5, 97.8, 75.3, 61.5, 74.7, and 86.6%. To prove MOFs’ catalytic action, the control experiments were carried out without compounds 1 or 2. The results showed that the efficiency was 80.5% with crystals 1 and 70.4% without them. Besides, catalysts 2 helped to improve the photocatalytic efficiency from 62.9 to 97.8%. The results showed that compounds 1 and 2 had the best degradability to RB and MB, respectively. The photocatalytic reactions started when a photocatalyst absorbed photons with energies exceeding its band gap energy (3.5 eV for compound 1 and 2.7 eV for compound 2) (Fig. 4). Compounds 1 and 2 have little or no absorption of these dyes. The absorption efficiencies of compounds 1 and 2 after 24 h for the six dyes were found to be 3.1, 0, 9.1, 21.8, 0.3, and 0% for compound 1; 5.8, 2, 4.1, 30.0, 0, and 12.4% for compound 2.

Kubelka–Munk plot as a function of the energy of the compounds 1 (1) and 2 (2) at room temperature.
In order to explore the effect of different concentrations of dye solution, we chose MB with compound 2 as a representative reaction for the study. Five mg of compound 2 were placed in 6 mL of 4, 6, 8, 10, and 12 ppm MB aqueous solution with 30% H2O2, respectively. We measured UV absorbance spectra. As shown in Fig. 5, with increasing time, the degradation degree gradually increases.

Kinetics of photodegradation of the MB dye with different low concentrations in compound 2.
Thus, two coordination polymers {[Co1/2(bpba)(H2O)] ⋅ H2O}n (1) and {[Ni1/2(bpba)(H2O)]⋅2H2O}n (2) were prepared under hydrothermal conditions. Photocatalysis of compounds 1 and 2 on six dyes was studied, and the results showed that compounds 1 and 2 had the best degradability to rhodamine B and methyl blue, respectively.
REFERENCES
H. Furukawa, K. E. Cordova, M. O’Keeffe, and O. M. Yaghi, Science 341, 1230444 (2013).
C. Wang, D. M. Liu, and W. B. Lin, J. Am. Chem. Soc. 135, 13222 (2013).
Y. J. Cui, B. Li, H. J. He, et al., Acc. Chem. Res. 49, 483 (2016).
H. X. Liang, X. L. Jiao, C. Li, and D. R. Chen, J. Mater. Chem. A 6, 334 (2018).
H. X. Deng, S. Grunder, K. E. Cordova, et al., Science 336, 1018 (2012).
M. Kandiah, M. H. Nilsen, S. Usseglio, et al., Chem. Mater. 22, 6632 (2010).
A. Carne-Sanchez, I. Imaz, K. C. Stylianou, and D. Maspoch, Chem. Eur. J. 20, 5192 (2014).
J. Wang, Q. Zhong, Y. H. Xiong, et al., Appl. Surf. Sci. 483, 1158 (2019).
Y. Y. Lu, W. W. Zhan, Y. He, et al., ACS. App. Mater. Inter. 6, 4186 (2014).
D. Y. Lee, D. V. Shinde, E. K. Kim, et al., Microp. Mesop. Mater. 171, 53 (2013).
P. Mahata, M. Prabu, and S. Natarajan, Inorg. Chem. 47, 8451 (2008).
Y. Chen, Z. O. Wang, Z. G. Ren, et al., Cryst. Growth Des. 9, 4963 (2009).
J. P. Zhang, Y. Y. Lin, W. X. Zhang, and X. M. Chen, J. Am. Chem. Soc. 127, 14162 (2005).
W. H. Huang, X. J. Luan, X. Zhou, et al., CrystEngComm 15, 10389 (2013).
C. Y. Guo, Y. H. Zhang, Y. Guo, et al., Chem. Commun. 54, 252 (2018).
Y. Zhang, W. Ju, X. Xu, et al., CrystEngComm 16, 5681 (2014).
C. A. Martinez-Huitle and E. Brillas, Appl. Catal. B 87, 105 (2009).
E. Brillas and C. A. Martinez-Huitle, Appl. Catal. B 166, 603 (2015).
X. Zhuang, Y. Wan, C. M. Feng, et al., Chem. Mater. 21, 706 (2009).
M. Rahmani, M. Kaykhaii, and M. Sasani, Spectroc. Acta Pt. A 188, 164 (2018).
F. Wang, F. L. Li, M. M. Xu, et al., J. Mater. Chem. A 3, 5908 (2015).
V. Vaiano, G. Iervolino, D. Sannino, et al., Appl. Catal. B 188, 134 (2016).
S. Khanchandani, S. Kumar, and A. K. Ganguli, ACS Sustain. Chem. Eng. 4, 1487 (2016).
M. R. S. Kebria, M. Jahanshahi, and A. Rahimpour, Desalination 367, 255 (2015).
X. P. Li, Y. B. Chen, X. Y. Hu, et al., J. Membr. Sci. 471, 118 (2014).
X. Huang, X. W. Bo, Y. X. Zhao, et al. Bioresour. Technol. 165, 116 (2014).
F. Q. Wang, C. M. Wang, Z. C. Yu, et al. RSC Adv. 5, 70086 (2015).
J. D. Xiao, L. L. Han, J. Luo, et al., Angew. Chem. Int. Ed. 57, 1103 (2018).
X. Z. Fang, Q. C. Shang, Y. Wang, et al., Adv. Mater. 30, 1705112 (2018).
J. L. Wang, C. Wang, and W. B. Lin, ACS Catal. 2, 2630 (2012).
H. M. Zhao, Q. S. Xia, H. Z. Xing, et al., ACS Sustain. Chem. Eng. 5, 4449 (2017).
L. L. Wen, J. B. Zhao, K. L. Lv, et al., Cryst. Growth Des. 12, 1603 (2012).
G. J. Xu, Y. H. Zhao, K. Z. Shao, et al., CrystEngComm 11, 1842 (2009).
L. Qin, H. Z. Chen, J. Lei, et al., Cryst. Growth Des. 17, 1293 (2017).
G. M. Sheldrick, Acta Crystallogr. 71, 3 (2015).
Bruker, APEX2, SAINT, and SADABS (Bruker AXS Inc., Madison, WI, 2009).
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (no. 21601045) and the China Postdoctoral Science Foundation (no. 2016M601768), as well as by the Fundamental Research Funds for the Central Universities (JZ2019HGTB0059, S201910359319), as well as by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Joint Laboratory of Atmospheric Pollution Control, and Jiangsu Engineering Technology Research Center of Environmental Cleaning Materials.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yue Dou, Li, GY., Ma, XR. et al. Two Co/Ni(II)-Based Coordination Polymers for Degradation of Dyes. Crystallogr. Rep. 66, 1262–1267 (2021). https://doi.org/10.1134/S1063774521070233
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521070233