
Abstract
In recent years, microRNAs have been used as cancer markers in clinical testing. In this study, an electrochemical biosensor was developed based on nanomaterials and the hybridization chain reaction (HCR) for highly sensitive and selective detection of the target miR-378 using K3Fe(CN)6/K4Fe(CN)6 as a redox indicator. MiR-378 is located on human chromosome 5q32 and serves as an important gene regulatory locus. Recent studies have found that aberrant expression of miR-378 is associated with cervical cancer, breast cancer, lung cancer, and other diseases. HCR is a simple and efficient isothermal amplification technique that does not require enzyme mediation and can be performed at room temperature. Evaluations of the specificity, stability, and sensitivity of the developed sensor revealed its ability to rapidly and specifically detect miR-378 in two hours with a detection limit as low as 100 cells in a volume of 400 µL, thereby indicating that this device could provide a novel approach for clinical diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
MicroRNAs (miRNAs) are a class of small non-coding RNAs approximately 18–24 nucleotides in size that play a diverse range of important regulatory roles in cells [1–3]. Recent studies have shown that numerous diseases, including lung, cervical, and breast cancers, are associated with high expression of miRNAs [4–6]. Consequently, these RNAs could serve as valuable biomarkers for disease diagnosis, treatment, and rehabilitation. Almost all existing miRNA detection approaches, such as northern blotting [7], in situ hybridization, microarray chip technology, and reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) [8, 9], are based on hybridization of the target miRNA with a capture probe, which comprises a complementary nucleotide sequence. However, all these methods have certain drawbacks. For example, whereas RT-qPCR, currently the gold standard for miRNA analysis, has both high selectivity and sensitivity, the procedure is time-consuming and requires highly qualified personnel and large instruments. Consequently, there is a need for an alternative simple and rapid method for detecting specific miRNA sequences that can be readily performed in non-specialized laboratories.
Given the specific characteristics of short miRNA sequences and their typically low abundance, effective signal amplification is an essential prerequisite. Among the methods of nucleic acid amplification commonly used at present are ligase chain reaction (LCR) [10, 11], loop-mediated isothermal amplification (LAMP) [12, 13], strand displacement amplification (SDA) [14, 15], and rolling circle amplification (RCA) [16, 17]. The LCR method although also has stringent requirements for primer design and necessitates close attention to the enzyme environment. Although the LAMP method technique has the advantages of being simple and rapid and does not require any special instrumentation, it also has stringent requirements for primer design and laboratory testing. The SDA method is a novel method for thermostatically amplifying DNA, which is based on the combined action of two enzyme types, a restriction endonuclease and DNA polymerase. Whereas this method is highly sensitive, the target sequence preparation procedure is nevertheless complex, thereby limiting its application. RCA is an isothermal nucleic acid amplification technique that enables not only linear amplification of target sequences but also signal amplification with high efficiency and low detection limits. In this regard, the hybridization chain reaction (HCR) [18, 19] has recently emerged as a simple and efficient isothermal amplification technique that overcomes the drawbacks of the hybridization techniques mentioned above. It is based on a point-mediated chain substitution reaction that does not require enzyme mediation, is relatively inexpensive, and can be performed at room temperature.
Since their early development in the 1980s, electrochemical biosensors [20, 21] have gradually evolved and matured. These devices essentially comprise the target substance (e.g., antigen, nucleic acid, or antibody) as a sensitive element fixed to an electrode, which converts the biological signal of the substance to a detectable electric signal output, such as current or resistance, thereby enabling specific recognition of the target biomolecules [22–24]. They have the advantages of miniaturization, high sensitivity, low cost, and good specificity, and have received considerable attention in multiple fields, including materials, chemistry, biology, and medicine [25–27].
In recent years, nanomaterials have been increasingly used in electrochemical sensors due to their large specific surface areas and good optical, electrical, magnetic, and mechanical properties. Given their excellent biocompatibility and chemical inertness, gold nanoparticles (AuNPs) are extensively used as nanomaterials for electrode modification [28, 29]. Notably, they can strongly combine with nucleic acids, antibodies, and other biomolecules, enhancing the electron transfer rate for electrochemical signal amplification.
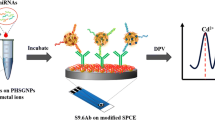
In this study, a strategy was developed for amplifying and detecting miR-378 using HCR and electrochemical biosensors. Based on the characteristics of low content and high homology of miR-378, the article proposed using the HCR reaction for cyclic amplification of miR-378, employing the principle of base complementary pairing. The hybridization product is then incubated on the working electrode. Due to the different structures presented on the electrode, electrochemical detection using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) is utilized to obtain different electrical signals, enabling efficient and sensitive specific detection of miR-378. This method, which is inexpensive, simple, and does not require any enzymes, can quantitatively detect miRNA-378 at the picomolar level, highlighting its novel potential for the detection of miRNA-378.
EXPERIMENTAL
Apparatus. UV-Vis spectra were collected on a Shimadzu 3600 Plus UV-Vis spectrophotometer. Size distribution of gold nanoparticles was tested using a Malvern Nano ZS90 Particle Size Potentiometer (Britain) through dynamic light scattering. The characterization of the nanoparticles was performed using a Hitachi High-Technologies Corporation transmission electron microscope (model H-7650, Japan). Gel electrophoresis apparatus from Liuyi Biotechnology Co., Ltd. (Beijing, China). Gel imaging was carried out using the Shanghai Tanon Company gel image (model Tanon 1600). Electrochemical measurements were taken using a CHI 660C electrochemical workstation (Shanghai, China) with a three-electrode system consisting of a gold plate electrode (5.0 mm), a platinum wire electrode, and a reference electrode of Ag/AgCl (sat. KCl). Electrochemical experiments were performed in a 2.5 mM K3Fe(CN)6 : K4Fe(CN)6 (1 : 1) solution containing 0.5 M KCl using CV and DPV. CV was performed at a scanning potential from –0.2 to 0.6 V at a scan rate of 30 mV/s. All electrodes were purchased from Chenhua Instrument Company (Shanghai, China). DPV was performed at a scanning potential from 0 to 0.6 V with an increment potential of 4 mV, while the amplitude, pulse width, and pulse period were set to 0.05 V, 0.05, and 0.5 s, respectively. The parameters for the CV method experiment are shown below.
Materials and chemicals. Agarose regular for electrophoresis use grade, all synthetic DNA strands, HPLC-purified RNA were purchased from Sangon Biotech Co., Ltd. (Nanjing, China). Their corresponding sequences are included in Table 1. Diethylpyrocarbonate (DEPC)-treated water was purchased from Sangon Biotech Co., Ltd. as DNA or RNA dissolved solution. Phosphate-buffered saline containing 0.2 M NaH2PO4 and 0.2 M Na2HPO4 was used as electrode activation buffer, and these were purchased from Nanjing Chemical Reagent Co., Ltd. (Nanjing, China). 6-Mercapto-1-hexanol (MCH) was purchased from Bide Pharmatech Ltd. (China). Tris (2‑chloroethyl) phosphate (TCEP) was purchased from Meryer (Shanghai) Chemical Technology Co., Ltd. All remaining reagents were purchased from Nanjing Chemical Reagent Co., Ltd. (Nanjing, China), and all liquid reagents were analytically pure.
Construction of biosensors. Preparation of glassy carbon electrodes. Bare glassy carbon electrodes (GCEs) were sequentially polished with alumina polishing powders of 1.0, 0.3, and 0.05 μm, and then cleaned with distilled water. Thereafter, the GCEs were cleaned ultrasonically with ethanol, purified water, and acetone solutions, each for approximately 2 min. After rinsing the electrodes with purified water, they were dried at room temperature. The GCEs were then immersed in 0.5 M sulfuric acid for 30 s for activation. Electrochemical tests, including CV and DPV measurements, were performed.
Preparation of gold nanoparticles. Nanogold was prepared by mixing 90 mL of distilled water with 10 mL of a 10 mM HAuCl4 solution in a three-necked flask and heating to 100°C under reflux with vigorous stirring. Following boiling, 5.2 mL of 75 mM trisodium citrate was added to initiate the reaction, and after approximately 3 min, the color of the solution changed from pale yellow to deep red. The solution was then refluxed for 40 min and cooled to room temperature with stirring. All glassware was cleaned with dilute nitric acid and thereafter thoroughly rinsed with distilled water before use.
Surface modification of glassy carbon electrodes. The surfaces of the GCEs were modified by dropping a 1 g/L solution of poly(diallyldimethylammonium) chloride(PDDA) containing 0.5 mol/L NaCl onto the electrode surface. PDDA is a cationic surfactant, and chloride ions dissociate after dissolution in water, leaving only a positively charged polymer, which can be used to adsorb negatively charged ions. The treated GCEs were subsequently submerged in the PDDA solution for 6 h to adsorb further positive charges on the electrode surface and were thereafter rinsed three times with purified water to remove the unstable PDDA. Subsequently, the prepared spherical nanogold particles were coated onto the electrode surface with PDDA and left overnight.
Connection of sulfhydryl DNA probes to gold nanoparticles. To attach sulfhydryl DNA (SH-DNA) probes to the AuNPs, 1 mM TCEP in NaAc−HAc buffer pH 5.2 was initially added to centrifuged SH-DNA, and the resulting mixture was allowed to react for 1 h. This step aimed to reduce the number of disulfide bonds in the sulfhydryl DNA. Subsequently, Tris-HAc buffer pH 8.8 and 1 M NaCl were added, mixed well in a vortex shaker, centrifuged, and allowed to react dropwise on the GCE for 24 h for Au–S bonding. Prior to electrochemical testing, the electrodes were rinsed with deionized water and then immersed in an aqueous solution of 1 mM MCH for 1 h to displace nonspecifically adsorbed aptamer molecules and to passivate the electrode surface.
Cell culturing and extraction of miRNA-378. HeLa cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere. During the exponential phase of growth, cells were collected and washed twice with phosphate-buffered saline (pH 7.4), and thereafter suspended in 1 × CHAPS lysis buffer (1 × 106 cells). The suspension was shaken at 4°C for 30 min and centrifuged at 10 000 rpm for 30 min at 4°C. The resulting supernatant was collected and stored at –80°C until used for further analysis [30–32].
Electrochemical detection of hybridized DNA. Two stem-loop structural probes, HP1 and HP2, were designed based on HCR. Initially, HP1 was designed according to the base sequence of miR-378. Bases on HP1 were selected to be complementary paired with miR-378 and form a stem-loop structure by itself. The secondary structure of HP1 was simulated and screened using the website [33]. Subsequently, the structure of the stem-loop HP2 was designed based on HP1. The number of bases hybridizing between HP1 and HP2 was ensured to be greater than the number of hybridization bases between miR-378 and HP1. The secondary structure of HP2 was simulated and screened accordingly. A set of stem-loop sequences with the best experimental results was selected by agarose gel electrophoresis experiments from the multiple sets of sequences designed. In the absence of the target miR-378, the two probes HP1 and HP2 can exist stably. However, in the presence of miR-378, the secondary structure of hairpin HP1 is opened to form a miR-HP1 double chain, and the sticky end of the stem end of HP1 is exposed, thereby functioning as a promoter to open hairpin HP2, the base part of which is identical to the target sequence. Based on kinetic and thermodynamic factors, HP2 displaces miR-378 and finally forms the long double-stranded HP1-HP2 structure. The displaced target is continuously recycled in the solution, contributing to signal amplification.The hybridization reaction mixture containing HP1 (10 µM, 15 µL) and HP2 (10 µM, 10 µL) was heated at 95°C for 5 min, cooled slowly to room temperature, and then added to the target RNA (miRNA-378,10 µM, 10 µL) and 1 × hybridization buffer (20 mM Tris-HCl, 1 mM EDTA, 5 mM MgCl2·6H2O, and 50 mM NaCl; pH 8.0). To amplify the target RNA, this mixture (40 µL) was incubated at 37°C for 60 min, after which it was dropped onto the surface of the SH-DNA/GCE/Au electrodes for 2 h. The electrodes were then rinsed three times with double-distilled water and subjected to electrochemical testing (Fig. 1).

Electrochemical sensor construction and detection principle.
RESULTS AND DISCUSSION
Characterization of nanogold. The prepared AuNPs were analyzed using a UV-Vis spectrophotometer. As shown in Fig. 2, the maximum absorption peak was observed at 520 nm, consistent with the surface plasmon resonance peak of these nanoparticles. The prepared nanogold was measured again under UV light after 30 days, revealing a negligible shift in the characteristic peak, indicating good stability of the prepared AuNPs.

UV absorption spectra of nanogold.
The fine morphology of the prepared AuNPs was characterized using transmission electron microscopy. As shown in Fig. 3a, the AuNPs had uniform spherical shapes. A size distribution plot is shown in Fig. 3b, constructed by calculating the diameters of all particles using the Image-Pro Plus labeling map. The diameters of these particles ranged mostly between 11 and 16 nm, with an average particle size and relative standard deviation of 12.98 nm and 15.38%, respectively. These findings indicate the presence of relatively uniformly sized AuNPs. The zeta potential of AuNPs prepared using the sodium citrate reduction method was –38.2 mV (Fig. 4). The negative charge of these particles can be ascribed to the adsorption of citrate and its carboxylate oxidation products on the nanoparticle surface. This low zeta potential of AuNPs contributes to their good stability.

Gold nanoparticle morphology: (a) a transmission electron micrograph of nanogold particles, (b) nanoparticle size distribution.

The zeta potential of nanogold.
Testing of the miR-378 electrochemical sensor. HP1 and HP2 probes with hairpin structures were designed for miR-378 HCR amplification with reference [33], and the HCR of miR-378 was confirmed using agarose gel electrophoresis. The experimental steps were as follows: firstly, a washed and dried electrophoretic gel-making mold was prepared, and a comb was inserted. 1.2 g of agarose was weighed in a beaker using an electronic balance, and 30 mL of 1 × Tris-acetate-EDTA (TAE) buffer was added. The mixture was heated with an electric stove until boiling and transparent, then the stove was switched off. After cooling to about 60°C, 2 µL of 4S Gelred nucleic acid dye was added, stirred well, and the gel solution was poured slowly into the mold to spread it evenly. The gel was left at room temperature for 30 min until completely solidified, then the comb was gently pulled out, and the gel plate was taken out and placed into the electrophoresis tank. 1 × TAE buffer was added to submerge the gel plate completely. For spotting, 2 µL of 6×loading buffer was mixed with 10 µL of DNA sample, and then 10 µL of the mixture was added into the gel well. After dispensing, the electrophoresis instrument was turned on, with the constant current at 120 mA and voltage controlled between 80 and 120 mV. Electrophoresis was timed for 60 min. After electrophoresis, the gel plate was taken out and placed into the UV imaging system to observe the position of the DNA bands, and pictures were taken for storage. Generally, DNA with a small number of bases migrated relatively fast, further away from the spotting hole. When DNA is hybridized, the number of bases increases, and the migration rate slows, closer to the sample wells. As shown in Fig. 5c, lane 1 is a Marker, 50–500 bp in length, serving as an electrophoretic marker indicating the position of the single-stranded DNA/RNA intervals. Here HP1 and HP2 are stem-looped, and stem-looped DNA moves at a faster rate than single-stranded DNA, so the Marker can only be used as a positive control to see where the miR-378 bands are located, as well as to verify electrophoretic manipulation without problems.

The structure of DNA probes: (a) HP1, (b) HP2, (c) PAGE experiment confirmation of the nucleic acid reactions: lane 1, Marker; lane 2, HP1; lane 3, HP2; lane 4, miR-378; lane 5, HP1 + HP2; lane 6, HP1 + HP2 + miR-378.
HP1 and HP2 each existed stably in a hairpin structure, and in the absence of miR-378, they were unable to open each other to form a long double-stranded structure. Conversely, in the presence of the target miR-378, HCR was triggered, resulting in the formation of a long double-stranded HP1-HP2 structure, as indicated by the gel band with a slower rate of migration in lane 6 of Fig. 5c.
To verify that SH-DNA had bound to the AuNPs and hybridized with the HCR products on the electrode surface, the CV responses of the variegated states of the electrodes were analyzed. As shown in Fig. 6, the control bare GCE and the experimental bare GCE showed a pair of redox peaks of [Fe(CN)6]3–/4– around 0.21 and 0.27 V (curves GCE). A significant increase in the peak current of the GCE/Au was observed, attributable to the excellent conductivity and large surface area of the AuNPs (curves AuNPs/GCE). Upon successfully immobilization of SH-DNA on the GCE/Au surface, a reduction in the peak current occurred due to the mutual repulsion between the phosphate molecules on the DNA surface and [Fe(CN)6]3–/4– (curves SH-DNA/AuNPs/GCE). To eliminate nonspecific adsorption and background staining, MCH blocking was performed, further reducing the peak current (curves MCH/SH-DNA/AuNPs/GCE). In response to further modifications, the control and experimental electrodes were found to have similar electrical properties, indicating the stability of the modified electrodes, beneficial for detecting miR-378. The experimental electrode exhibited a significant reduction in the peak current, attributed to the formation of a long double-stranded hybrid of SH-DNA and HCR products (curves with target/MCH/SH-DNA/AuNPs/GCE), while the peak current of the control electrode showed a slight reduction associated with the partial hybridization of SH-DNA with HP1 in the absence of miR-378 (curves without target/MCH/SH-DNA/AuNPs/GCE). The DPV method is a unidirectional current and evaluates the magnitude of the current similar to cyclic voltammetry, with the (c) and (d) plots corresponding to the (a) and (b) plots, respectively, displaying the same current trend.

Cyclic voltammetry verification of the application of electrochemical biosensors for (a) the control and (b) experimental electrodes. Differential pulse voltammetry verification of the application of electrochemical biosensors for (c) the control and (d) experimental electrodes.
Specificity and stability of the developed HCR-based electrochemical biosensor. To determine whether the HCR-based electrochemical biosensor developed herein could be applied to obtain an accurate disease diagnosis, it was important to establish whether this biosensor can specifically detect miR-378. To this end, the constructed sensors were used to sequentially detect miR-21, miR-155, and target miR-378, and their specificities were determined based on the respective peak current response values. As shown in Fig. 7a, the concentration of the samples was kept consistent in the experiment. The blank comprised HP1 + HP2 + DEPC water, miR-155 was the sample containing HP1 + HP2 + miR-155, miR-21 was the sample containing HP1 + HP2 + miR-21, and miR-378 referred to the sample containing HP1 + HP2 + miR-378. This method was employed to check the specificity of the experimental procedure. Only the presence of miR-378 resulted in a substantial change in the current response value, indicating that the constructed sensor had good selectivity.

(a) The specificity of sensors measured using the cyclic voltammetry method; (b) electrochemical tests were done to check the stability of the electrodes numbered 1, 2, and 3, respectively, by immersing them in purified water at the end of the experiment for 1, 3, 5, 7 days.
The stability of the biosensor was also assessed using three parallel testing electrodes for miR-378. The biosensors were immersed in purified water and stored at 4°C. These biosensors were evaluated at different time points, with the respective peak currents shown in Fig. 7b. After 7 days, the error bars of the values obtained for the three electrodes were 0.05, 0.14, and 0.09, and when measured after 3, 5, and 7 days, the current values were 95.4, 90.5, and 93.2% of the respective initial values, indicating the good stability of the sensors.
Cellular assays. On the basis of query results obtained from the DepMap website [34], miRNA-378 was found to exhibit elevated expression in HeLa cells. Accordingly, to evaluate the accuracy of the developed biosensors, they were used to detect different concentrations in HeLa cell lysates. As shown in Fig. 8, the results clearly demonstrated that the higher the HeLa cell concentration, the more significant the changes in peak current, indicating the sensitivity of these biosensors to the content of miR-378. Moreover, it was established that the detection limit was as low as 100 HeLa cells, indicating that this device could provide a new method for clinical diagnosis.

The detection performance of biosensors at different HeLa cell concentrations. The relative rate of change relative to the blank was tested by cyclic voltammetry with different amounts of HeLa cells added at the same concentration of HP1 and HP2 in the same volume.
CONCLUSIONS
In this study, an electrochemical biosensor was developed for the detection of miRNA-378, a tumor marker, in cells. To construct these biosensors, glassy carbon electrodes were initially modified with gold nanoparticles, providing binding sites for DNA-specific fixation via Au-S bonds. Using an electric biosensor based on HCR cyclic amplification of the target miRNA-378, a detection limit as low as 100 HeLa cells was achieved. Furthermore, evaluations of the specificity and stability of the constructed biosensor indicated its excellent electrochemical performance. This biosensor shows considerable promise for providing new perspectives and methods for clinical marker detection. Although electrochemical biosensors have the advantages of low cost, high sensitivity, and convenience, their detection signals can be unstable, which challenges the reproducibility and specificity of experimental results. The development of nanomaterials and advancements in science and technology have pushed sensor development in the direction of miniaturization and intelligence. In the future, more electrochemical sensors with special properties and advantages will continue to emerge and find practical applications.
REFERENCES
Ranjbari, S., Rezayi, M., Arefinia, R., Aghaee-Bakhtiari, S.H., Hatamluyi, B., and Pasdar, A., Talanta, 2023, vol. 255, p. 124247. https://doi.org/10.1016/j.talanta.2022.124247
Kasturi, S., Eom, Y., Torati, S.R., and Kim, C., J. Ind. Eng. Chem., 2021, vol. 93, p. 186. https://doi.org/10.1016/j.jiec.2020.09.022
Kokkinos, C., Nanomaterials, 2019, vol. 9, p. 1361. https://doi.org/10.3390/nano9101361
Wang, J., Wang, D., and Hui, N., Bioelectrochemistry, 2020, vol. 136, p. 107595. https://doi.org/10.1016/j.bioelechem.2020.107595
Weng, C., Li, X., Lu, Q., Yang, W., Wang, J., Yan, X., Li, B., Sakran, M., Hong, J., Zhu, W. and Zhou, X., Microchim. Acta, 2020, vol. 187, p. 575. https://doi.org/10.1007/s00604-020-04548-5
Rezaei, H., Motovali-Bashi, M., and Radfar, S., Anal. Chim. Acta, 2019, vol. 1092, p. 66. https://doi.org/10.1016/j.aca.2019.09.037
Trayhurn, P., Proc. Nutr. Soc., 1996, vol. 55, p. 583. https://doi.org/10.1079/pns19960051
Emery, S.L., Erdman, D.D., Bowen, M.D., Newton, B.R., Winchell, J.M., Meyer, R.F., Tong, S.X., Cook, B.T., Holloway, B.P., McCaustland, K.A., Rota, P.A., Bankamp, B., Lowe, L.E., Ksiazek, T.G., Bellini, W.J., and Anderson, L.J., Emerg. Infect. Dis., 2004, vol. 10, p. 311. https://doi.org/10.3201/eid1002.030759
Corman, V.M., Landt, O., Kaiser, M., Molenkamp, R., Meijer, A., Chu, D.K.W., Bleicker, T., Bruenink, S., Schneider, J., Schmidt, M.L., Mulders, D.G.J.C., Haagmans, B.L., van der Veer, B., van den Brink, S., Wijsman, L., Goderski, G., Romette, J.-L., Ellis, J., Zambon, M., Peiris, M., Goossens, H., Reusken, C., Koopmans, M.P.G., and Drosten, C., Eurosurveillance, 2020, vol. 25, p. 23. https://doi.org/10.2807/1560-7917.Es.2020.25.3.2000045
Liu, Z.J., Yang, L.Y., Lu, T.C., Liang, Y.Q., Liu, M.M., Zhong, G.X., Lin, X.H., Huang, P.F., and Chen, J.Y., Sens. Actuators, B, 2022, vol. 368, p. 132096. https://doi.org/10.1016/j.snb.2022.132096
Yang, L.Y., Xu, X.W., Lin, Y., Ye, C.L., Liu, W.Q., Liu, Z.J., Zhong, G.X., Xu, Y.F., Lin, X.H., and Chen, J.Y., Anal. Chem., 2023, vol. 95, p. 5331. https://doi.org/10.1021/acs.analchem.2c05421
Gonzalez-Lopez, A., Cima-Cabal, M.D., Rioboo-Legaspi, P., Costa-Rama, E., Garcia-Suarez, M.D., and Fernandez-Abedul, M.T., Anal. Chem., 2022, vol. 94, p. 13061. https://doi.org/10.1021/acs.analchem.2c02127
Marangoni, J.M., Ng, K.K.S., and Emadi, A., Micromachines, 2023, vol. 14, p. 472. https://doi.org/10.3390/mi14020472
Zhang, R., Zhang, M.R., Ding, Q., Yuan, R., and Yuan, Y.L., Anal. Chim. Acta, 2023, vol. 1238, p. 340609. https://doi.org/10.1016/j.aca.2022.340609
Ouyang, P., Qing, Y., Zou, S.H., Fang, C.X., Han, J.L., Yang, Y.X., Li, H.Y., Wang, Z.C., and Du, J., Biochem. Eng. J., 2022, vol. 186, p. 108576. https://doi.org/10.1016/j.bej.2022.108576
Ding, Z.S., Yang, S., Wang, J., Zhao, Z.Y., Xu, H.Q., Chen, Z.G., Liu, Z.Y., Wang, Y.X., Bao, J., Chang, K., and Chen, M., ChemElectroChem, 2023, vol. 10, p. e202300257. https://doi.org/10.1002/celc.202300257
You, J., Park, H., Lee, H., Jang, K., Park, J., and Na, S., Biosens. Bioelectron., 2023, vol. 224, p. 115078. https://doi.org/10.1016/j.bios.2023.115078
Dirks, R.M.and Pierce, N.A., Proc. Natl. Acad. Sci. U. S. A., 2004, vol. 101, p. 15275. https://doi.org/10.1073/pnas.0407024101
Ge, Z.L., Lin, M.H., Wang, P., Pei, H., Yan, J., Sho, J.Y., Huang, Q., He, D.N., Fan, C.H., and Zuo, X.L., Anal. Chem., 2014, vol. 86, p. 2124. https://doi.org/10.1021/ac4037262
Smutok, O. and Katz, E., Biosensors, 2023, vol. 13, p. 44. https://doi.org/10.3390/bios13010044
Mohammadpour-Haratbar, A., Boraei, S.B.A., Zare, Y., Rhee, K.Y., and Park, S.J., Biosensors, 2023, vol. 13, p. 80. https://doi.org/10.3390/bios13010080
Kimmel, D.W., LeBlanc, G., Meschievitz, M.E., and Cliffel, D.E., Anal. Chem., 2012, vol. 84, p. 685. https://doi.org/10.1021/ac202878q
Luo, L., Wang, L., Zeng, L., Wang, Y., Weng, Y., Liao, Y., Chen, T., Xia, Y., Zhang, J., and Chen, J., Talanta, 2020, vol. 207, p. 120298. https://doi.org/10.1016/j.talanta.2019.120298
Yan, Q.H., Zhi, N., Yang, L., Xu, G.R., Feng, Q.G., Zhang, Q.Q., and Sun, S.J., Sci. Rep., 2020, vol. 10, p. 10607. https://doi.org/10.1038/s41598-020-67394-8
Maduraiveeran, G., Sasidharan, M., and Ganesan, V., Biosens. Bioelectron., 2018, vol. 103, p. 113. https://doi.org/10.1016/j.bios.2017.12.031
Nunez, C., Jose Trivino, J., and Arancibia, V., Talanta, 2021, vol. 223, p. 121702. https://doi.org/10.1016/j.talanta.2020.121702
de Brito, A.R., de Jesus, R.S., Tavares, I.M.D., Silva, F.N., Santana, N.B., Ferrao, S.P.B., Bilal, M., Santos, A.D., Salay, L.C., de Oliveira, J.R., and Franco, M., Surf. Interfaces, 2021, vol. 22, p. 100839. https://doi.org/10.1016/j.surfin.2020.100839
Khan, I., Saeed, K., and Khan, I., Arab. J. Chem., 2019, vol. 12, p. 908. https://doi.org/10.1016/j.arabjc.2017.05.011
Ma, C., Lu, D.N., Gan, H.H., Yao, Z.Y., Zhu, D.Z., Luo, J.Y., Fu, Q., and Kurup, P., Talanta, 2023, vol. 253, p. 124009. https://doi.org/10.1016/j.talanta.2022.124009
Ma, F., Wang, T.T., Jiang, L.H., and Zhang, C.Y., Anal. Chim. Acta, 2019, vol. 1053, p. 122. https://doi.org/10.1016/j.aca.2018.11.058
Zhang, B., Shi, L., Liu, W., Li, B.X., and Jin, Y., Analyst, 2020, vol. 145, p. 6992. https://doi.org/10.1039/d0an01291b
Ma, Y.X., Mao, G.B., Wu, G.Q., Fan, J., He, Z.K., and Huang, W.R., Sens. Actuators, B, 2020, vol. 304, p. 127385. https://doi.org/10.1016/j.snb.2019.127385
OligoAnalyzer Tool, Integrated DNA Technologies, Inc. https://sg.idtdna.com. Accessed March 1, 2024.
The Broad Institute’s Cancer Program. The Cancer Dependency Map Project (DepMap). https://depmap.org/portal/. Accessed March 1, 2024.
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors of this work declare that they have no conflicts of interest.
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Q., Fan, B., Wang, S. et al. A Hybridization Chain Reaction-Based Electrochemical Biosensor for Highly Sensitive and Selective Detection of miR-378. J Anal Chem 79, 1329–1337 (2024). https://doi.org/10.1134/S1061934824700692
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934824700692