
Abstract
Modification of MFI-type zeolites with tin involves impregnating the zeolite with an SnCl2 solution in excess NaCl, calcination (to fix the tin oxide), and water washing (to remove excess NaCl). Loading platinum on the tin-modified support results in a highly active (5–10 mol of C3H6 per mol of Pt per second), selective (96–98% towards propylene), and stable catalyst for dehydrogenation of propane. Due to the high dispersion of tin, which serves as an anchor for platinum, the average Pt particle size is 1.2 nm. The dispersion of tin is facilitated not only by the molecular dilution of its precursor with sodium chloride but also by the redispersion of tin across the surface due to the formation of volatile SnCl4 during calcination.

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Propylene is extensively used for the production of monomers, polymers, and basic organic chemicals [1]. A well-known technique for its direct production is propane dehydrogenation (PDH) [2]. To achieve significant conversion in this equilibrium-type process, high temperatures are required (typically about 600°C). The highest activity in PDH has been achieved in the presence of platinum-based catalysts [3]. Given the harsh process conditions, these catalysts need to provide adequate resistance to coking and to the agglomeration of the active metal [4]. For this purpose, additional metal promoters that can affect the dispersion of platinum and change its electronic properties are generally introduced into the catalysts [5]. The most common promoter for Pt-based PDH catalysts, including those implemented on a commercial scale, is tin [6]. The behavior of Pt–Sn catalytic systems in PDH has been studied comprehensively. Their performance depends on many factors, the most important of which are support type, Sn/Pt ratio, metal introduction method, and preparation conditions.
Alumina is often used as a support for Pt–Sn catalysts due to its high specific surface area, acid–base properties, and thermal stability [7]. Although γ-Al2O3 [8] or θ-Al2O3 [9] are used conventionally, mesoporous Al2O3 [10] and alumina with a significant content of coordinatively unsaturated Al3+ [11] have been found to appreciably improve the catalytic performance due to their stronger metal–support interaction. The preparation of an active and stable catalyst further requires thorough acidity control to ensure, on the one hand, a strong metal–support interaction and, on the other, the absence of residual acid sites responsible for side reactions and for coke deposition [12].
Unlike alumina, other supports such as SiO2, SBA, or silicalite do not provide an adequate level of interaction with Sn and Pt, causing the agglomeration of platinum and facilitating the reduction of tin to the metallic state and, thus, the formation of markedly less active Pt–Sn alloys [13, 14]. In order to fix the metals on the surface, direct reduction of a Pt–Sn/SiO2 catalyst precursor with hydrogen at 800°C has been proposed [15]. Under these conditions, the alloy Pt3Sn preferentially formed; this alloy provided high catalytic activity and stability. Another approach is incorporating aluminum into SBA-15 [16], thus generating acid sites on the support surface; these acid sites prevent the active phase from agglomeration.
Zeolites appear to be almost ideal supports for PDH catalysts [17]. Among them, MFI-type aluminosilicate zeolites have been investigated most thoroughly. Although the presence of aluminum in zeolite makes it capable of interacting with tin and platinum [18], acid sites also catalyze side reactions such as cracking, oligomerization, and aromatization, thus impairing both the selectivity and stability of the catalyst. A number of approaches have been developed to suppress the side reactions. One effective technique is to adjust the acidity by adding alkaline or alkaline-earth metals [19–21]. Another approach is the encapsulation of Sn and Pt during the synthesis of Al-free silicalite-1 [22]. This technique allows for uniform dispersion of Pt–Sn clusters in zeolite channels, thus achieving high levels of activity, propylene selectivity, and catalytic stability. Yet another acidity suppression approach, which simultaneously immobilizes tin on the support surface, is dealumination—a process that removes aluminum from the zeolite lattice, thus forming silanol nests. The Pt–Sn/BEA catalysts prepared by this technique have achieved activity and stability levels that are among the highest ever reported for PDH [23, 24].
The catalytic performance of Pt–Sn/zeolite catalysts has been further improved by changing the shape of crystals and agglomerates [25], as well as by using hierarchical porous structures [26]. Increasing the zeolite external surface area improves the ability of the catalyst to disperse coke, thus extending its lifetime. Furthermore, the stability of Pt–Sn/MFI catalysts has been markedly enhanced by increasing the acidity of the zeolite external surface [27], which provides better metal dispersion.
The influence of tin on the catalytic performance is manifested both in geometric and electronic effects [28]. Various factors affect the formation of Pt–Sn particles on the MFI surface during catalyst preparation. When the catalyst is calcined in air at 500 °C, the interaction between Pt and Sn is strengthened without noticeable agglomeration of particles [29]. The sequence of metal introduction also plays an important role in the Pt–Sn system properties. It is preferable to first introduce tin and then platinum because good dispersion of tin on the surface enhances the dispersion of platinum and prevents the formation of nonselective Pt particles bound directly to the support [27, 30]. Another important consideration is the choice of an optimal Sn/Pt atomic ratio. Increasing the amount of tin promotes its reduction and the formation of tin-rich Pt–Sn alloys, thus reducing the catalytic activity and stability [31].
In our previous research, we have found that involving large amounts of NaCl in the zeolite impregnation with tin and platinum precursors not only effectively suppresses the zeolite acidity but also promotes the metal dispersion [20, 27]. We referred to the catalysts prepared by this method as ICW (impregnation–calcination–washing) catalysts. When investigating the effects of the metal introduction sequence, the best performance was exhibited by the catalysts that were successively co-impregnated with SnCl2 and NaCl, calcined, and impregnated with H2PtCl6. The procedure was finished by water washing to remove excess NaCl. In the present study, we investigated in detail a modified catalyst preparation sequence in which NaCl was washed off prior to platinum introduction. A special focus was on the tin dispersion method and on the role of tin as an anchor for platinum, as well as on the effects of the Sn/Pt atomic ratio on the activity, selectivity, and stability of the synthesized catalysts.
EXPERIMENTAL
Reagents and catalysts. A series of MFI-type zeolites with nominal SiO2/Al2O3 molar ratios of 50, 80, 280, and 716 (Zeolyst International) were used in the H-form after calcination at 600°C. AR-grade NaCl, SnCl2·2H2O, and H2PtCl6·6H2O were also used for catalyst preparation. Propane (99.92%) was purchased from BK Group, Russia. The physicochemical properties of the initial zeolites are described in [20, 27].
Catalyst preparation procedure (example): An impregnating solution was prepared by dissolving 0.024 g of SnCl2·2H2O and 0.39 g of NaCl in 3 mL of 0.2M HCl. The resultant solution and 5 g of a zeolite powder were preheated separately in a water bath to 70°C. The solution and zeolite were then mixed and held for 1 h under intermittent stirring at 70°C. The mixture was dried at 150°C for 1 h and calcined in air in a muffle furnace at 500°C. The obtained powder was mixed with 25 mL of distilled water using a magnetic stirrer at 80°C for 0.5 h. The product was filtered without additional washing, dried at 150°C for 1 h, and calcined at 500°C for 1 h. The sample was further impregnated with a mixture of 1.33 g of an H2PtCl6 solution (Pt 9.4 g/L) and 1.66 g of water. After drying at 150°C for 1 h, the sample was calcined again at 500°C. The calcination involved heating at a rate of 10°C/min to 500°C and holding at this temperature for 1 h. In accordance with the nominal amounts of the metals added, the catalyst sample was designated as (3Na0.5Sn)w(0.25Pt).
All the prepared samples were labeled (3NaxSn)w(yPt), where the indices “3”, “x”, and “y” are the nominal weight percentages of Na, Sn, and Pt, respectively. The indication of more than one metal in parentheses pointed to their co-impregnation. The letter “w” denotes the water washing step as part of the procedure. Each impregnation or washing step was followed by calcination. By default, calcination was carried out at 500°C. Otherwise, the temperature value was indicated as a subscript: e.g., (1Sn)400. The initial MFI zeolites were labeled MFI-M, where M is the SiO2/Al2O3 molar ratio. MFI-80 was used by default.
Characterization of catalysts. Quantitative elemental analysis was performed by energy dispersive X-ray fluorescence spectroscopy (ED-XRF) using a Thermo Scientific ARL Perform'x instrument.
The porous properties were examined by low-temperature nitrogen adsorption using a Micromeritics ASAP 2010 instrument. The specific surface area was evaluated by the Brunauer–Emmett–Teller (BET) method with a reproducibility of ±3%.
IR spectra of adsorbed pyridine (Py-IR) were recorded on a Nicolet Protégé 460 FTIR spectrometer in the range of 400–4000 cm–1 with a resolution of 4 cm–1. The experiments were carried out in a vacuum IR cell. The calcined samples (1Sn)400 and HMFI-80 were examined in accordance with the following standard procedure. The samples (disks 1.6 cm in diameter with a density of about 10 mg/cm2) were first evacuated in the IR cell at 400°C for 1 h. Pyridine adsorption was carried out at 150°C and 2 Torr Py for 30 min. The adsorption cycle was followed by desorbing pyridine at 150°C for 15 min, after which spectra were recorded. For the uncalcined samples (1Sn)80 and (1Sn)150, their evacuation, as well as the pyridine adsorption/desorption, were performed at 80 and 150°C, respectively. Brønsted acid sites (BAS) and Lewis acid sites (LAS) were quantified according to [32] based on peak areas of 1550 and 1450 cm–1, respectively.
The phase composition of the samples was identified by powder X-ray diffraction analysis (XRD) on a TongDa TD-3700 diffractometer equipped with CuKα irradiation.
The size distribution of platinum particles was characterized by transmission electron microscopy (TEM) using FEI Osiris and JEOL JEM2100 UHR instruments. These instruments had autoemission sources at an accelerating voltage of 200 keV. The diameter of a circle equal to that of the particle was taken as the equivalent particle diameter. The average particle diameter was calculated as \(\bar D = \sum {D_i^2} {\rm{/}}\sum {{D_i}} ,\) where Di is the equivalent particle diameter.
The Pt dispersion was measured by CO pulse chemisorption using a USGA-101 analyzer. About 200 mg of a sample was placed in a quartz microreactor 4 mm in inner diameter (ID). The sample was heated to 200°C at a rate of 10°C/min in a helium flow (10 mL/min) and held at 200°C for 0.5 h. The gas flow was switched from He to H2, and the sample was heated in H2 at 10°C/min to 570°C, followed by holding at this temperature for another 30 min. The reactor was cooled in H2 to 200°C, purged with He at 200°C for 0.5 h, and finally cooled to room temperature in a continuous He flow. For the chemisorption analysis, 0.25 mL pulses of carbon monoxide (CO) diluted to 9.7% in He were injected into the helium flow at 3-min intervals. This was continued until the chemisorption sites reached saturation. The CO concentration at the reactor outlet was monitored by a thermal conductivity detector (TCD). The amount of chemisorbed CO was derived from the TCD signal areas.
The Pt dispersion was calculated by the equation:
where V is the volume of the adsorbed CO (mL); AW is the atomic weight of Pt (g/mol); SF is the stoichiometric factor (assumed to be equal to 1); and W is the Pt weight in the sample (g).
Catalytic tests were carried out in a 5-mm ID tubular quartz reactor at atmospheric pressure. The sample powder was pressed and crushed to 0.5–1.1 mm particles. A weighed amount of the catalyst (0.05 g) was loaded into the reactor between two layers of quartz wool, heated in nitrogen to 300°C at 15 mL/min, then in H2 to 570°C at 10 mL/min, and held at 570°C for 0.5 h. The flow was then switched from H2 to propane with a flow rate of 13 mL/min (WHSV 28 h–1). The PDH reaction was conducted using pure propane without hydrogen or an inert gas. The reaction products were analyzed online on a Chromatec Crystal 5000 gas chromatograph equipped with a 25-m KCl-Al2O3 capillary column and a flame ionization detector (FID).
The deactivation constant Kd (h–1) was derived from the equation:
where t is the reaction time (h); Xstart is the propane conversion after 1 h; and Xend is the propane conversion at the end of the test run.
Higher constants correspond to a more rapid deactivation of the catalyst.
RESULTS AND DISCUSSION
Physicochemical properties of catalysts. The textural properties, acidity, and chemical composition of the initial zeolites MFI-50, MFI-80, MFI-280, and MFI-716 are described in our previous studies [20, 27]. A series of ICW catalysts (3NaxSn)w(yPt) with nominal Pt content of 0.1–0.5 wt % and Sn content of 0.1–1.0 wt % were prepared in accordance with sequence (Eq. (1)).

The chemical composition of the samples prepared from MFI-80 is presented in Table 1. The sodium content measured in samples 1–12 was far below 3% due to the NaCl wash-off procedure. On the other hand, the residual sodium content slightly exceeded the theoretical value (about 1%) determined by the concentration of aluminum-bound acid sites in MFI-80, thus indicating some presence of NaCl in the synthesized catalysts. The Pt content was comparable to its nominal value. At the same time, the Sn content in all samples was about half the nominal amount added to prepare the catalyst.
A decrease in the tin content was observed as early as during the drying step at 80°C and continued to 300°C, after which this trend became markedly slower. Importantly, the tin content was insensitive to the presence of sodium chloride (Fig. 1a), but clearly depended on the concentration of acid sites in the initial zeolite (Fig. 1b). Specifically, the amount of tin remaining on the zeolite surface increased with the acid site concentration.

Tin content: (a) in samples (1.0Sn) and (3Na1.0Sn) calcined at different temperatures; and (b) in sample (3Na1.0Sn)w(0.5Pt) calcined at 500°C prepared from MFI zeolites differing in SiO2/Al2O3 ratio as a function of BAS concentration.
Figure 2 and Table 2 provide the Py-IR spectroscopy data regarding the effects of the calcination temperature on the concentration and composition of acid sites for (1Sn) compared to the initial HMFI-80. The spectrum of pyridine adsorbed on (1Sn)80 after evacuation at 80°C shows peaks at 1547 cm–1 (BAS) and 1454 cm–1 (LAS), as well as a shoulder at 1446 cm–1 (H2-bonded Py). In the range sensitive to the site type, we see peaks at 1623 (tetrahedral aluminum), 1615 (tin LAS), and 1600 cm–1 (H2-bonded Py). The evacuation at 150°C had no effect on the spectrum except that the 1446 and 1600 cm–1 peaks (weakly-bound Py) disappeared.

IR spectra of pyridine adsorbed on HMFI-80, (1Sn)80, (1Sn)150, and (1Sn)400.
It is worth noting that the BAS and LAS concentrations increased (compared to the initial zeolite) after the tin incorporation and drying at 80°C, but both concentrations declined during the further heating. Sample (1Sn)400, which was calcined in air at 400°C and evacuated in the cell at the same temperature, exhibited a decrease in the BAS and LAS concentrations and a slight shift of the tin peak from 1615 to 1610 cm–1. It is most likely that two types of tin sites were available on the surface, one of which was removed after the calcination. It is fair to posit that these effects were interrelated, and some tin was present on the surface (e.g., as HSnCl3), thus providing both Brønsted and Lewis acidity.
Although the BAS concentrations were equal in (1Sn)400 and HMFI-80, the LAS concentration was higher in (1Sn)400. This indicates that tin was present as oxide particles on the surface rather than interacting with BAS.
Tin(II) chloride is a strong reducing agent and is easily hydrolyzed in aqueous solutions. To prevent hydrolysis, acidified solutions (most often, hydrochloric acid solutions) are typically used for impregnation. In this study, the impregnating solutions for tin introduction were prepared from 0.2M HCl. During air drying at 70°C, Sn(II) is oxidized to Sn(IV), which—in the presence of HCl and NaCl—forms volatile SnCl4 with a boiling point of 114°C. The evaporation of SnCl4 during the drying and calcination was presumably responsible for the drop in the tin concentration. A similar decrease in tin content has been reported in prior research [33]: after hydrothermal treatment of PtSnNa/ZSM-5 at 650°C, the tin concentration decreased from 0.84 to 0.31%.
We previously demonstrated that the presence of a large amount of NaCl in the impregnating solution together with SnCl2 favors a more uniform tin dispersion on the zeolite surface [27]. Tin(IV) chloride is even more prone to hydrolysis and is reactive with silanol groups and BAS on a zeolite surface. When passing through zeolite pores, SnCl4 vapors interact with the support’s active sites, resulting in additional tin dispersion. Therefore, an increase in the BAS concentration favors the retention of tin in higher concentrations (Fig. 1b). Thus, tin dispersion is facilitated by two processes: the molecular dilution of the tin precursor with sodium chloride; and the formation of volatile SnCl4 during calcination, this compound favoring the redispersion of tin across the zeolite surface.
The electronic images of (3Na1Sn)w(0.5Pt) (Fig. 3) clearly show that the Pt particles were distributed within a narrow size range of 0.6–1.8 nm with an average size of 1.2 nm. We attribute this good dispersion of platinum to the high tin dispersion prior to the platinum introduction. As an anchor for platinum, tin is known to promote Pt dispersion throughout the surface [34]. The interaction between SnCl4 vapors and the zeolite’s hydroxyl groups allows for almost atomic tin dispersion and potentially generates a large number of Sn sites to subsequently retain platinum even despite a small total tin content. Although the resolution of the TEM instrument was insufficient to detect single platinum and tin atoms, the elemental mapping images show that both platinum and tin were highly dispersed.

Electronic images of (3Na1Sn)w(0.5Pt): (a) TEM micrograph; (b) HAADF-STEM image; (c) particle size distribution histogram (based on 124 measurements); and (d–i) elemental mapping images.
The specific surface area of MFI-80 (387 m2/g) slightly decreased after the tin addition (352 m2/g for (1Sn)500). Nonetheless, the catalyst (3Na0.5Sn)w(0.25Pt), prepared according to sequence (1), had a specific surface area of 373 m2/g, i.e., similar to the initial value. It is reasonable to interpret these data as follows. Impregnating the zeolite with the SnCl2 solution in the absence of NaCl caused non-uniform tin dispersion with large particles being generated. A portion of these particles was able to block the zeolite pores, resulting in the lowest BET surface area for (1Sn)500. The co-impregnation with SnCl2 and a large amount of NaCl provided a markedly more uniform tin dispersion across the surface, and therefore, the zeolite textural properties changed only slightly after excess NaCl was washed off and platinum was introduced.
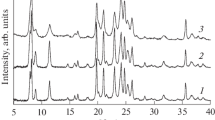
The XRD pattern of (0.5Pt) at 39.8° contains a reflection attributed to Pt(111) (Fig. 4), which points to the presence of large Pt(0) crystals. The lack of this reflection in the patterns of (3Na)w(0.5Pt) and (3Na1Sn)w(0.5Pt) indicates that these samples had small Pt particles and that sodium and tin were involved in their dispersion.

XRD patterns of initial zeolite and 0.5Pt-based samples.
The Pt dispersion in Pt–Sn catalysts measured by CO pulse chemisorption may be noticeably lower then calculated from TEM data for the same samples. The point is that a major portion of platinum could be blocked during the generation of Pt–Sn nanoparticles, and/or change in the electronic properties of platinum could weaken the Pt–CO interaction [35]. Nonetheless, the catalysts can be quantitatively compared within a series of similar samples. Figure 5 shows the correlation between the platinum dispersion and the tin and platinum content as a regression surface represented by a 2nd order polynomial. The correlation was plotted based on twelve points, five of which were hidden under the surface. The platinum dispersion increased with a decrease in the tin content, clearly due to the increase in the number of Pt sites accessible to CO. The lowest dispersion was observed at the highest Sn/Pt ratios. However, the Sn/Pt ratio optimal for catalytic performance does not necessarily correspond to a maximum dispersion.

Effects of Sn content and Pt content on Pt dispersion in (3NaxSn)w(yPt). R2 = 0.95.
Catalytic performance. Figure 6 shows the deactivation curves for the (3NaxSn)w(yPt) series of ICW catalysts. The initial propane conversion increased with Pt content. Given that the platinum dispersion also rises with its content (Fig. 5), the catalytic activity is clearly determined by the amount of accessible platinum. The tested catalysts exhibited propylene selectivity up to 97.5%; this selectivity dropped to 92% in the initial reaction step for the catalysts with the lowest nominal tin content. Increasing the Sn content led to an increase in the propylene selectivity. As the tin content increased, the catalytic activity first rose, then declined. Therefore, there is an optimum Sn/Pt ratio to maximize the catalytic activity and selectivity. In general, these observations are consistent with prior research on the role of tin in Pt–Sn/MFI systems [27, 31].

Effects of Sn content and Pt content in (3NaxSn)w(yPt) on activity, selectivity, and yield of propylene. PDH reaction conditions: 570°C; 0.1 MPa; WHSV 28 h−1.
Two major causes are known to lie behind the deactivation of platinum catalysts: coke deposition on the platinum surface and the agglomeration of platinum particles [17]. We previously demonstrated that the activity loss of the Pt–Sn/MFI type of ICW catalysts during the reaction was caused by coking, whereas the slight decline in their initial activity after oxidative regeneration served as evidence of a change in the size or composition of their Pt–Sn particles [27]. Regarding the catalysts synthesized in the present study, the deactivation constants did not clearly correlate with the Sn/Pt ratio (Fig. 7a). Nonetheless, the graph of deactivation constant versus tin and platinum content shows a distinct minimum within the concentration ranges of 0.2–0.3% Sn and 0.3–0.5% Pt (Fig. 7b). In other words, the ICW catalysts (3NaxSn)w(yPt) with an Sn/Pt atomic ratio of 0.6 to 2.2 turned out to be the most stable.
We previously proposed a comparison of dehydrogenation catalysts using the index of productivity (IP) [27]:
where SA is the specific activity defined as moles of propylene produced per mole of Pt atoms per second (s–1); and Kd is the deactivation constant (h−1).

Effects of Sn and Pt content on catalytic stability. Deactivation constant (h–1) of (3NaxSn)w(yPt) as a function of: (a) Sn/Pt ratio; and (b) Sn content and Pt content. R2 = 0.90.
Table 3 summarizes the data on the Pt–Sn/MFI catalysts for propane dehydrogenation reported to date in comparison with some catalysts prepared in the present study. For the best Pt–Sn/MFI catalysts reported elsewhere, the productivity index after 8 h of reaction amounts to 150–230. The best catalyst tested in this study, namely (3Na0.5Sn)w(0.25Pt), achieved an IP value of 404.
CONCLUSIONS
A series of ICW catalysts (i.e., catalysts prepared by the impregnation–calcination–washing method) that were designated as (3NaxSn)w(yPt) exhibited high activity and selectivity in the dehydrogenation of propane. When tin is introduced along with NaCl, its additional dispersion is facilitated both by the molecular dilution of tin chloride with sodium chloride and by the redispersion of tin across the surface due to the formation of volatile SnCl4 during calcination in air. After removing excess NaCl by water washing, tin is no longer bound to BAS, and the finely dispersed tin oxides promote the generation of platinum particles within a narrow size range with an average diameter of 1.2 nm. The Pt dispersion steadily increases with a decrease in the tin content; at the same time, as the tin content increases, the catalyst activity passes through a maximum. The Pt–Sn ICW catalysts achieved their highest stability (indicated by a deactivation constant of 0.011–0.020 h–1 at a propane WHSV of 28 h–1) within the concentration ranges of 0.2–0.3% Sn and 0.3–0.5% Pt. The catalyst (3Na0.5Sn)w(0.25Pt) exhibited a significantly higher performance than that ever reported for Pt–Sn/MFI catalysts for propane dehydrogenation.
REFERENCES
Pisarenko, E.V., Ponomarev, A.B., Smirnov, A.V., Pisarenko, V.N., and Shevchenko, A.A., Theor. Found. Chem. Eng., 2022, vol. 56, no. 5, pp. 687–722. https://doi.org/10.1134/S0040579522050335
Nawaz, Z., Rev. Chem. Eng., 2015, vol. 31, no. 5, pp. 413–436. https://doi.org/10.1515/revce-2015-0012
Martino, M., Meloni, E., Festa, G., and Palma, V., Catalysts, 2021, vol. 11, no. 9, Art. 1070. https://doi.org/10.3390/catal11091070
Hu, Z.-P., Yang, D., Wang, Z., and Yuan, Z.-Y., Chin. J. Catal., 2019, vol. 40, no. 9, pp. 1233–1254. https://doi.org/10.1016/S1872-2067(19)63360-7
Deng, L., Wang, J., Wu, Z., Liu, C., Qing, L., Liu, X., Xu, J., Zhou, Z., and Xu, M., J. Alloys Compd., 2022, vol. 909, Art. 164820. https://doi.org/10.1016/j.jallcom.2022.164820
Sattler, J.J.H.B., Ruiz-Martinez, J., Santillan-Jimenez, E., and Weckhuysen, B.M., Chem. Rev., 2014, vol. 114, no. 20, pp. 10613–10653. https://doi.org/10.1021/cr5002436
Zhao, D., Lund, H., Rodemerck, U., Linke, D., Jiang, G., and Kondratenko, E.V., Catal. Sci. Technol., 2021, vol. 11, pp. 1386–1394. https://doi.org/10.1039/D0CY01980A
Yu, C., Ge, Q., Xu, H., and Li, W., Appl. Catal. A, 2006, vol. 315, no. 2, pp. 58–67. https://doi.org/10.1016/j.apcata.2006.08.038
Lee, M.-H., Nagaraja, B.M., Natarajan, P., Truong, N.T., Lee, K.Y., Yoon, S., and Jung, K.-D., Res. Chem. Intermed., 2016, vol. 42, no. 1, pp. 123–140. https://doi.org/10.1007/s11164-015-2370-1
Zhang, Y., Zhou, Y., Shi, J., Zhou, S., Sheng, X., Zhang, Z., and Xiang, S., J. Mol. Catal. A: Chem., 2014, vol. 381, pp. 138–147. https://doi.org/10.1016/j.molcata.2013.10.007
Shi, L., Deng, G.-M., Li, W.-C., Miao, S., Wang, Q.-N., Zhang, W.-P., and Lu, A.-H., Angew. Chem. Int. Ed., 2015, vol. 54, no. 47, pp. 13994–13998. https://doi.org/10.1002/anie.201507119
Jang, E.J., Lee, J., Jeong, H.Y., and Kwak, J.H., Appl. Catal. A, 2019, vol. 572, pp. 1–8. https://doi.org/10.1016/j.apcata.2018.12.024
Feng, J., Zhang, M., and Yang, Y., Chin. J. Chem. Eng., 2014, vol. 22, nos. 11–12, pp. 1232–1236. https://doi.org/10.1016/J.CJCHE.2013.07.001
Kaylor, N. and Davis, R.J., J. Catal., 2018, vol. 367, pp. 181–193. https://doi.org/10.1016/j.jcat.2018.09.006
Deng, L., Shishido, T., Teramura, K., and Tanaka, T., Catal. Today, 2014, vol. 232, pp. 33–39. https://doi.org/10.1016/j.cattod.2013.10.064
Duan, Y., Zhou, Y., Zhang, Y., Sheng, X., and Xue, M., Catal. Lett., 2011, vol. 141, pp. 120–127. https://doi.org/10.1007/s10562-010-0445-6
Sun, M.-L., Hu, Z.-P., Wang, H.-Y., Suo, Y.-J., and Yuan, Z.-Y., ACS Catal., 2023, vol. 13, no. 7, pp. 4719–4741. https://doi.org/10.1021/acscatal.3c00103
Treesukol, P., Srisuk, K., Limtrakul, J., and Truong, T.N., J. Phys. Chem. B, 2005, vol. 109, no. 24, pp. 11940–11945. https://doi.org/10.1021/jp0511348
Zhang, Y., Zhou, Y., Qiu, A., Wang, Y., Xu, Y., and Wu, С., Acta Phys.-Chim. Sin., 2006, vol. 22, no. 6, pp. 672–678. https://doi.org/10.1016/S1872-1508(06)60026-0
Ponomaryov, A.B., Smirnov, A.V., Pisarenko, E.V., and Shostakovsky, M.V., Microporous Mesoporous Mater., 2022, vol. 339, Art. 112010. https://doi.org/10.1016/j.micromeso.2022.112010
Bai, L., Zhou, Y., Zhang, Y., Liu, H., and Tang, M., Catal. Lett., 2009, vol. 129, pp. 449–456. https://doi.org/10.1007/s10562-008-9822-9
Liu, L., Lopez-Haro, M., Lopes, C.W., Rojas-Buzo, S., Concepcion, P., Manzorro, R., Simonelli, L., Sattler, A., Serna, P., Calvino, J.J., and Corma, A., Nat. Catal., 2020, vol. 3, pp. 628–638. https://doi.org/10.1038/s41929-020-0472-7
Ivanushkin, G.G., Smirnov, A.V., Kots, P.A., and Ivanova, I.I., Petrol. Chem., 2019, vol. 59, pp. 733. https://doi.org/10.1134/S0965544119070077
Xu, Z., Yue, Y., Bao, X., Xie, Z., and Zhu, H., ACS Catal., 2020, vol. 10, no. 1, pp. 818–828. https://doi.org/10.1021/acscatal.9b03527
Wang, Y., Zhou, Y., Zhou, S., He, Q., and Zhang, Y., China Pet. Process. Petrochem. Technol., 2020, vol. 22, no. 1, pp. 87–97.
Zhou, S., Zhou, Y., Zhang, Y., Sheng, X., and Zhang, Z., J. Appl. Chem. Biotechnol., 2016, vol. 91, no. 4, pp. 1072–1081. https://doi.org/10.1002/jctb.4686
Ponomaryov, A.B., Smirnov, A.V., Pisarenko, E.V., and Shostakovsky, M.V., Appl. Catal. A: General, 2024, vol. 673, Art. 119588. https://doi.org/10.1016/j.apcata.2024.119588
Feng, B., Wei, Y.-C., Song, W.-Yu., and Xu, C.-M., Pet. Sci., 2022, vol. 19, no. 2, pp. 819–838. https://doi.org/10.1016/j.petsci.2021.09.015
Zhang, Y., Zhou, Y., Li, Y., Wang, Y., Xu, Y., and Wu, С., Catal. Commun., 2007, vol. 8, pp. 1009–1016.
Liu, H., Zhou, Y., Zhang, Y., Sheng, X., Zhang, Z., and Zhou, S., China Pet. Process. Petrochem. Technol., 2013, vol. 15, no. 4, pp. 54–62.
Zhang, Y., Zhou, Y., Qiu, A., Wang, Y., Xu, Y., and Wu, С., Catal. Commun., 2006, vol. 7, no. 11, pp. 860–866.
Tamura, M., Shimizu, K., and Satsuma, A., Appl. Catal. A., 2012, vols. 433–434, pp. 135–145. https://doi.org/10.1016/j.apcata.2012.05.008
Zhang, Y., Zhou, Y., Yang, K., Li, Y., Wang, Y., Xu, Y., and Wu, С., Microporous Mesoporous Mater., 2006, vol. 96, no. 1, pp. 245–254. https://doi.org/10.1016/j.micromeso.2006.07.003
Pham, H.N., Sattler, J.J.H.B., Weckhuysen, B.M., and Datye, A.K., Catal. ACS Catal., 2016, vol. 6, no. 4, pp. 2257–2264. https://doi.org/10.1021/acscatal.5b02917
Cortright, R.D. and Dumesic, J.A., J. Catal., 1994, vol. 148, pp. 771–778. https://doi.org/10.1006/jcat.1994.1263
Zhang, Y., Zhou, Y., Huang, L., Xue, M., and Zhang, S., Ind. Eng. Chem. Res., 2011, vol. 50, no. 13, pp. 7896–7902. https://doi.org/10.1021/ie1024694
Liu, L., Lopez-Haro, M., Lopes, C.W., Li, C., Concepcion, P., Simonelli, L., Calvino, J.J., and Corma, A., Nat. Mater., 2019, vol. 18, pp. 866–873. https://doi.org/10.1038/s41563-019-0412-6
Zhu, J., Osuga, R., Ishikawa, R., Shibata, N., Ikuhara, Y., Kondo, J.N., Ogura, M., Yu, J., Wakihara, T., Liu, Z., and Okubo, T., Angew. Chem., Int. Ed., 2020, vol. 59, no. 44, pp. 19669–19674. https://doi.org/10.1002/anie.202007044
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation within governmental order (project no. 075-00277-24-00).
The phase compositions were characterized using equipment supported by the national project “Science and Universities”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest requiring disclosure in this article.
Additional information
Publisher's Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ponomaryov, A.B., Smirnov, A.V., Shostakovsky, M.V. et al. Modification of Zeolites with Tin to Synthesize Pt–Sn/MFI Catalysts for Propane Dehydrogenation. Pet. Chem. (2024). https://doi.org/10.1134/S096554412404008X
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S096554412404008X