
Abstract
The chemical and electrochemical syntheses of polyaniline (PANI) were performed in a sulfuric electrolyte in the presence of the vanadate anion (VA). The resulting composite materials based on PANI and VA (PANI–VA) were characterized by IR spectroscopy, X-ray diffractometry, SEM, and elemental analysis. In the electrochemical trials, conditions were indicated under which the composite material retained 96% of its electrochemical capacitance (C) and the cycling of PANI–VA in the range of potentials extended to the anodic and cathodic regions did not lead to the degradation of the polymer material over 50 cycles.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 INTRODUCTION
Currently, methods for the synthesis and study of materials that are potential components of energy storage products [1–3] are being actively developed. Carbon materials (carbon nanotubes, graphenes) and metal oxides are widely used, which can expand the window of potentials for charge-discharge processes and provide high specific surface area to increase the double-layer capacitance. The main disadvantages of these materials are relatively low intrinsic specific capacitance and energy density [4–7]. Several methods can be used to improve their characteristics: the use of new composites as components, e.g., materials that provide stable double-layer capacitance, create high pseudocapacitance, and allow the expansion of the window of potentials. Promising candidates for supercapacitor electrodes are composite materials based on polyaniline (PANI) and oxide compounds of metals with variable valence, which show high specific capacitance [5, 8]. Some transition metal oxides such as indium, vanadium, and tungsten oxides are pseudocapacitance materials with a large window of potentials, which can thus be used as an alternative to carbon-based electrodes. Among these, vanadium oxide compounds are of particular interest [9]. Presumably, vanadates can show high pseudocapacitance due to several oxidation states of vanadium [10], which allows various redox processes on its surface. The oxidation potential of the V+5/V+4 system depends on pH of the medium and can be increased by increasing the solution acidity [11]. Vanadium oxides proved effective at potentials from ‒1 to +0.9 V [12, 13]. PANI as a component of composite materials is well defined because of easy synthesis and high stability in air. It can be assumed that combination of these two components in one material will open up an opportunity of creating a new composite high-capacitance electrode element for a supercapacitor. This paper discusses the use of vanadic acid (VA) as an electroactive participant in the chemical and electrochemical syntheses (ESs) of PANI–VA, the properties of the resulting materials, and conditions for their effective operation.
2 EXPERIMENTAL
The electrochemical synthesis of PANI was performed in a potentiodynamic mode at potentials from –0.2 to +0.7 V (relative to Ag/AgCl) in a three-compartment glass electrochemical cell (the electrolyte volume used was 15 mL) at room temperature (21–22°C). The spaces of the working and auxiliary electrodes were separated with a porous glass partition. The working solutions used as electrolytes are presented in Table 1. The sample obtained from E1 is designated as PANI-S; the samples obtained from electrolytes with VA additions, are denoted accordingly as variants of PANI–VA with indications of the synthetic procedure. The working electrode was the surface of a glassy carbon (GC) rod with a diameter of 5 mm (S = 0.2 cm2) sealed in a glass tube. The auxiliary electrode was a GC plate of 1 × 5 cm. The product was synthesized in large quantities for physicochemical studies on a GC plate of 1 × 5 cm. All the potentials are given relative to Ag/AgCl. The solutions were prepared from freshly distilled water with pH 6.7. To measure pH of the prepared electrolytes, a portable Checker pH meter (Hanna, Portugal) was used with a measurement accuracy of ±0.2 pH. The electrochemical tests of the resulting materials were performed on a PS-7 potentiostat (Elins, Russia) with computer programming. The digital processing of the electrochemical data was performed on a computer.
The electron microscopic studies were performed on a Supra 25 field emission scanning electron microscope (Zeiss) with an INCA energy dispersive X-ray analyzer (Oxford Instruments) to determine the element composition of the samples obtained in the chemical synthesis. The resolution of the images was 1–2 nm. The IR spectra were recorded on a Perkin Elmer Spectrum 100 Fourier IR spectrometer equipped with a frustrated multiple total internal reflection (FMTIR) attachment with a germanium prism in the range 4000–675 cm–1. The dry samples were placed on the prism of the attachment and pressed using a mechanical presser. A comparative characteristic of the structure of the obtained preparations was evaluated by X-ray diffractometry using an ARL X’TRA instrument (Switzerland). The samples in powder form were ground in vaseline oil and placed in a fused quartz cell.
3 RESULTS AND DISCUSSION
3.1 Formation of PANI–VA
Vanadium in the oxidation state +5 can form various vanadium salts (vanadates), both monomeric (meta, ortho, pyro) and polymeric (hexa, deca, dodeca). It can also form complex compounds with various ions and organic ligands. These forms of vanadate ions equilibrate in solution, one of the forms prevailing in each pH range [14, 15]. Vanadic acids are nearly as weak as acetic acid. The potential of the transition of vanadium from the highest oxidation state, +5, to the next state, +4, and further strongly depends on pH of the medium and can be increased by increasing the acidity of solution. The reduction of V+5 is also observed when the vanadate solution interacts with some reducing agent in a sulfuric medium [11, 14]. In our case, this one-electron reducing agent can be the An molecule, which becomes the start of the polymerization chain during the one-electron oxidation to the radical cation. The possibility of this catalytic effect and the peculiarities of PANI–V2O5 bonding, which led, e.g., to the development of a new promising cathode material, were indicated in [16, 17]. Thus, a reduction of V+5 to V+4 with one-electron oxidation of An can take place in a solution of An and VA at pH ≤ 4, leading to the formation of PANI, which eventually settles from solution as a dark precipitate (PANI‒VA(a)1), whose properties will be discussed below. The filter cake was washed with distilled water and dried in a vacuum gun at 40°C for 24 h. The element analysis of the precipitate allows us to identify it as [С5.7Н5N(VО3)0.3(SО4)0.3 · 2Н2О].
3.2 Electrochemical Synthesis of PANI–VA
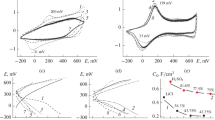
The electrochemical synthesis of PANI in an electrolyte containing VA is performed (until precipitation is completed) in the presence of a V+5 initiator of ES in electrolyte. As already mentioned, the chemical synthesis of PANI–VA also starts in this electrolyte, and its rate can be decreased by maintaining pH ≤ 4. Figure 1 shows examples of the first cycles of ES performed in a potentiodynamic mode in E1 (CV1) and E6 (CV2). According to Fig. 1, the cyclic voltammetry (CV) characteristic CV2, in contrast to CV1, shows PANI formation already on the first cathodic and then anodic branch, as evidenced by the appearance of the reduction peak of the product at 23 mV and oxidation peak at 166 mV in the first cycle on this CV2 curve. On the CV1 curve, the first cycles do not show the formation of any An oligomerization products. In addition, according to these CV curves, the initial Eox of An lies at 670 mV in the E1 electrolyte (CV1), while the initial Eox of An in the E6 electrolyte (CV2) is 110 mV lower. It was also found that in sulfuric and hydrochloric acids, the ES of PANI in the presence of VA occurs differently.

First cycles of ES of PANI from E1 (CV1) and E6 (CV2). 50 mV/s.
The ES of PANI in both acids are shown in Fig. 2. The conditions of interaction of VA with An were fulfilled for both acids. The E3 solution (500 mL) was kept in air for a month (An : VA = 100). The solution remained yellow transparent and did not change color all this time. A month later, the solution was divided into two parts, and sulfuric acid was added to one part to pH 4 (E3(a) solution), and hydrochloric acid was added to the other part to pH 4 (E3(b) solution). This pH value was chosen in order to slow down the precipitation and enable the simultaneous chemical synthesis and the ES of PANI in the same solution. The E3(a) solution began to darken within a few minutes after the addition of sulfuric acid, and a dark precipitate slowly began to settle in it (PANI–VA(a) 1). The ES was performed in this solution in parallel with the start of precipitation in the potentiodynamic mode on the GC electrode. The progress of ES is shown in Fig. 2a (CV1). Active growth of the polymer coating of the electrode starts from the first cycle and continues until the ES is stopped. A coating (PANI–VA(a)2) was obtained in 15 cycles, the final CV of which is shown in the upper insert of Fig. 2a. The dashed curve (CV2) in the same figure shows the ES of PANI–VA(a)3 in E6, where the ratio is An : VA = 1500. The CV1 and CV2 show the first cycles of the ES. The start of ES on both CV curves is recorded as an increase in the CV area, and the peaks of polymerization products appear already in the first cycle. The total amount of PANI obtained within 10 cycles in E6 is shown on CV (bottom insert).

(a) ES from E3(a) in the presence of H2SO4 at pH 4 (first cycle of CV1). Development of CV response during the ES of PANI–VA(a)2 over 15 cycles (upper insert). The start of ES from E6 (first cycle of CV2) and development of the CV response during the ES of PANI–VA(a)3 (lower insert) over 10 cycles. 50 mV/s. (b) ES from E3(b) in the presence of HCl at pH 4 (CV1 and CV2, the first two cycles of ES). Development of the CV response during the ES of PANI–VA(b)2 over 50 cycles (insert).
In the E3(b) solution with a hydrochloric acid addition (also to pH 4), there were no visible changes for a few minutes; the color of the solution remained yellow. When the solution began to darken, a precipitate (PANI–VA(b)1) began to slowly settle from it; at the same time, the ES shown in Fig. 2b was performed. In the first cycle, shown in the main figure (Fig. 2b), there is an oxidation peak at 676 mV, as in the previous experiment, which most likely belongs to the oxidation of V+4 to V+5. Then, starting from the second CV, there are no peaks corresponding to the oxidative transformations of the vanadium component, possibly because the cathodic branch of CV has no reduction peak of V+5. Then the ES occurred very slowly to the end. A thin coating (PANI–VA(b)2) was obtained within 50 cycles; by the 50th cycle, the ES stopped almost completely, probably because of the formation of a polymer with high resistance. This overall picture is indicated on the CV in Fig. 2b (top insert). The CV of the formed coating resembles that of the polymer subjected to overoxidation in the anodic region with formation of numerous irreducible quinoid groups and all possible crosslinks between the chains [18].
As a result, two (in sulfuric acid) and one (in hydrochloric acid) polymer coatings were obtained in each of the described ES. The amount of the obtained polymers can be judged from the area of CV of both ESs. It is quite obvious that the oxidation of An and the ES itself are more active in electrolytes with sulfuric acid, all other things being equal. The different behaviors of the compounds and the rate and amount of PANI are possibly explained by the fact that acidification of the aqueous solution of VA and An with sulfuric acid [19] results in sulfovanadyl (VO2)2SO4, for which, as reported by the authors, vanadium transitions from the highest oxidation state +5 to the lowest state +2 via all the intermediate oxidation states are facilitated when the sodium vanadate solution reacts with any reducing agent of the corresponding strength in the sulfuric medium. In our case, the reducer for the V+5/V+4 transition is the An molecule, and for the next steps of vanadium reduction, the reducing agent may be the applied potential. The oxidation of An and the ES itself are more active in electrolytes with sulfuric acid, all other conditions being equal. It should also be noted that the ES of PANI was performed in the E3 electrolyte, which did not contain a mineral acid with pH 5.2. This ES stopped almost completely within the first few cycles, and the polymer did not accumulate.
3.3 Physicochemical Study of PANI–VA
The micrographs show that the polymers obtained from E1 (PANI-S) (Fig. 3a), E6, and E3 (PANI–VA(a)3 and PANI–V(a)1) (Figs. 3b and 3c, respectively) have diverse morphology. The PANI-S sample obtained in E1 is a fairly homogeneous mixture of globular particles with clear-cut edges with a diameter of 20–50 nm. The PANI–VA(a)3 particles obtained in E6 (at a ratio of An : VA = 1500) (Fig. 3b) are much larger (~50–100 nm), but also represent a rather loose mass. The PANI–VA(a)2 particles obtained in E3 (An : VA = 100) (Fig. 3c) coalesced; this can lead to a decrease in the surface area. An interesting possibility found for PANI is that it can be formed on the surface of VA by simply dropping the E1 solution onto the surface of VA. A black spot immediately forms at the site of contact of E1 with VA, while around it, the remaining VA material remains yellow (black-and-white insert, Fig. 3d). If the formed polymer is carefully removed, the micrograph shows the picture shown in Fig. 3d. The polymer itself is a gelatinous mass. In the presence of a large excess of VA, active aggregation of PANI tubes probably takes place due to the vanadium oxide compounds, which cover them in excess, as reported in [18, 19]. As noted in these publications, similar aggregation of PANI tubes in the presence of vanadium oxide compounds led to an abrupt decrease in the electrochemical capacitance (C), due to which the authors revealed the dependence of the amount of the vanadium oxide compound added to the polymer in order to preserve its stable electrochemical characteristics. The elemental analysis of our material, obtained after dropping E1 on VA, showed a small content of the carbon component (PANI); this confirms the formation of oligomers on the VA surface, the bulk of which was VA (it was very difficult to separate them in this case). The micrographs (Figs. 3e, 3f, and 3g) show the SEM images of VA (Figs. 3e and 3f) and VA surface (Fig. 3g) partially covered with the polymer particles that formed on it. An element of the deposit is enlarged and shown in the insert in the upper right corner. It can be seen that this product is a low-structured gel-like mass, which is obtained with a significant excess of VA, while the vanadium base itself retains its crystal structure. Also note that on an enlarged scale, all the materials in question (a, b, c) retain spongy morphology that allows the electrolyte to freely penetrate in the material.

SEM images: (a) PANI-S; (b) PANI–VA(a)3; (c) PANI–VA(a)2; (e, f) VA; (g) VA with the PANI element that formed on the surface; (d) general view of VA with the coating element shown in (g); insert: black-and-white image of VA on which the An solution was dropped.
The IR spectrum of PANI–VA (spectra 2–6, Fig. 4) contains the 1485 and 1577 cm–1 bands responsible for the vibrations of benzoid and quinoid groups (by analogy with the spectrum of PANI-S; curve 7, Fig. 4) [20, 21]. In the range 1100–1300 cm–1, the spectrum contains the bands that were attributed to various vibrations of C–N, N=Q=N, and other bonds [21]. The IR spectrum of PANI-S (curve 7, Fig. 4) contains a wide band in the range 1000–1100 cm–1 characteristic of the sulfo anion. The vibrations at 745 and 980 cm–1 indicate that the polymer has vanadium oxide compounds; the 745 cm–1 band indicates the presence of the V–O bond [19], and the 980 cm–1 band suggests the presence of the V=O bond [16 , 22]. The latter two bands are characteristic of the spectrum of VA (spectrum 1, Fig. 4), and their presence in the spectra of PANI–VA indicates that this anion is incorporated in the polymer. For nanocomposites for which it was known that they included V2O5, it was assumed [21] that, according to the intensity ratio of the 745 and 980 cm–1 bands, the morphology of the composite can change. The decrease in the intensity ratio of the V=O/V–O bands indicated increased amorphism, as in the case of PANI–VA(b)1 (spectrum 3, Fig. 4). In addition, an increase in the intensity of the V–O band suggests the formation of ortho-vanadate anions with an increased content of the V–O bond (V(O)(OH)3) compared to the meta-vanadate anion in which the V=O bands are more intense (V(O)2(OH). The slope of the IR spectra of the PANI–VA(a)1 (spectrum 2) and PANI–VA(a)3 (spectrum 6) samples suggests higher electron conductivity of these polymers compared with that of the others. The elemental analyses also confirm that all the samples contain vanadium; in PANI–VA(b)1, the amount of vanadium significantly increased relative to the calculated amount. The compositions of the products were calculated for various PANI–VA samples based on these analyses (Table 2).

IR spectra of (1) VA, (2) PANI–VA(a)1, (3) PANI–VA(b)1, (4) and (5) PANI–VA obtained by dropping E1 on VA (with 5 and 12% C), (6) PANI–VA(a)3, and (7) PANI-S.
These observations were confirmed by the XRD analysis of the compounds. Figure 5 shows the diffraction patterns of the PANI–VA samples. The diffraction pattern of VA (curve 1, Fig. 5) confirms the SEM image of VA (Figs. 4e and 4f). The compound has a crystal structure with the main angle 2θ = 11.3°, which means that the distance between the main planes of crystalline VA is almost the same over the entire mass of the substance. All the samples obtained on the surface of VA at its large excess also retain their crystal structure (curve 3, Fig. 5). PANI–VA(a)1, obtained from a solution with a ratio of An : VA = 100, revealed an amorphous structure; i.e., there were no diffraction peaks responsible for the angles between the crystal planes (curve 6, Fig. 5). The diffraction pattern of PANI–VA(b)1 (curve 2, Fig. 5) is interesting as the sample has fewer crystal formations than VA and is not so amorphous as PANI–VA(a)1. When ES is performed in the electrolyte with HCl, the resulting polymer contains a reasonably large amount of the vanadium component according to the elemental analysis. Probably as a result of this, the polymer has high resistance, which, in turn, slows down the formation of the composite, as mentioned above. During the elemental analysis, the PANI–VA(b)1 sample was burnt, and the diffraction pattern resembled that of the V2O5 sample after burning [21]. Figure 5 (curve 5) shows the diffraction pattern of the standard PANI-S sample; in the SEM micrograph, this a homogeneous, loose amorphous mass without any crystal formations. However, two PANI samples (curves 5 and 6, Fig. 5) are different. The center of the amorphous peak of PANI–VA(a)1 lies at 2θ = 20.6°, while for PANI-S, the angle is 18.2°. This difference in the interplanar distances of the two PANI samples probably means that the distance between the chains in PANI–VA(a)1 is smaller than in PANI-S. The reason for this may be the aggregation interactions of the ─N∙∙∙H∙∙∙O=V type inside the PANI–VA(a)1 polymer [18, 19, 22], which have already been mentioned when considering the morphology of the polymer materials. It should be emphasized once again that the solution with VA and An at pH 5.2 at any ratio of components can stand for a long time (for several months) without changing its (yellow) color and without precipitation of any products of dimerization, oligomerization, and polymerization of An. The main conclusion drawn from the series of previous syntheses was that in a sulfuric medium at definite pH values, V+5 can initiate the polymerization of PANI; i.e., it becomes an oxidant for the An molecule and can act as initiator of ES. Likewise, the redox processes were facilitated and the structural and electrochemical characteristics improved in the presence of V+5 in other syntheses of conducting polymer composite materials with V+5 oxide compounds [23, 24]. Importantly, the An : VA ratio should be carefully chosen to preclude the aggregation of polymer chains, as indicated on the SEM image, in order to preserve the active surface of the material to the maximum possible extent.

Diffraction patterns of (1) VA, (2) PANI–VA(b)1, (3) PANI on the surface of VA with 12% C, (4) VA oxide after burning PANI–VA(b)1, (5) PANI-S, and (6) PANI–VA(a)1.
3.4 Electrochemical Studies of Polymers
Thus, the ES of PANI–VA was performed in three variants in the potentiodynamic mode on a GC electrode from the electrolytes: (1) E3, (2) E3 with addition of sulfuric acid to pH 4, and (3) E3 with addition of hydrochloric acid to pH 4. In (2), the An : VA ratio was 100 or 1500.
In this discussion of the results of the chemical and electrochemical syntheses, it can be concluded that variant 2 allows us to obtain high-quality composite materials at An : VA = 100 (PANI–VA(a)1) in the chemical synthesis and at An : VA = 1500 (PANI–VA(a)3) in the electrochemical synthesis. The electrochemical coating of PANI–VA(a)3 was studied in various electrolytes: (1) E2, (2) E4, and (3) E5. Figure 6 shows the results of testing PANI–VA(a)3 in the potential and charge-discharge cycling modes. The characteristics of the polymer in all these electrolytes were studied, namely, retention of stability during the cycling and electrochemical capacitance (C). The capacitance C was calculated by the equation С = it/m∆V, where i is the discharge current (A), t is the discharge time (s), m is the mass of the composite (g), and ∆V is the range of potentials used (V). Figure 6a shows the results of testing PANI–VA(a)3 in E2. Judging by the area of CV, the polymer quickly loses its activity during the cycling. The calculation of C from the charge-discharge characteristics showed that C decreased by 29%, from 138 to 97 F/g, after 20 charge–discharge cycles. The C value drastically decreased by 23% (from 138 to 103 F/g) during the first 10 discharge cycles and by 6% during the next 10 discharge cycles. In the E4 electrolyte (Fig. 6b), PANI–VA(a)3 showed good electrochemical activity with preservation of the peaks of the redox transformations of the polymer during all the 20 cycles, but during long-term cycling, ∆Е increased from 200 to 420 mV. During the charge–discharge tests, the polymer retained 81% of its activity. The C value decreased from 193 to 157 F/g during 20 charge–discharge cycles. As in the previous case, C mainly decreased during the first 10 charge-discharge cycles (by 13%, from 193 to 168 F/g); by the end of the 20th cycle, the C values were almost stabilized. Finally, the testing this polymer in E5 at pH 1.2 (CV1, Fig. 6c) showed excellent stability of this electrode in the system. During the cycling and charge–discharge trials, the loss of electrochemical activity was insignificant, ~4%; ∆E increased by only 10 mV over 50 cycles. The C values decreased by 2.5%, from 255 to 242 F/g after 50 charge–discharge cycles. Against the background of the first two electrolytes, in the E5 electrolyte, the initial and highest (of all these variants) C values did not change and were reproduced in each subsequent cycle in the charge–discharge processes throughout all 50 cycles. These facts indicate that the polymer tests in the “native” system in the presence of “native” anions, which formed the polymer morphology, lead to stable electrochemical functioning of the system and are important factors for its reliable and effective operation despite higher pH values than in E4. The cycling of PANI with preserved initial electrochemical characteristics without overoxidation of the polymer is stable in the potential range from –0.2 to +0.7 V (Ag/AgCl) [25, 26]. The present study revealed that an increase in the cycling range of PANI–VA(a)3 into the anodic region by 0.3 V (CV(b), Fig. 7) allows us to observe fairly stable behavior of the composite material during 50 cycles. The ∆Е value slightly increases by 20 mV and the anodic oxidation shifts (LE → E) by 80 mV. This happens within the first cycles in the changed range of potentials; thereafter, CV is well reproduced, and there are no peaks of PANI overoxidation. This material was also stable during the charge–discharge testing in E5 (charge–discharge curves in the insert, shown after CV(a) and CV(b), Fig. 7). During the cycling in the potential range extended to the anodic region after 50 charge-discharge processes, the C value changed insignificantly, from 255 to 242 F/g. The composite material was also quite stable in the potential range extended even more widely, from –1 to +1 V (CV(c), Fig. 7). According to Fig. 7, 20 cycles in the extended anodic and cathodic regions showed good stability of the composite material and reproducibility of its characteristics. The calculation of the charge-discharge curves (insert, Fig. 7) revealed that the cycling of PANI–VA(a)3 in the potential range from –0.2 to +1 V at a current density of 0.5 mA/g led to the C value slightly increased to 265 F/g. The obtained data can be compared with C = 443 F/g published for a similar system at a window of potentials of 1.6 V and current density of 0.5 mA/cm2 [18].

Results of electrochemical tests: 20 cycles (50 mV/s) and 20 charge–discharge processes at I = 500 μA of the PANI–VA(a)3 composite material in (a) E2 (m = 0.0014 g), (b) E4 (m = 0.001 g), and (c) E5 (m = 0.0014 g). The loss was 23% (Fig. 6a) and 13% (Fig. 6b) over the first 10 cycles; over the second 10 cycles, the loss was 6% in both figures.

Results of electrochemical tests of the PANI–VA(a)3 composite material. 50 cycles (50 mV/s) in E5 at potentials in the ranges: (a) –0.2…+0.7 V; (b) –0.2 …+1 V, and (c) –1…+1 V and 50 charge–discharge processes with i = 500 μA (insert).
4 CONCLUSIONS
(1) In electrolytes containing vanadate anions (at pH ≤ 4), V+5 oxidizes the An molecule, leading to the start of the chemical synthesis of PANI and accelerates the ES of PANI.
(2) The incorporation of the vanadate anion into PANI changes the structure of the polymer, bringing together the polymer chains due to their aggregation at the NH–O–V bonds; with a large excess of vanadate anions, however, this can lead to a significant reduction in the surface area.
(3) The electrochemical study of the composite material obtained in ES at a ratio of An : VA = 1500 showed that this polymer was quite stable during the cycling in an electrolyte containing vanadate ions.
(4) When the potential range is extended to the anodic region by 0.3 V and to the cathodic-anodic region by 0.8 V during the cycling of PANI–VA(a)3, the operation of the composite electrode remains stable within a few dozen cycles. After 50 cycles, the electrochemical capacitance decreased by 5%; there were no peaks of overoxidation of PANI.
REFERENCES
Fan, Z., Yan, J., Wei, T., Zhi, L., Ning, G., Liand, T., and Wei, F., Asymmetric supercapacitors based on graphene/MnO2 and activated carbon nanofiber electrodes with high power and energy density, Adv. Funct. Mater., 2011, vol. 21, p. 2366.
Wu, X.-L., Jiang, L.-Y., Cao, F.-F., Guo, Y.-G., and Wan, L.-J., LiFePO4 Nanoparticles embedded in a nanoporous carbon matrix: Superior cathode material for electrochemical energy-storage devices, Adv. Mater., 2009, vol. 21, p. 2710.
Peng, C., Zhang, S., Zhou, X., and Chen, G.Z., Unequalisation of electrode capacitances for enhanced energy capacity in asymmetrical supercapacitors, Environ. Sci., 2010, vol. 3, p. 1499.
Lu, X., Yu, M., Zhai, T., Wang, G., Xie, S., Liu, T., Liang, C., Tong, Y., and Li, Y., High energy density asymmetric quasi-solid-state supercapacitor based on porous vanadium nitride nanowire anode, Nano Lett., 2013, vol. 13, p. 2628.
Wang, G., Zhang, L., and Zhang, J., A review of electrode materials for electrochemical supercapacitors, Chem. Soc. Rev., 2012, vol. 41, p. 797.
Yu, G., Hu, L., Vosgueritchian, M., Wang, H., Xie, X., Mc Donough, J.R., Cui, X., Cui, Y., and Bao, Z., Solution-processed graphene/MnO2 nanostructured textiles for high-performance electrochemical capacitors, Nano Lett., 2011, vol. 11, p. 2905.
Xiao, J. and Yang, S., Sequential crystallization of sea urchin-like bimetallic (Ni, Co) carbonate hydroxide and its morphology conserved conversion to porous NiCo2O4 spinel for pseudocapacitors, RSC Adv., 2011, vol. 1, p. 588.
Wang, G., Lu, X., Lingm, Y., Zhai, T., Wang, H., Tong, Y., and Li, Y., LiCl/PVA gel electrolyte stabilizes vanadium oxide nanowire electrodes for pseudocapacitors, ACS Nano, 2012, vol. 11, p. 10296.
Wu, C., Feng, F., and Xie, Y., Design of vanadium oxide structures with controllable electrical properties for energy applications, Chem. Soc. Rev., 2013, vol. 42, p. 5157.
Zhu, J., Cao, L., Wu, Y., Gong, Y., Liu, Z., Hoster, H.E., Zhang, Y., Zhang, S., Yang, S., Yan, Q., Ajayan, P.M., and Vajtai, R., Building 3D structures of vanadium pentoxide nanosheets and application as electrodes in supercapacitors, Nano Lett., 2013, vol. 13, p. 5408.
Pletnev, R.N., Gubanov, V.A., and Fotiev, A.A., YaMR v oksidnykh soedineniyakh vanadiya (NMR of Vanadium Oxide Compounds), Moscow: Nauka, 1979.
Mak, W.F., Wee, G., Aravindan, V., Gupta, N., Mhaisalkarand, S.G., and Madhavi, S., High-energy density asymmetric supercapacitor based on electrospun vanadium pentoxide and polyaniline nanofibers in aqueous electrolyte, J. Electrochem. Soc., 2012, vol. 159, p. A1481.
Qu, Q., Zhu, Y., Gao, X., and Wu, Y., Core–shell structure of polypyrrole grown on V2O5 nanoribbon as high performance anode material for supercapacitors, Adv. Energy Mater., 2012, vol. 2, p. 950.
Fotiev, A.A., Trunov, V.K., and Zhuravlev, V.D., Vanadaty dvukhvalentnykh metallov (Bivalent Metal Vanadates), Moscow: Nauka, 1985.
Pelletier, O., Davidson, P., Bourgaux, C., Coulon, C., Regnault, S., and Livage, J., A Detailed study of the synthesis of aqueous vanadium pentoxide nematic gels, Langmuir, 2000, vol. 16, p. 5295.
Wu, C.G., De Groot, D.C., Marcy, H.O., Schindler, J.L., Kannewurf, C.R., Liu, Y.-J., Hirpo, W., and Kanatzidis, M.G., Redox intercalative polymerization of aniline in V2O5 xerogel. The postintercalative intralamellar polymer growth in polyaniline/metal oxide nanocomposites is facilitated by molecular oxygen, Chem. Mater., 1996, vol. 8, p. 1992.
Ferreira, M., Huguenin, F., Zucolotto, V., Pereira da Silva, J. Ed., Cordoba de Torresi, S.I., Temperini, M.L.A., Torresi, R.M., and Oliveira, Os.N., Electroactive Multilayer Films of Polyaniline and Vanadium Pentoxide, J. Phys. Chem. B, 2003, vol. 107, p. 8351.
Hao, Q., Lei, W., Xia, X., Yan, Zh., Yang, X., Lu, L., and Wang, X., Exchange of counterions in electropolymerized PANI film, Electrochim. Acta, 2010, vol. 55, p. 632.
Bai, M.-H., Liu, T.-Yu., Luan, F., Liand, Ya., and Liu, X.-X., Electrodeposition of vanadium oxide–polyaniline composite nanowire electrodes for high energy density supercapacitors, J. Mater. Chem. A, 2014, vol. 2, p. 10882.
Ragupathy, D., Gopalan, A.I., Lee, K.-P., and Manesh, K.M., Electro-assisted fabrication of layer-by-layer assembled poly(2,5-dimethoxyaniline)/phosphotungstic acid modified electrode and electrocatalytic oxidation of ascorbic acid, Electrochem. Commun., 2008, vol. 10, p. 527.
Trchova M., Moravkova Z., Blaha, M., and Stejskal, Ja., Raman spectroscopy of polyaniline and oligoaniline thin films, Electrochim. Acta, 2014, vol. 122, p. 28.
Huguenin, F., Ticianelli, E.A., and Torresi, R.M., XANES study of polyaniline–V2O5 and sulfonated polyaniline–V2O5 nanocomposites, Electrochim. Acta, 2002, vol. 47, p. 3179.
Shouji, E. and Buttry, D.A., New organic-inorganic nanocomposite materials for energy storage applications, Langmuir, 1999, vol. 15, p. 669.
Huguenin, F., Prado Gambardella, M.T., Torresi, R.M., Cordoba Torresi, S.I., and Buttryc, D.A., Chemical and electrochemical characterization of a novel nanocomposite formed from V2O5 and poly(N-propane sulfonic acid aniline), a self-doped polyaniline, J. Electrochem. Soc., 2000, vol. 47, p. 2437.
Geniès, E.M., Lapkowski, M., and Penneau, J.F., Cyclic voltammetry of polyaniline: interpretation of the middle peak, J. Electroanal. Chem. Interfacial Electrochem., 1988, vol. 249, p. 97.
Kieffel, Y., Pierre Travers, J., Ermolieff, A., and Rouchon, D., Thermal aging of undoped polyaniline: Effect of chemical degradation on electrical properties, J. Appl. Polymer Sci., 2002, vol. 86, p. 395.
ACKNOWLEDGMENTS
We are grateful to A.M. Kolesnikova for consultations and technical assistance with this work.
Funding
This study was performed under state assignment (state ID nos. 0089-2019-0010 and 0089-2014-0022, Institute of Problems of Chemical Physics, Russian Academy of Sciences).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by L. Smolina
Rights and permissions
About this article

Cite this article
Abalyaeva, V.V., Dremova, N.N. Electrochemical Behavior of Polyaniline in the Presence of the Vanadate Anion. Russ J Electrochem 55, 850–859 (2019). https://doi.org/10.1134/S1023193519090027
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193519090027