
Abstract
This study aims to identify miRNAs and their potential targets involved in regulating the defense response of cotton verticillium wilt in different resistant Gossypium hirsutum varieties. RNA libraries and degradome libraries from control samples and infected roots of G. hirsutum (resistant cotton variety ‘Zhongmian-49’ and susceptible cotton variety ‘Junmian-1’) were constructed. High-throughput small RNA sequencing was used to identify and analyze miRNAs from two different resistant G. hirsutum varieties under different stress modes. Degradome sequencing was used to detect cleaved target genes. The real-time reverse transcription-PCR (qRT-PCR) was used to verify the miRNAs and detect target genes related to growth, development and resistance. A total of 69 known miRNAs and 330 novel miRNAs were identified in all libraries. By comparing the expression levels of miRNAs in control samples and V. dahliae stress libraries, we found that there are 23 differentially expressed miRNAs in ‘Zhongmian-49’, and 62 differentially expressed miRNAs in ‘Junmian-1’. In addition, 615 degradation sites were identified in 615 target genes by degradome analysis. Bioinformatics analysis further showed that genes might be involved in the regulation of many cellular and molecular processes in the growth, development and resistance process of cotton. Finally, qRT-PCR further confirmed 8 target genes with potential roles in cotton growth and resistance regulation. This study shows that the resistant and susceptible varieties of G. hirsutum have different responses to V. dahliae inoculation at the miRNA level, and that miRNA may contribute to the successful defence of the resistant cultivars.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Verticillium wilt is a severe vascular disease widely distributed all over the world [1]. Verticillium dahliae is the main pathogen of verticillium wilt and a devastating plant pathogenic fungus. V. dahliae has strong pathogenicity and a wide range of hosts, and can survive in the soil as microsclerotia for decades [2]. Gossypium hirsutum is also known as upland cotton, and it accounts for more than 95 percent of the global cotton crop. However, there is currently no fungicide that can eradicate verticillium wilt of G. hirsutum after plants have been affected [3–5]. Cotton is widely grown due to the high economic value of its fiber [6]. Fungal infection hinders the growth and production of cotton. The study found that cotton fields with cotton verticillium wilt disease accounted for 58.2% of cotton fields in Xinjiang (the main cotton production area), and cotton fields with disease index above 5.0 accounted for 28.1% [7]. Therefore, the prevention and control of G. hirsutum verticillium wilt is the key to increasing cotton production.
With the deepening of research, there are more and more small RNAs (sRNAs), including small interfering RNA (siRNAs) and microRNAs (miRNAs). MiRNAs are a type of endogenous sRNA that primarily regulates gene expression at the transcriptional and post-transcriptional levels in plants and animals via mRNA cleavage or translational repression, thereby regulating various life activities [8–10]. In plants, miRNAs play a vital role in a variety of regulatory pathways and participate in almost all developmental processes, including leaf [11], flower [12], stem [13] and root development [14]. There is growing evidence that miRNAs play a role in the regulation of abiotic and biotic stress responses, as well as disease resistance [15–18].
In this study, through high-throughput sequencing analysis and degrading group sequencing to profile the miRNA and its target mRNA in different resistant G. hirsutum subjected to biological (V. dahliae) stress. These findings reveal a putative miRNA-mediated regulatory network that plays a key role in the response of this important crop to biological stress.
MATERIALS AND METHODS
Plant materials and stress treatments. The cotton samples were provided by the Economic Crop Research Institute of Xinjiang Academy of Agricultural Sciences. The G. hirsutum (including resistant cotton variety ‘Zhongmian-49’ and susceptible cotton variety ‘Junmian-1’) [19, 20] were delinted with concentrated sulfuric acid, sterilized with 10% sodium hypochlorite for 3 minutes, and then washed with sterile water 6 times and soaked in distilled water overnight. The seeds were wrapped in sterile moist germination paper and placed in a 28°C constant temperature incubator to moisturize and cultivate until germination. When the radicle was about 0.5 cm long, the seeds were sown in paper cups filled with a mixture of vermiculite: nutritional soil (3 : 2) in culture room for 16 h/8 h at 28°C for day/night.
V. dahliae (V991) was provided by Huazhong Agricultural University. The activated V. dahliae strains were cultured to Czapek’s liquid medium with shaking for 2–3 days (180 rpm, 26°C). Conidia were collected by centrifugation and washed with sterile water. The final concentration was adjusted to 1 × 107 cfu/mL with sterile distilled water using a haemocytometer (Marienfeld, Germany). The cotton seedlings were inoculated according to the previously described method [21]. Cotton seedlings grown for 3 weeks were uprooted, and the roots were gently rinsed with sterile distilled water. Then, the cotton roots were immersed in the V. dahliae conidia suspension (5 mL per seedling) for 2 minutes, and the seedlings were replanted in paper cups filled with a mixture of vermiculite: nutritional soil (3 : 2). The cotton roots were harvested 24, 48 and 72 h after inoculation, and then the harvested cotton roots were stored in liquid nitrogen. The control plants were treated with distilled water and sampled in the same way.
RNA isolation, library construction and sequencing. Four small RNA libraries were constructed using total RNA extracted from resistant cotton variety ‘Zhongmian-49’ (S01-S03: control samples; S04-S06: V. dahliae stress) and susceptible cotton variety ‘Junmian-1’ (S07-S09: control samples; S10-S12: V. dahliae stress) roots. Total RNA was extracted using an RNA kit (EASYspin Universal Plant RNA Kit AidLab, China) based on the manufacturer’s instructions. The NanoDrop 2000 UV-Vis spectrophotometer (NanoDrop, Wilmington, United States) was used to detect the purity of RNA samples (OD260/280 is approximately equal to 2.0; OD260/230 ≥ 1.8). Polyacrylamide gel electrophoresis was used to separate the sRNA from 1.5 g of total RNA, and the 5'RNA adaptor and 3'RNA adaptor were ligated using T4 RNA Ligase 1 and T4 RNA Ligase 2 (truncated), respectively. The cDNA was synthesized via reverse transcription, followed by PCR amplification and PAGE gel separation of target DNA fragments. After gel extraction, a small RNA library was obtained.
Qubit 2.0 fluorometer (Qubit 2.0, Life Technologies, ThermoFisher Scientific, United States) was used to detect the concentration of the library after the library was constructed. The library concentration was diluted to 1 ng/μL, and the size of the insert was measured using an Agilent 2100 bioanalyzer (Agilent Technologies, United States). Moreover, the effective concentration of the library was accurately quantified using real-time reverse transcription-PCR (qRT-PCR) method to ensure the quality of the library. After the library was qualified, HiSeq 2500 (Illumina, United States) was used for high-throughput sequencing.
Identification of conserved and novel miRNAs. First, clean reads were obtained by filtering the original readings of the small RNA library to remove low-quality reads (Length < 18 nt or > 30 nt reads, reads with 3'adaptor, and reads with N content ≥ 10%). To obtain unannotated reads containing miRNA, clean reads were aligned with the Silva, GtRNAdb, Rfam, and Repbase databases to filter out ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and other non-coding RNA (ncRNA). The miRDeep2 software (http://www.mdc-berlin.de/rajewsky/miRDeep, the last date of access: September 30, 2020) was used to analyze clean reads, identify miRNAs as known and novel miRNAs and determine the expression of miRNAs. Besides, according to the known miRNA, novel miRNA and gene sequence information, the TargetFinder software is used to predict the target gene.
Differential expression analysis. The expression patterns of miRNAs were compared between two samples subjected to control samples and V. dahliae stress to determine the differentially expressed miRNAs. The Fold Change ≥ 2 was used as the threshold for screening differentially expressed miRNAs.
The BLAST software (https://blast.ncbi.nlm.nih. gov/Blast.cgi, the last date of access: September 30, 2020) was used to compare the predicted target gene sequence with the NR (https://ftp.ncbi.nlm.nih.gov/ blast/db/FASTA/, the last date of access: September 30, 2020), Swiss-Prot (http://www.gpmaw.com/html/ swiss-prot.html, the last date of access: September 30, 2020), Gene Ontology (GO) (http://geneontology.org/, the last date of access: September 30, 2020), COG (http://www.ncbi.nlm.nih.gov/COG, the last date of access: September 30, 2020), Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp, the last date of access: September 30, 2020), KOG (http://www.ncbi.nlm.nih.gov/KOG, the last date of access: September 30, 2020), Pfam (http://pfam. xfam.org, the last date of access: September 30, 2020) database to obtain the annotation information of the target gene. Then, GO hierarchical enrichment analysis and KEGG pathway enrichment analysis were performed on the target genes of differentially expressed miRNAs.
Degradome analysis. Four degradation libraries were constructed according to the methods described previously [22]. In short, the mRNA was first captured with Oligo (dT) magnetic beads, and then the 5'RNA adaptor was connected to the cleavage product. The purified ligation product was reverse transcribed into cDNA, and then digested with enzyme MmeI and amplified by PCR. The purified PCR products were sequenced on Illumina Hiseq 2500 sequencing system. Clean tags and cluster tags were obtained after jointing and filtering the original tags. The cluster Tags were compared with the genome (https://www.ncbi.nlm. nih.gov/genome, the last date of access: September 30, 2020) and Rfam library (http://rfam.sanger.ac.uk, the last date of access: September 30, 2020) to obtain the statistical results of the comparison with the genome and the annotation information of ncRNA. The ncRNAs were annotated after cluster Tags were compared with the genome and Rfam library. The unannotated sequence would be used for degradation site analysis. The degradation site was detected by Cleaveland software, and the condition was set to P value < 0.05.
QRT-PCR analysis. To validate the existence and expression of the identified miRNAs and target genes, 8 target genes corresponding to 3 known miRNAs and 7 novel miRNAs were selected for qRT-PCR analysis. TransScript II All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) kit was used to reverse transcription of miRNA into cDNA. The Histone-3 (Accession NO. AF024716) was used as an internal reference. The qRT-PCR reaction was performed using the Top Green qRCR SuperMix (China Trans). The total volume of the reaction was 32.8 μL, including 2 μL Template (diluted cDNA), 10 μL 2 × TransStart Top Green qPCR SuperMix (TransGen, Beijing (Beijing, China), 0.4 μL (10 μmol/μL) for each primer, and 20 µL ddH2O. The reaction conditions were 94°C for 2 minutes, and then 45 cycles, respectively, at 94°C for 5 s, 60°C for 15 s, and 72°C for 10 s. The 2–ΔΔCt method was used to calculate relative gene expression levels.
RESULTS
High-Throughput Sequencing of sRNAs in Cotton
In order to characterize the sRNA profiles of the two cotton varieties under control samples and V. dahliae stress, 12 sRNA libraries were constructed. A total of 194937832 reads were obtained (Table 1). Most readings (>90%) are high-quality clean readings. As shown in Table 1, a total of 33.83 million clean reads were obtained, and at least 7.94 million clean reads were retrieved for each sample. In all evaluation stages, the size distribution of sRNAs is similar.
In the ‘Zhongmian-49’ samples, more than 80% (30709303 total sRNAs) were common between control samples and V. dahliae stress (Fig. 1a), but only 11.65% of the unique sequences overlapped were the same in control samples and V. dahliae stress (Fig. 1b). In the ‘Junmian-1’ samples, only 79% (30033343 total sRNAs) were common between control samples and V. dahliae stress (Fig. 1c), but 12.96% of the unique sequences overlapped were the same in control samples and V. dahliae stress (Fig. 1d). Therefore,most of the expressed sRNAs were common between control samples and V. dahliae stress,but the types of expressed sRNAs (unique sequences) were significantly different. The location of the sequenced sRNAs also provide evidence of sRNA expression.

Sequence comparison between control samples and Verticillium dahliae stress. (a) Total clean reads in ‘Zhongmian-49’ (2739542 total sRNA). (b) The unique small RNA sequences in ‘Zhongmian-49’ (5105855 unique sRNA). (c) Total clean reads in ‘Junmian-1’ (37835703 total sRNA). (d) The unique small RNA sequences in ‘Junmian-1’ (7728739 unique sRNA).
Identification of Known and Novel miRNAs
A total of 69 known miRNAs and 330 novel miRNAs were obtained by Illumina deep-sequencing method from all libraries (Table 2). The length of known miRNAs and novel miRNAs were mostly concentrated at 21-nt, followed by 24-nt (Fig. 2). This size distribution is consistent with what has been reported in other organisms [23]. The first base at the 5'end of miRNAs in plants has a strong preference for uracil (U), while the first base at the 5' end of the 24-nt miRNAs sequence has a clear preference for thymine (A) [24, 25], This situation also appeared in our research results. It can be seen from Fig. 3 that the first base of most miRNAs sequence was biased towards U, while the sequence of 24-nt and 25-nt miRNAs was more biased towards A. The cutting site specificity of cytoplasmic Dicer enzymes may be the reason for the bias of nucleotide composition at different positions [26].

Size distribution (nucleotide length) of identified miRNAs. (1) Control samples in ‘Zhongmian-49’—known miRNAs; (2) control samples in ‘Zhongmian-49’—novel miRNAs; (3) ‘Zhongmian-49’ after Verticillium dahliae stress—known miRNAs; (4) ‘Zhongmian-49’ after V. dahliae stress—novel miRNAs; (5) control samples in ‘Junmian-1’—known miRNAs; (6) control samples in ‘Junmian-1’—novel miRNAs; (7) ‘Junmian-1’ after V. dahliae stress—known miRNAs; (8) ‘Junmian-1’ after V. dahliae stress—novel miRNAs.

The percentage of the first nucleotide at the 5′ end of identified miRNAs. A—adenine, U—uracil, C—cytosine, G—glycine.
Differential Expression of miRNAs
We compared S01-S03 and S04-S06 (S01_S02_S03_vs_S04_S05_S06), as well as S07-S09 and S10-S12 (S07_S08_S09_vs_S10_S11_S12), to identify the differences in expression of known and novel miRNAs in control samples and V. dahliae stress (Table 3). S01_S02_S03_vs_S04_S05_S06 and S07_S08_S09_vs_S10_S11_S12 reflect the differences in responsiveness of the two cotton varieties under V. dahliae stress conditions. As shown in Table 2, there are 23 differentially expressed miRNAs in S01_S02_S03_vs_S04_S05_S06. Among them, 10 miRNAs including 2 known miRNAs and 8 novel miRNAs were up-regulated, while 13 miRNAs including 4 known miRNAs and 9 novel miRNAs were down-regulated. Meanwhile, 62 miRNAs were differentially expressed in S07_S08_S09_vs_S10_S11_S12. Among them, 5 known miRNAs and 9 novel miRNAs were up-regulated, and 5 known miRNAs and 43 novel miRNAs were down-regulated. In addition, two novel miRNAs (D05-25287 and 4734-44138) were up-regulated in S01_S02_S03_vs_S04_S05_S06 and S07_S08_S09_vs_S10_S11_S12. Six miRNAs including 2 known miRNAs (miR7501 and miR7509) and 4 novel miRNAs (A03-3323, A05-5874, D05-25947 and 159474-48525) were all down-regulated in S01_S02_S03_vs_S04_S05_S06 and S07_S08_S09_ vs_S10_S11_S12. Only one miRNAs (7264-45089) was up-regulated in S01_S02_S03_vs_S04_S05_S06 and down-regulated in S07_S08_S09_vs_S10_S11_S12. In addition, further analysis revealed that microRNA 7264-45089 belongs to the MIR9560 family.
Identification of Target Genes of Differentially Expressed miRNAs
In order to better understand the function of m-iRNAs, The TargetFinder software was used to predict target genes. Among all 399 miRNAs, 45 miRNAs out of 69 known miRNAs predicted 229 target genes, and 214 out of 330 novel miRNAs predicted 731 target genes. Because plant miRNAs typically have strong support for their target genes that perform critical functions. The BLAST software was used to compare the predicted target gene sequence with different databases to obtain the annotation information of the target genes. Of the 867 predicted targets, 241 can be annotated in the COG database, 403 can be annotated in the GO database, and 239 can be annotated in the KEGG database (Table 4). To clearly understand the regulatory role of miRNAs in V. dahliae stress, the target genes of differentially expressed miRNAs in S01_S02_S03_vs_S04_S05_S06 and S07_S08_S09_ vs_S10_S11_S12 were analyzed based on GO terminology classification. As shown in Fig. 4, the five most important biological processes in S01_S02_S03_ vs_S04_S05_S06 were “metabolic process”, “single-organism process”, “cellular process”, “response to stimulus” and “cellular component organization or biogenesis”. In S07_S08_S09_vs_S10_S11_S12, the order of the first four biological processes was the same, followed by “biological regulation”. The main cell components of S01_S02_S03_vs_S04_S05_S06 and S01_S02_S03_vs_S04_S05_S06 were “cell”, “cell part” and “extracellular region”. Regarding molecular functions, in S01_S02_S03_vs_S04_ S05_S06 and S01_S02_S03_vs_S04_S05_S06, all genes were clustered into “catalytic activity”, “binding”, “transporter activity” and “electronic carrier activity”.

GO function classification according to cellular component, molecular function and biological process of all miRNA targets between control samples and Verticillium dahliae stress. (a) ‘Zhongmian-49’; (b) ‘Junmian-1’.
Degradome Sequencing Analysis
Through the degradome sequencing method, a total of 113.25M clean readings were obtained after filtering. Among them, 6503350 (44.90%) readings matched perfectly with the genome and Rfam library. The cleaveland software 4.0 predicted the degradation sites and cleavage products, and 615 degradation sites were identified in 615 target genes. Based on the abundance of degradation sites and the abundance of transcripts, target genes were classified into category 0, 1, 2, 3, and 4. In this study, only categories 0 and 1 were found to have raw read, of which category 0 had the most raw read at the cleavage position.
Then 106 genes targeted by miRNAs were annotated through GO analysis. The targets were classified into 16 biological process, 66 molecular functions, and 24 cellular components (Supplementary Table S1). “Metabolic process”, “cellular process” and “single-organism process” were the most abundant among the biological processes. For cell components, the most common categories were “cell”, “cell part” and “organelle”. Among molecular functions, the most abundant molecular function categories were “catalytic activity” and “binding”. Next, the KEGG Pathway database was used to further classify target genes, and the results show that target genes are expected to participate in 77 pathways, including “Metabolic pathways” and “Biosynthesis of secondary metabolites”.
Confirmation of Predicted miRNAs and Target Genes by qRT-PCR
Two novel miRNAs and two known miRNAs were chosen for qRT-PCR analysis to validate the existence and expression patterns of the predicted miRNAs in control and Verticillium-inoculated cotton roots. The qRT-PCR results for those miRNAs were basically consistent with the sequencing data, confirming the alterations in miRNA expression in response to V. dahliae infection (Fig. 5).

Relative expression analysis of miRNAs by qRT-PCR analysis and high-throughput sequencing. (a) ghr-miR7484b, (b) ghr-miR7501, (c) unconservative_A05_587, (d) unconservative_D05_25287. *P < 0 .05, **P < 0.01.
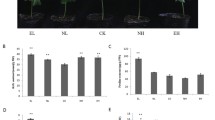
Furthermore, 3 known and 7 novel miRNAs corresponding to 8 target genes (evm.TU.Gh_A03G1932, evm.TU.Gh_D06G1480, evm.TU.Gh_A06G1102, evm.TU.Gh_A12G0659, evm.TU.Gh_D01G1421, evm.TU.Gh_D10G0671, evm.TU.Gh_D10G2348 and evm.TU.Gh_D05G3876) related to growth, development and resistance were also selected for qRT-PCR analysis (Table 5). The qRT-PCR results of these target genes were shown in the Fig. 6. After the two cotton varieties were subjected to V. dahliae stress, the expression levels of five differentially expressed miRNAs target genes including evm.TU.Gh_A03G1932, evm.TU.Gh_D06G1480, evm.TU.Gh_A06G1102, evm.TU.Gh_A12G0659 and evm.TU.Gh_D01G1421 were lower than that in control samples. Evm.TU.Gh_D10G0671 and evm.TU.Gh_D05G3876 were both expressed at higher levels in ‘Zhongmian-49’, while evm.TU.Gh_D10G2348 had a higher expression level in ‘Junmian-1’.

QRT-PCR validation of 8 target genes. (a) ‘Zhongmian-49’; (b) ‘Junmian-1’. (1) Control samples in ‘Zhongmian-49’; (2) ‘Zhongmian-49’ after Verticillium dahliae stress; (3) control samples in ‘Junmian-1’; (4) ‘Junmian-1’ after V. dahliae stress.
DISCUSSION
V. dahliae is a soil-borne fungus that causes devastating vascular dysfunction on more than 200 plant species, including many economically important crops such as cotton and tomatoes [27]. At present, no cotton varieties have been discovered to be resistant to verticillium wilt, however most Gossypium barbadense L. (G. Barbadense) varieties exhibit substantial benefits in verticillium wilt resistance [28, 29]. Zhang et al. used G. hirsutum and G. barbadense as models to identify miRNAs and their targets related to the regulation of the defense response of Verticillium dahliae [23]. However, no studies have evaluated the miRNA functions related to the regulation of the defense response of Verticillium dahliae in G. hirsutum. Therefore, in this study, two resistant G. hirsutum species ‘Junmian-1’ (a susceptible cotton variety) and ‘Zhongmian-49’ (a resistant cotton variety) were used as models to study miRNA functions related to cotton verticillium wilt defense response regulation. Four sRNA libraries and four degradation group libraries were constructed from cotton roots inoculated with simulation (control group) and V. dahliae (experimental group). Thus, the miRNA and its target genes that may be related to V. dahliae stress in G. hirsutum were discovered through analysis.
Through deep sequencing, 69 known miRNAs and 330 new miRNAs were detected in ‘Junmian-1’ and ‘Zhongmian-49’. Compared with previous studies, there are significantly fewer miRNAs and more novel miRNAs in this study, which may be caused by different cotton varieties. In addition, 23 differentially expressed miRNAs were detected in S01_S02_S03_ vs_S04_S05_S06, of which about 43.5% of miRNAs were up-regulated and 56.5% of miRNAs were down-regulated. A total of 62 differentially expressed miRNAs were detected in S07_S08_S09_vs_S10_S11_S12, 22.6% of miRNAs were up-regulated, and 77.4% of miRNAs were down-regulated. Through GO annotation of the target genes of differentially expressed miRNAs, it is found that the GO terms with the most significant enrichment of genes in S01_S02_S03_ vs_S04_S05_S06 and S07_S08_S09_vs_S10_S11_S12 were basically similar. Differentially expressed miRNAs target genes were mainly involved in cellular components (“cell”, “cell part” and “extracellular region”), biological processes (“metabolic process”, “cellular process”, “single-organism process”, “response to stimulus” and “cellular component organization or biogenesis”) and molecular functions (catalytic activity, binding, transporter activity and electronic carrier activity). This indicates that the interaction between V. dahliae and cotton also requires the assistance of genes related to these items. Although the GO entries for target gene enrichment in the two varieties of cotton are very similar, the number of target genes differs greatly. The number of target genes in the susceptible cotton variety ‘Junmian-1’ subjected to the V. dahliae stress (129 target genes) was much larger than that of the resistant cotton variety ‘Zhongmian-49’ subjected to the V. dahliae stress (69 target genes). This may partly explain why V. dahliae can cause disease in susceptible cotton.
In this study, degradome analysis revealed that different miRNAs can regulate the same target gene and that one miRNA can detect multiple target genes at the same time. There are a large number of differentially expressed miRNAs involved in growth and development and resistance between S01_S02_S03_ vs_S04_S05_S06 and S07_S08_S09_vs_S10_S11_S12. The regulation of miRNAs and their targets were analyzed through a variety of biological information annotated and analyzed. And from the identified target genes, 8 target genes related to growth, development and resistance were selected for qRT-PCR. The results showed that evm.TU.Gh_D10G0671 and evm.TU.Gh_D05G3876 were expressed at higher levels in the resistant variety ‘Zhongmian-49’ after V. dahliae stress, and expressed at lower levels in the susceptible variety ‘Junmian-1’ after V. dahliae stress. The evm.TU.Gh_D10G2348 had a higher expression level after V. dahliae stress in ‘Junmian-1’, while ‘Zhongmian-49’ showed a tendency to inhibit expression level after V. dahliae stress. This may be caused by ‘Zhongmian-49’ is a kind of resistant cotton variety and ‘Junmian-1’ is a kind of susceptible cotton variety.
The evm.TU.Gh_D10G2348, evm.TU.Gh_ D10G0671 and evm.TU.Gh_D05G3876 are members of MAPK, ATP-dependent RNA helicase and NPR1, respectively. Studies have shown that NPR1 plays a key role in various defense networks [30]. It is reported that the defense response mechanism mediated by NPR1 by Fusarium graminearum in wheat [31], and Botrytis cinerea, Sclerotinia sclerotiorum, Alternaria radicina, Erysiphe heraclei, and Xanthomonas hortorum in carrots [32] were evolutionarily conserved. Furthermore, the overexpression of NPR1 can confer broad-spectrum resistance to pathogens of other important crop species. In this study, evm.TU.Gh_ D05G3876 (a member of NPR1) was expressed at a higher level in ‘Zhongmian-49’, which indicates that the drug-resistant variety ‘Zhongmian-49’ has certain resistance to V. dahliae.
Mitogen-activated protein kinase (MAPK) is a conserved serine/threonine protein kinase signaling system ubiquitous in plants [33, 34]. MAPK plays an important role in regulating plant growth and development, conducting and responding to various biotic and abiotic stress signals. For example, Arabidopsis MPK4 is involved in the regulation of photosynthesis, growth and immune defense of plants [33]. AtMPK12 is activated by auxin and regulates auxin response genes, acting as a negative regulator in the auxin signal transduction pathway [35]. Therefore, it can be concluded that evm.TU.Gh_D10G2348 is involved in the regulation of cotton disease resistance.
CONCLUSION
In conclusion, 12 sRNA libraries and 4 degradome libraries were constructed from control samples and V. dahliae stress susceptible cotton variety ‘Junmian-1’ and resistant cotton variety ‘Zhongmian-49’ for deep sequencing in this study. A large number of miRNAs were identified in both species, including 330 novel miRNAs and 69 known miRNAs. In response to V. dahliae inoculation, we identified 23 and 65 differentially expressed miRNAs in the resistant cotton variety ‘Zhongmian-49’ and susceptible cotton variety ‘Junmian-1’, respectively, and 62 differentially expressed miRNAs when comparing the resistant cotton variety ‘Zhongmian-49’ and susceptible cotton variety ‘Junmian-1’. At the same time, 615 genes were detected by degrading genome sequencing. The differential expression of miRNAs under V. dahliae stress conditions between the two varieties suggests that the miRNA machinery in the two G. hirsutum varieties may be different. The results provide a foundation for understanding the miRNA-dependent V. dahliae stress response in G. hirsutum.
REFERENCES
Li, J.J., Zhou, L., Yin, C.M., Zhang, D.D., Klosterman, S.J., Wang, B.L., Song, J., Wang, D., Hu, X.P., Subbarao, K.V., Chen, J.Y., and Dai, X.F., The Verticillium dahliae Sho1-MAPK pathway regulates melanin biosynthesis and is required for cotton infection, Environ. Microbiol., 2019, vol. 21, p. 4852.
Banno, S., Ikeda, K., Saito, H., Sakai, H., Urushibara, T., Shiraishi, T., and Fujimura, M., Characterization and distribution of two subtypes of Verticillium longisporum isolated from cabbage fields in Japan, J. Gen. Plant Pathol., 2015, vol. 81, p. 118.
Deketelaere, S., Tyvaert, L., França, S.C., and Höfte, M., Desirable traits of a good biocontrol agent against verticillium wilt, Front. Microbiol., 2017, vol. 8, p. 1186.
Wang, Y., Liang, C., Wu, S., Zhang, X., Tang, J., Jian, G., Jiao, G., Li, F., and Chu, C., Significant improvement of cotton verticillium wilt resistance by manipulating the expression of gastrodia antifungal proteins, Mol. Plant, 2016, vol. 9, p. 1436.
Zhang, Y., Wang, X., Rong, W., Yang, J., Li, Z., Wu, L., Zhang, G., and Ma, Z., Histochemical analyses reveal that stronger intrinsic defenses in Gossypium barbadense than in G. hirsutum are associated with resistance to Verticillium dahliae, Mol. Plant Microbe Interact., 2017, vol. 30, p. 984.
Majumdar, A., Selection of raw materials in textile spinning industry using fuzzy multi-criteria decision making approach, Fibers Polym., 2010, vol. 11, p. 121.
Erdogan, O., Sezener, V., Ozbek, N., Bozbek, T., Yavas, I., and Unay, A., The effects of verticillium wilt (Verticillium dahliae Kleb.) on cotton yield and fiber quality, Asian J. Plant Sci., 2006, vol. 5, p. 867.
Llave, C., Xie, Z., Kasschau, K.D., and Carrington, J.C., Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA, Science, 2002, vol. 297, p. 2053.
Carrington, J.C. and Ambros, V., Role of microRNAs in plant and animal development, Science, 2003, vol. 301, p. 336.
Voinnet, O., Origin, biogenesis, and activity of plant microRNAs, Cell, 2009, vol. 136, p. 669.
Palatnik, J.F., Allen, E., Wu, X., Schommer, C., Schwab, R., Carrington, J.C., and Weigel, D., Control of leaf morphogenesis by microRNAs, Nature, 2003, vol. 425, p. 257.
Rhoades, M.W., Reinhart, B.J., Lim, L.P., Burge, C.B., Bartel, B., and Bartel, D.P., Prediction of plant m-icroRNA targets, Cell, 2002, vol. 110, p. 513.
Mallory, A.C., Dugas, D.V., Bartel, D.P., B and artel, B., MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs, Curr. Biol., 2004, vol. 14, p. 1035.
Guo, H.S., Xie, Q., Fei, J.F., and Chua, N.H., MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for arabidopsis lateral root development, Plant Cell, 2005, vol. 17, p. 1376.
Katiyar-Agarwal, S., Morgan, R., Dahlbeck, D., Borsani, O., Villegas, A., Jr., Zhu, J.K., Staskawicz, B.J., and Jin, H., A pathogen-inducible endogenous siRNA in plant immunity, Proc. Natl. Acad. Sci. U.S.A., 2006, vol. 103, p. 18002.
Navarro, L., Dunoyer, P., Jay, F., Arnold, B., Dharmasiri, N., Estelle, M., Voinnet, O., and Jones, J.D., A plant miRNA contributes to antibacterial resistance by repressing auxin signaling, Science, 2006, vol. 312, p. 436.
Padmanabhan, C., Zhang, X., and Jin, H., Host small RNAs are big contributors to plant innate immunity, Curr. Opin. Plant Biol., 2009, vol. 12, p. 465.
Zhang, X., Zhao, H., Gao, S., Wang, W.C., Katiyar-Agarwal, S., Huang, H.D., Raikhel, N., and Jin, H., Arabidopsis Argonaute 2 regulates innate immunity via miRNA393(*)-mediated silencing of a Golgi-localized SNARE gene, MEMB12, Mol. Cell, 2011, vol. 42, p. 356.
Wang, P., Su, L., Qin, L., Hu, B., Guo, W., and Zhang, T., Identification and molecular mapping of a Fusarium wilt resistant gene in upland cotton, Theor. Appl. Genet., 2009, vol. 119, p. 733.
Shi, Y., Zhang, B., and Liao, Y., Cotton-Zhongmian 49, Xinjiang Farm Res. Sci. Technol., 2013, vol. 36, p. 26.
Liu, S.Y., Chen, J.Y., Wang, J.L., Li, L., Xiao, H.L., Adam, S.M., and Dai, X.F., Molecular characterization and functional analysis of a specific secreted protein from highly virulent defoliating Verticillium dahliae, Gene, 2013, vol. 529, p. 307.
German, M.A., Pillay, M., Jeong, D.H., Hetawal, A., Luo, S., Janardhanan, P., Kannan, V., Rymarquis, L.A., Nobuta, K., German, R., De Paoli, E., Lu, C., Schroth, G., Meyers, B.C., and Green, P.J., Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends, Nat. Biotechnol., 2008, vol. 26, p. 941.
Zhang, Y., Wang, W., Chen, J., Liu, J., Xia, M., and Shen, F., Identification of miRNAs and their targets in cotton inoculated with Verticillium dahliae by high-throughput sequencing and degradome analysis, Int. J. Mol. Sci., 2015, vol. 16, p. 14749.
Mi, S., Cai, T., Hu, Y., Chen, Y., Hodges, E., Ni, F., Wu, L., Li, S., Zhou, H., Long, C., Chen, S., Hannon, G.J., and Qi, Y., Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide, Cell, 2008, vol. 133, p. 116.
Czech, B. and Hannon, G.J., Small RNA sorting: matchmaking for Argonautes, Nat. Rev. Genet., 2011, vol. 12, p. 19.
Chen, X., MicroRNA biogenesis and function in plants, FEBS Lett., 2005, vol. 579, p. 5923.
Masny, A., Zurawiez, E., Pruski, K., and Madry, W., Combining ability analysis in 10 strawberry genotypes used in breeding cultivars for tolerance to verticillium wilt, J. Am. Soc. Hortic. Sci., 2014, vol. 139, p. 275.
Xu, L., Zhu, L., Tu, L., Liu, L., Yuan, D., Jin, L., Long, L., and Zhang, X., Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry, J. Exp. Bot., 2011, vol. 62, p. 5607.
Song, R., Li, J., Xie, C., Jian, W., and Yang, X., An overview of the molecular genetics of plant resistance to the verticillium wilt pathogen Verticillium dahliae, Int. J. Mol. Sci., 2020, vol. 21, p. 1120.
Peng, J.L., Bao, Z.L., Ren, H.Y., Wang, J.S., and Dong, H.S., Expression of harpin(xoo) in transgenic tobacco induces pathogen defense in the absence of hypersensitive cell death, Phytopathology, 2004, vol. 94, p. 1048.
Makandar, R., Essig, J.S., Schapaugh, M.A., Trick, H.N., and Shah, J., Genetically engineered resistance to Fusarium head blight in wheat by expression of Arabidopsis NPR1, Mol. Plant Microbe Interact., 2006, vol. 19, p. 123.
Wally, O., Jayaraj, J., and Punja, Z.K., Broad-spectrum disease resistance to necrotrophic and biotrophic pathogens in transgenic carrots (Daucus carota L.) expressing an Arabidopsis NPR1 gene, Planta, 2009, vol. 231, p. 131.
Pitzschke, A., Djamei, A., Bitton, F., and Hirt, H., A major role of the MEKK1-MKK1/2-MPK4 pathway in ROS signaling, Mol. Plant, 2009, vol. 2, p. 120.
Ulm, R., Ichimura, K., Mizoguchi, T., Peck, S.C., Zhu, T., Wang, X., Shinozaki, K., and Paszkowski, J., Distinct regulation of salinity and genotoxic stress responses by Arabidopsis MAP kinase phosphatase 1, EMBO J., 2002, vol. 21, p. 6483.
Lee, J.S., Wang, S., Sritubtim, S., Chen, J.G., and Ellis, B.E., Arabidopsis mitogen-activated protein kinase MPK12 interacts with the MAPK phosphatase IBR5 and regulates auxin signaling, Plant J., 2009, vol. 57, p. 975.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 31860418], and the Special Project for Basic Scientific Activities of Non-profit Institutes Supported the Government of Xinjiang Uyghur Autonomous Region (grant no. KY2014025).
Author information
Authors and Affiliations
Contributions
Xiaorong Li and Fangyuan Chen contributed equally to this article.
Corresponding authors
Ethics declarations
Conflict of interests. The authors declare that they have no conflicts of interest.
Statement on the welfare of humans or animals. This article does not contain any studies involving humans or animals performed by any of the authors.
Additional information
ADDITIONAL INFORMATION
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations: GO—Gene Ontology; KEGG—Kyoto Encyclopedia of Genes and Genomes; MAPK—mitogen-activated protein kinase; miRNAs – microRNAs; ncRNA – non-coding RNA; rRNA—ribosomal RNA; sRNAs—small RNAs; siRNAs—small interfering RNA; snRNA—small nuclear RNA; snoRNA—small nucleolar RNA; tRNA—transfer RNA.
Supplementary Information
Rights and permissions
About this article

Cite this article
Li, X.R., Chen, F.Y., Wang, J. et al. Analysis of Different Resistant Gossypium hirsutum Inoculated with Verticillium dahliae Infection by Small RNA and Degradome Sequencing. Russ J Plant Physiol 69, 80 (2022). https://doi.org/10.1134/S1021443722040112
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S1021443722040112