
Abstract
Genetic diversity of the common vole Microtus arvalis Pall., 1778 of the obscurus form was examined by analyzing the variability of two markers, the cytochrome b gene (1143 bp) and the fragment of the mitochondrial DNA control region (800 bp). One hundred forty-five individuals were captured at 39 locations from the Crimean Peninsula in the west to northern Altai in the east. On the basis of the analysis of the cytochrome b gene sequences, all M. arvalis individuals of the obscurus form from the regions under consideration were assigned to the previously identified Sino-Russian clade, distributed in the main part of the range of the obscurus form. On the basis of concatenated sequences of the two mtDNA markers, the internal structure of the Sino-Russian clade was established and two subclades, Crimean and Eurasian, were identified. In the Eurasian subclade, related haplotypes form several geographically localized groups (Vyatka–Ural, Volga–Ural, Southern Ural, Southern Cis-Urals 1, and Southern Cis-Urals 2). The groups are connected with each other through a widespread set of poorly differentiated haplotypes found from the Cis-Urals to Western Siberia. Two haplotypes from the North Altai and northern foothills of the Altai Mountains are included in the Eurasian subclade. However, these haplotypes are separated from each other and from haplotypes of other groups by distances comparable with the intergroup distance for this subclade. Demographic analysis enables the conclusion that the history of the Sino-Russian clade of M. arvalis of the form obscurus has regional specificity. Haplotypes found in seven locations on the Crimean Peninsula are most likely the result of a single expansion event, whereas for the Eurasian subclade, several successive waves of expansion can be proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The common vole, Microtus arvalis s. str. (2n = 46), is a widespread species in Northern Eurasia. It belongs to a group of taxa of varying degrees of divergence, previously known as a single polytypic species M. arvalis s. l., but subsequently separated on the basis of karyological and hybridological data [1, 2]. M. arvalis s. str. is represented by two parapatric forms, between which there is a contact zone (western “arvalis” form and eastern “obscurus” form). The forms of M. arvalis s. str. were described in the second half of 20th century on the basis of the difference in their karyotypes. Having equal number of chromosomes in a diploid set, the forms differ in the total number of chromosomal arms [1, 3]. The taxonomic status of the two forms of common vole is controversial. Different authors consider them as chromosomal forms within the same species [1, 2, 4, 5], semispecies [6], or species [7, 8]. Regardless of which species conception the authors adhere to, in all studies, a rather high level of genetic divergence between the two cryptic forms of the common vole is recognized along with the preserved ability to successfully hybridize in natural conditions.
Phylogeographical analysis [8–11] showed that the history of contemporary arvalis and obscurus range formation was different. Despite the fact that each of the isolated forms occupies about half of the species range, for the arvalis form, six mitochondrial lineages were identified, while for the obscurus form, only two lineages were identified. In the contact zone of the arvalis and obscurus forms, located on the territory of East European Plain, their present-day hybridization takes place [6, 12, etc.] and, according to the latest data [13], the introgression of mitochondrial and nuclear DNA occurs.
Today, the phylogeographic structure of M. arvalis s. str. in the western part of the range has been reconstructed [8–11, 14–16]. It is suggested that the most probable regions of origin of all modern mitochondrial lineages assigned to the arvalis form are central Europe [10] or the Carpathian and Balkan refugia [15]. These reconstructions are based mainly on sequence analysis of the mitochondrial DNA (mtDNA) cytochrome b (cytb) gene. Analysis of partial sequence of the mtDNA control region (≈300 bp) showed that the results were generally consistent with the data on the cytb variability [10, 14]. At the same time, simultaneous sequence analysis of two markers in phylogenetic reconstructions made it possible to detail the intragroup structure of one of the mitochondrial lineages, distributed through the range of the arvalis form [14]. At present, the mtDNA variability of the obscurus form of M. arvalis s. str. is explored in two studies [8, 17]. It was demonstrated that, within the range of the obscurus form, two mitochondrial clades were present, the Middle Eastern according to [17], including South Caucasian according to [8], and Sino-Russian. As a possible region of origin of the latter clade, the Crimean Peninsula or the northern Altai was considered [8].
Phylogenetic and phylogeographic analyses were carried out using two mtDNA markers, cytb [8–11, 14, 15, 17] and the mtDNA CR [10, 14]. Both markers are widely used for phylogenetic reconstructions and are independent functional elements of the mtDNA. The cytb gene encodes the cytochrome b transmembrane protein, which is a component of the mitochondrial respiratory chain complex III [18]. The mtDNA CR is the noncoding region of mtDNA, also called the D-loop, containing the origin of replication of the mtDNA heavy strand [19].
In the present study, new data on the mtDNA variability in M. arvalis s. str., the obscurus form, from three regions within the distribution area of the Sino-Russian clade, specifically, the Crimean Peninsula, Urals and the adjacent plains, and Northern Altai, are presented.
The objective of this study was description of the genetic diversity, phylogenetic relationships, and geographical distribution of the mitochondrial DNA haplotypes of M. arvalis s. str, the obscurus form, and subsequent reconstruction of the history of contemporary population genetic diversity formation of M. arvalis s. str., the obscurus form, in the central part of Northern Eurasia.
MATERIALS AND METHODS
Specimens. Muscle tissue specimens (skeletal and/or heart muscle) were obtained from 172 M. arvalis s. l. captured at 40 locations in 2008–2016 (see the Appendix, Tables 1 and 2; the Appendix is placed in the electronic version of the journal; Fig. 1). Since in the studied regions M. arvalis s. str. and its sibling species M. rossiaemeridionalis Ognev, 1924 are found in sympatry, for all specimens, species identification was carried out using nuclear DNA markers [20]. Among 172 individuals, 145 were defined as M. arvalis s. str. and used to solve the tasks set in the present study. The remaining 27 individuals were identified as M. rossiaemeridionalis.

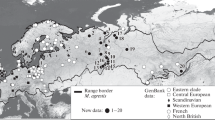
The range of M. arvalis s. str. (according to [25] with additions according to [17, 26–28]) and sampling sites of M. arvalis s. str., the obscurus form. The map above is the main range of M. arvalis s. str. (continuous outline), regions of the M. arvalis s. l. range where the presence of M. arvalis s. str. is not confirmed but possible [25, 28] (dotted line) and the contact zone of the arvalis and obscurus forms (dashed line). A–C, sampling areas. A, the Crimean Peninsula; B, the Urals and the adjacent plains; C, the southeast of Western Siberia and northern Altai. 1–46, sampling localities (see Appendix, Tables 1 and 2, electronic version of the journal).
For 145 individuals of M. arvalis s. str., complete cytb sequences (1143 bp) were obtained, and for 137 of them, partial sequences (800 bp) of the mtDNA CR were obtained (GenBank database accession nos. MG702966–MG703237) (Appendix, Table 1). Owing to the differences in the DNA quality in laboratory specimens (see the Laboratory methods section), for 8 out of 15 M. arvalis s. str. from the Crimean Peninsula, no mtDNA CR sequences have been obtained so far.
The analysis was performed using our own data and the data on M. arvalis s. str., the obscurus form, reported elsewhere, including the cytb sequences from 62 individuals [8, 9, 17, 21] (GenBank accession nos. AY220760–65, FR865393–429; KF839584–90; KX581042–52) and the mtDNA CR from 1 individual [22] (GenBank accession no. KP013595) (Appendix, Tables 1 and 2, Fig. 1). The phylogenetic reconstructions were performed using the mtDNA sequences of M. arvalis s. str., the arvalis form [9, 10] (GenBank accession nos. AY220766,74,77,88; AM991028,30,43, 49,70,89, 79) (Appendix, Table 1), M. rossiaemeridionalis [23, 24] (GenBank accession nos. Q015676; AY513819,21) (Appendix, Table 1), M. ilaeus Thomas, 1912 (=M. kirgisorum Ognev, 1950) [24] (GenBank accession nos. AY513809–10), and M. socialis Pall., 1773 [24] (GenBank accession nos. AY513829–31) as outgroup. The nomenclature of voles is given according to the data of the latest report on the taxonomy of mammals in Russia [5].
Laboratory methods. Total genomic DNA was isolated by the method of salt extraction from muscle tissue specimens fixed in 96% ethanol [29, 30].
Species identification of M. arvalis s. str. and M. rossiaemeridionalis was carried out with the help of the PCR identification method [20] in accordance with the recommended protocol.
PCR and subsequent sequencing of the mtDNA fragments was carried out with primers as follows. L7, L8, H12, and H6 for cytb [31]; Pro+, Phe–, micro8+, and micro4– for the mtDNA CR [32]. The PCR conditions and temperature protocols for the used primer pairs are given in the Appendix, Table 3.
In the case of satisfactory DNA quality and the possibility to amplify the 1200-bp fragments, PCR of the cytb-containing region was performed with L7 and H6 primers followed by sequencing in both directions. In the case of unsuccessful amplification of 1200-bp fragments, two PCRs with L7–H12 and L8–H6 primer pairs were run, followed by sequencing of each PCR product with L7 and H6 primers, respectively. PCR of the mtDNA CR-containing fragment was performed only in the case where amplification of 1000-bp fragments was available. This was associated with the location of the Pro+, Phe–, micro4–, and micro8+ annealing sites [32] and the impossibility of amplifying two overlapping fragments of smaller length with these four primers, as was done in the case of the cytb-containing fragment. Sequencing was performed using the Big Dye Terminator Cycle Sequencing Kit v. 3.1 (Applied Biosystems, United States) and the BrightDye® Terminator Cycle Sequencing Kit (NimaGen, Netherlands) according to the protocols of the manufacturers. Detection of sequencing results was carried out on an ABI Prism 3130 genetic analyzer (Applied Biosystems, United States) at the Institute of Plant and Animal Ecology, Ural Branch of the Russian Academy of Sciences, Yekaterinburg, and the Children’s Oncology and Hematology Center, Regional Children Hospital No. 1, Yekaterinburg.
The methods of data analysis. The chromatograms were analyzed using the BioEdit v. 7.2.0 (4.30.2013) software program [33]. Sequence alignment, calculation of genetic distances, and construction of phylogenetic trees with the neighbor joining (NJ) and maximum likelihood (ML) methods were carried out in the MEGA v. 6 software program [34]. Construction of phylogenetic trees using the Bayesian inference (BI) was carried out in the MrBayes v. 3.2.2 software program [35]. Searching for optimum models of nucleotide sequence evolution was performed in the MrModeltest 2.3 software program [36]. The construction of median-joining networks (MJN) was carried out in the Network v. 5.0.0.0 software program [37]. Assessment of nucleotide diversity and the tests for selective neutrality were performed in the Arlequin v. 3.1 [38] and DnaSP v. 5.10 [39] software programs. The marker congruence was evaluated using the partition homogeneity test (HOMPART) [40] in the PAUP v. 4.0b10 software package [41].
To construct the phylogenetic trees by the NJ method, Tamura’s 3-parameter model (T3P) was used to calculate intra- and intergroup distances [42]. To construct the tree by the ML method on the basis of the cytb and cytb + CR sequences, the general time reversible model (GTR) [43] with the proportion of invariant sites (+I) and gamma distribution (+G) normalization was chosen. When the phylogenetic tree on the basis of the cytb sequences was reconstructed using the BI method, a complex approach with the choice of the model for each of the three codon positions separately was used. For the first codon position, it was Kimura’s model (K80) [44] +I; for the second position, the Hasegawa, Kishino, and Yano model (HKY) [45]; and for the third position, GTR + G. When the phylogenetic tree was reconstructed using the Bayesian analysis on the basis of the cytb + CR sequences, a complex approach with the choice of a model for each of the three positions in the cytb codon and for the mtDNA CR separately was used. For the first codon position it was K80; for the second position, HKY; for the third position, GTR + G; and for the mtDNA CR, GTR + I + G. In the construction of phylogenetic trees by the ML and NJ methods on the basis of the cytb and cytb + CR sequences, statistical testing of the tree topology was carried out using the bootstrap analysis (1000 cycles). In the case of Bayesian inference of phylogenetic trees, two parallel analyses consisting of four Markov chains, each for 10 000 000 cycles, were run simultaneously, with sampling every 500th cycle and removing the first 5001 cycles as the burn-in stage. The differences in the choice of models for constructing trees by the ML and BI methods are associated with the impossibility of defining a complex model for constructing a phylogenetic tree by the ML method.
RESULTS
Analysis of the cytb sequences of M. arvalis s. str., the obscurus form, obtained in this study revealed 78 haplotypes, of which 70 were described for the first time. Taking into account the haplotypes presented earlier, the total number of cytb haplotypes of the obscurus form was 112. The number of cytb polymorphic sites was 159, of which 84 were informative for parsimony analysis, and the substitutions at 30 of these sites were nonsynonymous. Excluding the sequences belonging to the Middle Eastern clade, the number of haplotypes was 101; the number of polymorphic sites was 127, of which 52 were informative for parsimony analysis; and the substitutions at 25 sites were nonsynonymous.
Sequence analysis of the mtDNA CR of M. arvalis s. str., the obscurus form, revealed 77 haplotypes, of which 76 were first described in the present study. The number of polymorphic sites in the mtDNA CR was 71, with two of these having single nucleotide insertions/deletions, and 63 sites were informative for parsimony analysis.
In the analysis of the cytb + CR sequences of M. arvalis s. str., the obscurus form, 98 haplotypes were described for the first time. The number of polymorphic sites was 168, with two of these containing single nucleotide insertions/deletions, and 95 sites were informative for parsimony analysis.
Phylogenetic Reconstruction Inferred from the Cytochrome b Gene
Phylogenetic reconstruction using the sequences of M. arvalis s. str., the arvalis form, and M. rossiaemeridionalis, M. ilaeus, and M. socialis as outgroup showed that all haplotypes of M. arvalis s. str., the obscurus form, clustered into two clades (Middle Eastern and Sino-Russian). All cytb haplotypes described in this study were included in Sino-Russian clade (Appendix, Fig. 1).
Analysis of the phylogenetic tree showed that, within Sino-Russian clade, several groups could be identified, with five of these having rather clear geographic localization: group I, Crimean Peninsula; group II, from the Volga River in the west to the Southern and Middle Urals in the east; group III, northwestern provinces of China; group IV, represented in the localities of the south of the Ural region from the Talovskaya Steppe Site of the Orenburg Reserve (southwest of Orenburg oblast) in the west to the settlement of Zverinogolovskoye in Kurgan oblast (floodplain of the Tobol River) in the east; group V, a group of four haplotypes found in the localities of Kirov oblast (the northwestern part of the distribution area of M. arvalis s. str., the obscurus form).
Analysis of the median-joining network (Appendix, Fig. 2) supports the presence of these five groups within the Sino-Russian clade. In addition, analysis of the median-joining network showed that three of the five isolated groups (I, II, and III) were differentiated from each other and from other sequences much more strongly than groups IV and V.

Phylogenetic tree constructed using the cytb + CR sequences (1943 bp) of Sino-Russian clade of M. arvalis s. str., the obscurus form (BI topology shown). Values over the branches are a posteriori probabilities BI >0.7/bootstrap support ML >50/bootstrap support NJ >50. The subclades of the Sino-Russian clade and the groups within the Eurasian subclade are indicated (explanation in the text).
Phylogenetic Reconstruction Inferred from the mtDNA Control Region
The phylogenetic tree constructed on the basis of the available sequences of the mtDNA CR (800 bp) belonging to the Sino-Russian clade of M. arvalis s. str., the obscurus form (not represented), makes it possible to distinguish two groups within the clade. Group 1 includes haplotypes found only in the Crimean Peninsula and not found in other regions; group 2 consists of all other mtDNA CR sequences, slightly differentiated from one another, not forming a pronounced structure upon the construction of the phylogenetic tree, and distributed from the Taman Peninsula to the Altai.
The Crimean group of the mtDNA CR haplotypes corresponds to group I, isolated on the basis of the cytb data. No associations of the mtDNA CR haplotypes corresponding to groups II, IV, and V, isolated on the basis of the cytb sequence data, were observed. The difference between the mtDNA sequences assigned on the basis of the cytb analysis to group II and the closest mtDNA sequences assigned to the Sino-Russian clade reduces from 5–9 substitutions in the cytb analysis to 1–2 substitutions in the mtDNA CR analysis. The degree of differentiation of northwestern China voles (group III) in terms of the mtDNA CR remains uncertain because of the lack of sequences available for analysis.
Phylogenetic Reconstruction Based on Two mtDNA Markers
First, the congruence of the two mitochondrial markers under consideration was tested. The results of the partition homogeneity test (HOMPART, Paup) were not statistically significant (P = 0.761), which made it possible to exploit the combination of these markers in phylogenetic reconstruction.
The analysis showed that, within the Sino-Russian clade, six groups of haplotypes with different degrees of differentiation from each other could be distinguished (Fig. 2). Moreover, analysis of both the phylogenetic tree (Fig. 2) and the median-joining network (Fig. 3) showed that the most distant from the rest was the Crimean group of haplotypes. On the basis of considerable distance separating this group from the remaining haplotypes of the Sino-Russian clade in the median-joining network, two subclades within the Sino-Russian clade, the Crimean and Eurasian (Figs. 2, 3), were identified.

Median-joining network (MJN) of cytb + CR (1943 bp) haplotypes of Sino-Russian clade of M. arvalis s. str., the obscurus form. Gray circles are haplotypes; open circles are median vectors; the branch length is equivalent to the number of substitutions between haplotypes (the minimum branch length is one substitution); the diameter of gray circle is equivalent to the number of identical sequences assigned to a certain haplotype. Solid ellipses are groups of related haplotypes within the Eurasian subclade (see text); dotted line is the set of haplotypes serving as joints between the groups of the Eurasian subclade isolated from the results of the phylogenetic tree analysis (see text).
Within the Eurasian subclade, several groups of haplotypes can be distinguished (Figs. 2, 3). The Vyatka–Ural group includes all mtDNA sequences assigned to group V, identified on the basis of the cytb analysis (Kirov oblast), as well as some haplotypes of individuals from the Northern, Middle, and Southern Urals. The Southern Ural group includes the group IV sequences, identified from the results of the cytb analysis, and some of the previously undifferentiated sequences of individuals from the territory of the Southern Urals. The Volga–Ural group includes only the group II sequences, identified from the results of the cytb analysis. The Southern Cis-Ural 1 group includes the mtDNA sequences of individuals from the western slope of the Southern Urals (Ignatievskaya Cave and Kinzebulatovo); the Southern Cis-Ural 2 group includes the mtDNA sequences of individuals from the Buzuluksky Bor National Park, the Aituarskaya Steppe Site of the Orenburg Reserve, and from the vicinity of the Verblyuzhka Mountain (south and southwest of the Ural region). Figure 4 presents data on the distribution of different groups of the Eurasian subclade within the Ural region.

Distribution of groups of the Eurasian subclade across the localities within the Ural region. Locality numbers correspond to those in the Appendix Tables 1, 2. and in Fig. 1. On the map of the “Ural–Siberian Set,” circles indicate the localities where haplotypes of the Ural–Siberian set are found at the absence of haplotypes not assigned to any of the groups; squares indicate the localities where haplotypes that are not assigned to any of the groups are found at the absence of haplotypes of the Ural–Siberian set; rhombuses indicate the localities where haplotypes of the Ural–Siberian set and haplotypes not assigned to any of the groups are found.
As follows from the median-joining network analysis (Fig. 3), among haplotypes not included in any of the groups of the Eurasian subclade, identified in the analysis of the phylogenetic tree (Fig. 2), many haplotypes gravitate toward each other and form a starlike structure. In addition, there are several strongly differentiated single or paired haplotypes from the localities of the Middle and Southern Urals, Vyatka–Kama Cis-Urals, Buzuluksky Bor National Park, Altai krai, and the Altai Republic (northern Altai and northern foothills of the Altai). The haplotypes of the starlike structure gravitating toward each other (Fig. 3, dashed line) occupy the connecting position between all other groups of the Eurasian subclade. In the following, in the description of the results and their discussion, the name of “Ural–Siberian set of haplotypes” will be used to designate this set of close haplotypes, and it will be treated on a par with other groups of haplotypes identified within the Eurasian subclade on the basis of the analysis of the phylogenetic tree and median-joining network. The remaining, highly differentiated, haplotypes not included in any of the groups in Fig. 3 are separated from each other and from the haplotypes of other groups of the Eurasian subclade by distances comparable to those between groups of the Eurasian subclade.
Genetic Diversity and Demographic History of the Sino-Russian Clade
Evaluation of the genetic diversity indices was carried out for the Middle Eastern and Sino-Russian clades of M. arvalis s. str., the obscurus form, as well as for the subclades of the Sino-Russian clade with each of the mtDNA markers separately (Table 1). Using the cytb + CR sequences, the evaluation was carried out for the Sino-Russian clade as a whole and for all internal groupings identified in this clade earlier in the analysis (Table 1). Analysis of the variability of each of the mtDNA markers within the Sino-Russian clade showed the following. (1) The used mtDNA CR fragment was considerably more variable than the complete cytb sequence (the nucleotide diversity was 2 times higher at the sequences belonging to the Sino-Russian clade); (2) the Eurasian subclade was characterized by higher diversity at each of the markers examined, but in this case, the nucleotide diversity at the cytb sequences in the Eurasian subclade was 3 times higher than in the Crimean one, while at the mtDNA CR sequences, it was only 1.5 times higher.
The tests for selective neutrality, as well as the analysis of mismatch distribution at the cytb, mtDNA CR, and cytb + CR sequences, both in the Sino-Russian clade as a whole and in each of the subclades (Table 1, Appendix, Fig. 3) showed that, in the recent past, these groups went through the stage of increasing the effective population size, which could have been associated with the M. arvalis s. str. population expansion and resettlement across the modern range.
Analysis of the genetic diversity indices and selective neutrality tests for individual groups of the Eurasian subclade at the cytb + CR sequences (Table 1) showed that only in the case of the Southern Ural group and the Ural–Siberian set of haplotypes was it possible to speak of an increase in the effective population size in the recent past.
Comparison of intra- and intergroup distances (T3P) at the cytb + CR sequences (Appendix, Table 4) shows that the most differentiated is the Crimean group of haplotypes (the Crimean subclade), which supports the results of phylogenetic reconstructions. The average intergroup distance (T3P) between the Sino-Russian and Middle Eastern clades of M. arvalis s. str. at the cytb sequences was 0.0304 ± 0.0046.
DISCUSSION
Variability of Used mtDNA Markers
Variability analysis of both mtDNA markers in the Sino-Russian clade of M. arvalis s. str., the obscurus form, showed that, in general, within the used fragment of the mtDNA CR, the number of variable sites was lower (71 sites, or 8.875% of the total number of sites) than within the full cytb sequence (127 sites, or 11.11%). However, the number of parsimony informative sites in the mtDNA CR sequence was higher in both the total number and as a percentage of the number of variable sites (63 sites, or 88.73% of the number of variable sites) than in the full cytb sequence (52 sites, or 40.94% of the number of variable sites). In addition, the nucleotide diversity (Table 1) among the mtDNA CR sequences was approximately twice as high as among the cytb sequences, 8.43 × 10–3 ± 4.4 × 10–3 and 4.46 × 10–3 ± 2.4 × 10–3, respectively. A similar ratio between variable and parsimony informative sites upon comparison of cytb and the mtDNA CR is observed in other mammalian species, in particular, Siberian roe deer [46], and, apparently, is a consequence of the peculiarities of these two fragments (coding cytb and noncoding mtDNA CR).
Despite the fact that the results of the homogeneity test (the congruence of two mitochondrial markers examined) were not statistically significant, enabling application of both markers within the framework of one phylogenetic reconstruction, there are some differences between the phylogenetic reconstructions obtained with each of the markers separately. Thus, in phylogenetic reconstruction using both mtDNA markers within the Sino-Russian clade, each of these markers provides identification of unique differences between the mtDNA sequences. This situation differs from a previous attempt to use a combination of cytb and the mtDNA CR in phylogenetic reconstructions of common vole, performed by example of M. arvalis s. str., the arvalis form [10]. In that study, data on partial sequences of the mtDNA CR (304 bp), including one of the two variable regions (located at the 5' end of the mtDNA CR), were included in the analysis, along with complete cytb sequences (1143 bp). In that case, it was demonstrated that inclusion in the analysis of partial mtDNA CR sequences only repeated the results obtained in the analysis of the cytb sequences.
On the basis of the foregoing, it can be concluded that the use of only one of the two markers under consideration for the description of phylogeny, internal differentiation processes, and isolation of the groups of equal rank within the Sino-Russian clade of M. arvalis s. str., the obscurus form, is insufficient. It is desirable to use a combination of these markers for analysis within the given mitochondrial lineage.
Phylogenetic Reconstructions, Internal Structure, and Demographic History of the Sino-Russian Clade
Our data supported the existence within M. arvalis s. str., the obscurus form, of two previously identified [8, 17] mitochondrial clades (Middle Eastern and Sino-Russian) and the idea that the distribution area of the Sino-Russian clade occupies the largest part of the M. arvalis s. str. range, compared with all other clades, including clades of the arvalis form.
The inclusion of the mtDNA CR sequences in the analysis and the joint analysis at two mtDNA markers made it possible to describe the internal structure of the Sino-Russian clade, determined by the presence of groups of relative haplotypes differently distant from each other. The most differentiated within the Sino-Russian clade is the group of mtDNA sequences of individuals from the territory of the Crimean Peninsula, identified in our study on the basis of the analysis of two mtDNA markers in the Crimean subclade. All other mtDNA sequences of the Sino-Russian clade are grouped in the Eurasian subclade.
Comparison of the genetic distances between subclades and groups of the Sino-Russian clade (Appendix, Table 4) with the data on intergroup distances between mitochondrial clades of M. arvalis s. str. ([8], the results of our study) shows that the distances between subclades and groups of the Sino-Russian clade are two to ten times lower than those between the clades of M. arvalis s. str. In particular, the distance between the Middle Eastern and Sino-Russian clades (0.0304 ± 0.0046) is more than 2 times higher than the distance between the Eurasian and Crimean subclades of the Sino-Russian clade (0.012 ± 0.002) (Appendix, Table 4).
Comparison of the genetic distances between the clades of M. arvalis s. str. and subclades and groups of the Sino-Russian clade shows that differentiation of contemporary groups belonging to the Sino-Russian clade took place, apparently, much later than the formation of the phylogenetic structure of M. arvalis s. str. in the western part of the range. Moreover, full description of the range formation and the history of M. arvalis s. str. in the eastern part of the range requires the involvement in the analysis of the mtDNA sequences of fossil remains of M. arvalis s. str. from the territory of the contemporary Sino-Russian clade distribution.
Currently, the distribution boundaries of each of the two identified subclades of the Sino-Russian clade cannot be accurately described. On the basis of the available data (analysis of the cytb + CR sequences), it can be said that the Eurasian subclade is distributed in the central part of Northern Eurasia from Samara and Kirov oblasts in the west to the northern Altai in the east. However, phylogenetic reconstruction with the mtDNA CR showed that the sequence of this marker in an individual vole from the Taman Peninsula [22] belonged to a group of sequences that in the simultaneous analysis of two mtDNA markers formed the Eurasian subclade. This finding led to a suggestion that the Eurasian subclade distribution zone in the west could reach the Black Sea coast, North Caucasus, and the sympatry zone of the two forms of M. arvalis s. str. (arvalis and obscurus). The Crimean subclade, apparently, is the only group of M. arvalis s. str. distributed on the territory of the Crimean Peninsula. At the same time, the question whether this group is distributed outside the peninsula (in the southeast of the Ukraine and/or on the Taman Peninsula) cannot be solved without the involvement of new data on the mtDNA variability of M. arvalis s. str. on these territories. We suppose that the relatively high level of differentiation of the Crimean M. arvalis s. str. at the molecular markers can be more associated with the isolation of the Crimean subclade than with its earlier divergence. Inclusion of the data from the south of East European Plain in the analysis will make it possible to test this hypotheses.
Introduction of new data into the analysis is also necessary, because nonrandom range coverage and the use of data from parts of the range distant from each other can lead to misinterpretations and the appearance of artifacts of the analysis [47, 48], including the identification of nonexistent haplogroups owing to the lack of currently known intermediate haplotypes in the analysis [49].
At present, the Ural region is the most explored among the territories inhabited by the Sino-Russian clade, while other regions are studied to a much lesser degree. As mentioned above, the southern part of the East European Plain and the North Caucasus is the first region promising for further study. The importance of studying this region lies in the need for both assessing the distribution boundaries of the Sino-Russian clade and for testing the hypothesis of the Crimean subclade isolation.
The second promising region is the territory of Northwestern China and the adjacent territories of Altai and Northeastern Kazakhstan. It is the region where group III described at the cytb sequences and strongly differentiated (at the cytb + CR sequences) “Altai” haplotypes are distributed (Appendix, Fig. 1). The lack of data on the mtDNA CR sequences for individuals belonging to group III leaves open the question on the degree of its differences from the other groups. This is because the observed differences at only one mtDNA marker, as was demonstrated, cannot serve as an unambiguous criterion for identification of the groups of equal rank within the Sino-Russian clade and, consequently, are insufficient to describe the internal differentiation processes.
It is also necessary to further study the distribution of M. arvalis s. str., the obscurus form, since the authors of [25, 26, 28] reported considerably different data. In particular, the central part of Turkey [28] and the M. arvalis s. l. isolate located southwest of Lake Baikal can be considered as possible distribution areas of M. arvalis s. str., the obscurus form [25, 50]. In both regions, the presence of M. rossiaemeridionalis was proved (using cytogenetic and molecular genetic markers) [24, 51], which, however, cannot serve as evidence of the absence of M. arvalis s. str. in these regions because of the scarcity of published data.
The issue on the region of origin of the Sino-Russian clade also remains open. Neither the Crimean nor the Altai sequences occupy the basal position in relation to other sequences of the Sino-Russian clade on phylogenetic trees obtained from the cytb and cytb + CR sequences. Analysis of the cytb + CR showed that the basal tree position was occupied by the sequences of the Southern Ural group of haplotypes. However, this is not a sufficient reason to consider the south of the Urals as the region of origin of the Sino-Russian clade.
Analysis of the demographic history of both the Sino-Russian clade as a whole and the Crimean and Eurasian subclades separately showed that, with each of the mtDNA markers, the demographic history of these lineages can be described by the increase in the effective population size that could be the result of recent dispersal of the Sino-Russian clade through its contemporary distribution area. Further analysis of the demographic history conducted for individual groups of the Eurasian subclade showed (Table 1) that only for the Southern Ural group and the Ural-Siberian set of haplotypes was there evidence for the increase in effective population size. It can be suggested that the contribution of certain groups of the Eurasian subclade to the demographic and spatial expansion was not the same. This is indirectly supported by the distribution patterns of the groups in question in the Ural region (Fig. 4). Among the groups identified from the results of phylogenetic reconstruction, the Southern Ural and Vyatka–Ural groups, as well as the Ural–Siberian set of haplotypes, are the most widely distributed. The distribution zones of the Southern Ural and Vyatka–Ural groups slightly overlap only at the border of Middle and Southern Urals (Fig. 4). Moreover, the Southern Ural group is found exclusively in the localities of Southern Urals, while the Vyatka–Ural group steps a bit into the South Urals and is mainly distributed in the Middle and Northern Urals and Northern Cis-Urals. The Ural–Siberian set is widely distributed from the Southern Urals (the settlement of Aituar) to the northern boundary of M. arvalis s. str. in the Urals (the city of Ivdel). Three other groups (Volga–Ural, Southern Cis-Urals 1, and Southern Cis-Urals 2) are not widely distributed. They are found in the localities of Southern Urals, and only the Volga–Ural group in the north of its distribution zone gets into the Middle Urals (Fig. 4).
On the basis of spatial distribution patterns of the Eurasian subclade haplotype groups, a hypothesis was put forth.
The phylogeographic structure of the Eurasian subclade reflects at least two successive stages of the contemporary continuous range formation of M. arvalis s. str., the obscurus form: the stage of group differentiation and the stage of their subsequent expansion with the formation of contact zones. Moreover, higher genetic diversity of the M. arvalis s. str., the obscurus form, in the populations of the Southern Urals is determined by the fact that, among all groups under consideration, thedistribution area of only one group (Vyatka–Ural) is located mainly outside the Southern Urals. This is indirect evidence of a longer history of the existence of genetically successive populations of M. arvalis s. str. in the southern part of the Ural region.
CONCLUSIONS
(1) Variability analysis of two mtDNA markers (cytb and mtDNA CR) showed that, to describe the internal structure, the history of dispersal, and the processes of differentiation within the Sino-Russian clade of M. arvalis s. str., it was necessary to include both markers in the analysis, since each of them made a considerable contribution to phylogenetic reconstruction. Thus, in new phylogenetic reconstructions within the Sino-Russian clade, joint application of the cytb gene and mtDNA CR is advisable.
(2) Comparison of the genetic distances between the clades of M. arvalis s. str. and the subclades and groups of the Sino-Russian clade showed that colonization of the contemporary distribution area of Sino-Russian clade, as well as the differentiation of its contemporary groups, could have occurred later than in the western part of the range of M. arvalis s. str. (the distribution zone of the arvalis form). To confirm this hypothesis, mtDNA sequences from M. arvalis s. str. fossil specimens from the territory of the contemporary distribution of the Sino-Russian clade should be involved in the analysis.
(3) Phylogenetic reconstruction using two mtDNA markers (the cytb gene and mRNA CR) provided the description of the Crimean and Eurasian subclades within the Sino-Russian clade. Analysis of the demographic history of Sino-Russian clade as a whole, as well as of two subclades, showed that, in the recent past, they passed through the stage of the increase in effective population size.
(4) It was hypothesized that expansion of the Eurasian subclade consisted of at least two successive stages, at the first of which the differentiation of ancestral sequences of contemporary groups of the Eurasian subclade took place. Then followed the expansion of some of the formed groups. For the territories of the Northern, Middle, and Southern Urals, the highest genetic diversity (at molecular genetic markers) is characteristic of southern latitudes, which is indirect evidence of a longer history of the existence of genetically successive populations of the species in the Southern Urals.
(5) The data obtained suggest that promising regions for the reconstruction of phylogenetic relationships within the Sino-Russian clade and the history of formation of the eastern part of the range of M. arvalis s. str. are the southern part of the East European Plain and the Northwest of China with the adjacent territories of Altai and Northeast Kazakhstan.
REFERENCES
Malygin, V.M., Sistematika obyknovennykh polevok (Systematics of Common Voles), Moscow: Nauka, 1983.
Meier, M.N., Golenishchev, F.N., Radzhabli, S.I., and Sablina, O.L., Serye polevki fauny Rossii i sopredel’nykh territorii (Gray Voles of the Fauna of Russia and Adjacent Territories), in Tr. Zool. Inst. Ross. Akad. Nauk, 1996, vol. 232.
Meier, M.N., Orlov, V.M., and Skholl’, E.D., Sibling species in the Microtus arvalis (Rodentia, Cricetidae) group, Zool. Zh., 1972, vol. 51, pp. 724-738.
Mammal Species of the World: A Taxonomic and Geographic Reference, Wilson, D.E. and Reeder, D.A.M., Eds., JHU Press, 2005.
Abramson, N.I. and Lisovskii, A.A., Subfamily Arvicolinae, Mlekopitayushchie Rossii: sistematiko-geograficheskii spravochnik (Mammals of Russia: A Systematic and Geographical Reference Book), vol. 52 of Trudy Zoologicheskogo muzeya Moskovskogo gosudarstvennogo universiteta (Proceedings of Zoological Museum of the Moscow State University), 2012, pp. 127-141.
Lavrenchenko, L.A., Potapov, S.G., Bulatova, N.Sh., and Golenishchev, F.N., A genetic study of natural hybridization between two forms of the common vole (Microtus arvalis) with the use of molecular and cytogenetic methods, Dokl. Biol. Sci., 2009, vol. 426, nos. 1-6, pp. 222-224.
Malygin, V.M., Morphological criteria for determining of holotypes of species taxa of common voles (Microtus, Rodentia, Mammalia), Dokl. Biol. Sci., 1996, vol. 346, no. 2, pp. 282-286.
Tougard, C., Montuire, S., Volobouev, V., et al., Exploring phylogeography and species limits in the Altai vole (Rodentia: Cricetidae), Biol. J. Linn. Soc., 2013, vol. 108, no. 2, pp. 434-452.
Haynes, S., Jaarola, M., and Searle, J.B., Phylogeography of the common vole (Microtus arvalis) with particular emphasis on the colonization of the Orkney archipelago, Mol. Ecol., 2003, vol. 12, no. 4, pp. 951-956.
Tougard, C., Renvoise, E., Petitjean, A., and Quere, J.P., New insight into the colonization processes of common voles: inferences from molecular and fossil evidence, PLoS One, 2008, vol. 3, no. 10. e3532
Bužan, E.V., Förster, D.W., Searle, J.B., and Kryštufek, B., A new cytochrome b phylogroup of the common vole (Microtus arvalis) endemic to the Balkans and its implications for the evolutionary history of the species, Biol. J. Linn. Soc., 2010, vol. 100, no. 4, pp. 788-796.
Bulatova, N.Sh., Potapov, S.G., and Lavrenchenko, L.A., Genomic versus chromosomal polytypy in studies of mitochondrial and nuclear DNA markers in the Microtus arvalis group, Russ. J. Genet., 2010, vol. 46, no. 5, pp. 586-594. https://doi.org/10.1134/ S1022795410050121.
Gromov, A.R., Bulatova, N.Sh., Potapov, S.G., et al., Study of the hybridization zone of 46-chromosome forms of voles in the Vladimirskaya oblast, Struktura vida u mlekopitayushchikh (Species Structure in Mammals) (Proc. Conf.), Moscow: KMK, 2010, p. 29.
Fink, S., Excoffier, L., and Heckel, G., Mitochondrial gene diversity in the common vole Microtus arvalis shaped by historical divergence and local adaptations, Mol. Ecol., 2004, vol. 13, pp. 3501-3514.
Stojak, J., McDevitt, A.D., Herman, J.S., et al., Post-glacial colonization of eastern Europe from the Carpathian refugium: evidence from mitochondrial DNA of the common vole Microtus arvalis, Biol. J. Linn. Soc., 2015, vol. 115, no. 4, pp. 927-939.
Stojak, J., McDevitt, A.D., Herman, J.S., et al., Between the Balkans and the Baltic: phylogeography of a common vole mitochondrial DNA lineage limited to Central Europe, PLoS One, 2016, vol. 11, no. 12. e0168621
Mahmoudi, A., Darvish, J., Aliabadian, M., et al., New insight into the cradle of the grey voles (subgenus Microtus) inferred from mitochondrial cytochrome b sequences, Mammalia, 2017, vol. 81, no. 6, pp. 583-593.
Irwin, D.M., Kocher, T.D., and Wilson, A.C., Evolution of the cytochrome b gene of mammals, J. Mol. Evol., 1991, vol. 32, no. 2, pp. 128-144.
Clayton, D.A., Transcription and replication of animal mitochondrial DNAs, Int. Rev. Cytol., 1992, vol. 141, pp. 217-232.
Nekrutenko, A., Makova, K.D., Chesser, R.K., and Baker, R.J., Representational difference analysis to distinguish cryptic species, Mol. Ecol., 1999, vol. 8, no. 7, pp. 1235-1237.
Zhumabekova, B.K., Issakaev, Y.M., Surov, A.V., et al., INSDC (GenBank). https://www.ncbi.nlm.nih. gov/nuccore/?term=Microtus +obscurus + Zhumabekova. Accessed November 11, 2013.
Tkachenko, E.A., Witkowski, P.T., Radosa, L., et al., Adler hantavirus, a new genetic variant of Tula virus identified in Major’s pine voles (Microtus majori) sampled in southern European Russia, Infect., Genet. Evol., 2015, vol. 29, pp. 156-163.
Triant, D.A. and DeWoody, J.A., Accelerated molecular evolution in Microtus (Rodentia) as assessed via complete mitochondrial genome sequences, Genetica, 2006, vol. 128, no. 1, pp. 95-108.
Jaarola, M., Martínková, N., Gündüz, I., et al., Molecular phylogeny of the speciose vole genus Microtus (Arvicolinae, Rodentia) inferred from mitochondrial DNA sequences, Mol. Phylogenet. Evol., 2004, vol. 33, pp. 647-663.
Shenbrot, G.I. and Krasnov, B.R., An Atlas of the Geographic Distribution of the Arvicoline Rodents of the World (Rodentia, Muridae: Arvicolinae), Sofia: Pensoft, 2005.
Zagorodnyuk, I.V., Taxonomy and the distribution of Ukrainian gray voles (Rodentiformes: Arvicolini), in Mlekopitayushchie Ukrainy (Mammals of Ukraina), Kiev: Naukova Dumka, 1993, pp. 63-76.
Markova, E.A., Yalkovskaya, L.E., and Zykov, S.V., Taxonomic identity of voles of the “arvalis” group (genus Microtus, Arvicolinae, Rodentia) at the northern boundary of their distribution in the Urals, Dokl. Biol. Sci., 2010, vol. 432, nos. 1-6, pp. 212-215.
Demirsoy, A., Rodents of Türkiye: Türkiye Kemiricileri, Ankara: Meteksan, 2006.
Aljanabi, S.M. and Martinez, I., Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques, Nucleic Acids Res., 1997, vol. 25, no. 22, pp. 4692-4693.
Sibiryakov, P.A. and Amerkhanova, L.I., Assessment of the quality of DNA preservation in museum specimens of animal tissues, Ekologiya: fakty, gipotezy, modeli (Ecology: Facts, Hypotheses, Models) (Proc. Conf. Young Sci.), Yekaterinburg: Goshitskii, 2016, pp. 108-109.
Tougard, C., Delefosse, T., Hänni, C., and Montgelard, C., Phylogenetic relationships of the five extant rhinoceros species (Rhinocerotidae, Perissodactyla) based on mitochondrial cytochrome b and 12S rRNA genes, Mol. Phylogenet. Evol., 2001, vol. 19, no. 1, pp. 34-44.
Haring, E., Herzig Straschil, B., and Spitzenberger, F., Phylogenetic analysis of Alpine voles of the Microtus multiplex complex using the mitochondrial control region, J. Zool. System. Evol. Res., 2000, vol. 38, no. 4, pp. 231-238.
Hall, T.A., BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows95/98/NT, Nucleic Acids Symp. Ser., 1999, vol. 41, pp. 95-98.
Tamura, K., Stecher, G., Peterson, D., et al., MEGA6: molecular evolutionary genetics analysis version 6.0, Mol. Biol. Evol., 2013, vol. 30, pp. 2725-2729. doi 10.1093/molbev/mst197
Ronquist, F. and Huelsenbeck, J.P., MrBayes 3: Bayesian phylogenetic inference under mixed models, Bioinformatics, 2003, vol. 19, no. 12, pp. 1572-1574.
Nylander, J.A.A., MrModeltest, Version 2, Program distributed by the author, Uppsala: Evolutionary Biology Centre, Uppsala University, 2004, vol. 2.
Bandelt, H.-J., Forster, P., and Röhl, A., Median-joining networks for inferring intraspecific phylogenies, Mol. Biol. Evol., 1999, vol. 16, pp. 37-48.
Excoffier, L., Laval, G., and Schneider, S., Arlequin ver. 3.0: an integrated software package for population genetics data analysis, Evol. Bioinf. Online, 2005, vol. 1, pp. 47-50.
Librado, P. and Rozas, J., DnaSP v5: a software for comprehensive analysis of DNA polymorphism data, Bioinformatics, 2009, vol. 25, pp. 1451-1452.
Farris, J.S., Källersjö, M., Kluge, A.G., and Bult, C., Testing significance of incongruence, Cladistics, 1995, vol. 10, no. 3, pp. 315-319.
Swofford, D.L., PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods): Version 4.0b10, Sunderland: Sinauer Associates, 2002.
Tamura, K., Estimation of the number of nucleotide substitutions when there are strong transition—transversion and G + C-content biases, Mol. Biol. Evol., 1992, vol. 9, pp. 678-687.
Tavaré, S., Some probabilistic and statistical problems in the analysis of DNA sequences: some mathematical questions in biology—DNA sequence analysis, Lect. Math. Life Sci., 1986, vol. 17, no. 2, pp. 57-86.
Kimura, M., A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences, J. Mol. Evol., 1980, vol. 16, no. 2, pp. 111-120.
Hasegawa, M., Kishino, K., and Yano, T., Dating the human—ape splitting by a molecular clock of mitochondrial DNA, J. Mol. Evol., 1985, vol. 22, no. 2, pp. 160-174.
Zvychainaya, E.Yu., Danilkin, A.A., Kholodova, M.V., et al., Analysis of the variability of the control region and cytochrome b gene of mtDNA of Capreolus pygargus Pall., Biol. Bull., 2011, vol. 38, article 1134. https://doi.org/10.1134/S1062359011050189.
Abramson, N.I., Phylogeography: results, problems, perspectives, Vavilovskii Zh. Genet. Sel., 2007, vol. 11, no. 2, pp. 307-331.
Khrunyk, Yu.Ya., Bogdanov, V.D., Yalkovskaya, L.E., et al., The genetic diversity of burbot (Lota lota L., 1758) of Western Siberia (the analysis of the mtDNA control region polymorphism), Russ. J. Genet., 2017, vol. 53, no. 2, pp. 233-241. https://doi.org/10.1134/ S1022795417010082.
Polityko, Yu.K. and Sibiryakov, P.A., Variability of mitochondrial DNA of Arctic char (Salvelinus alpinus L., 1758) from the Polar Urals, Ekologiya. Genetika. Evolutsiya (Ecology. Genetics. Evolution) (Proc. Conf. Young Sci.), Yekaterinburg: Goshitskii, 2015, pp. 122-128.
Yigit, N., Hutterer, R., Krystufek, B., and Amori, G., Microtus arvalis, The IUCN Red List of Threatened Species, 2016. http://dx.doi.org/ 10.2305/IUCN.UK.2016-2.RLTS.T13488A22351133.en. Accessed Juni 10, 2016.
Demidovich, A.P. and Lipin, S.I., Biology of common vole in the Irkutsk oblast, Vestn. Irkutsk Gos. S-kh. Akad., 1997, no. 3, pp. 1-25.
ACKNOWLEDGMENTS
We thank Zoological museum of the institute of Plant and Animal Ecology, Ural Branch of Russian Academy of Sciences, Yekaterinburg and our collegs A.V. Borodin, T.V. Strukova, S.B. Rakitin, and D.A. Toporkova (Institute of Plant and Animal Ecology, Ural Branch of Russian Academy of Sciences, Yekaterinburg) for kindly providing collection specimens for the analysis and S.V. Zykov, M.A. Fominykh, L.I. Amerkhanova, E.V. Zinoviev, E.P. Izvarin, E.Yu. Zakharova, T.S. Oslina, A.O. Shkurikhin (Institute of Plant and Animal Ecology, Ural Branch of Russian Academy of Sciences, Ekaterinburg), and R.Yu. Dudko (Institute of Systematics and Ecology of Animals, Siberian Branch of Russian Academy of Sciences, Novosibirsk) for their help in organizing field studies and catching small mammals. Special thanks to the Joint Directorate of the Orenburg and Shaitan-Tau State Nature Reserves, personally to O.V. Soroka and inspectors of the Talovskaya Steppe, Aituirskaya Steppe, and Aschisay Steppe sites for assistance in organizing and carrying out small mammal catching in the Orenburg Reserve, and the Administration of the Buzuluksky Bor National Park for assistance in organizing catching on the territory of the national park. We also thank G.A. Tsaur and the staff of the Laboratory of Molecular Biology, Immunophenotyping, and Pathomorphology of the Children’s Oncology and Hematology Center, Regional Children Hospital No. 1 (Yekaterinburg) for the opportunity to use an ABI Prism 3130 genetic analyzer (Applied Biosystems, United States).
This study was supported by the Russian Foundation for Basic Research (grant no. 16-04-01486-a) and the Integrated Program of the Ural Branch of the Russian Academy of Sciences (grant no. 18-4-4-43).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by N. Maleeva
Rights and permissions
About this article

Cite this article
Sibiryakov, P.A., Tovpinets, N.N., Dupal, T.A. et al. Phylogeography of the Common Vole Microtus arvalis, the Obscurus Form (Rodentia, Arvicolinae): New Data on the Mitochondrial DNA Variability. Russ J Genet 54, 1185–1198 (2018). https://doi.org/10.1134/S1022795418100137
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795418100137