
Abstract
The effects of the p-toluenesulfonic acid monohydrate (TsOH∙H2O) concentration on the alkoxycarbonylation of cyclohexene over a Pd(PPh3)2Cl2–PPh3–TsOH∙H2O catalytic system were quantified at 363–383 K. Within this temperature range, the correlation between the cyclohexyl cyclohexanecarboxylate production rate and the TsOH∙H2O concentration was found to be similar to an S-shaped curve. Based on these data, as well as previous findings with regard to the effects of water and TsOH∙H2O concentrations on the cyclohexene methoxycarbonylation rate, the hydride mechanism for the alkoxycarbonylation process was updated by adding relevant ligand exchange reactions between ballast palladium complexes, specifically reactions that produce a palladium aqua complex. The accordingly-modified kinetic equation for cyclohexene alkoxycarbonylation with cyclohexanol and CO was found to be consistent with the experimental data. Effective constants were evaluated for the modified kinetic equation over the studied temperature range. A number of relevant parameters—namely, the effective activation energy and the changes in enthalpy, entropy, and Gibbs free energy during the ligand exchange between the complexes Pd(PPh3)2(C6H11OH)2 and Pd(PPh3)2(H2O)2—were further evaluated in light of the activated complex theory. This reaction was found to be nearly equilibrium at 373 K.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
The development of the unsaturated compounds alkoxycarbonylation with alcohols and CO has advanced considerably in recent decades. For this process, a wide variety of alkenes have been used, from ethylene [1, 2] to heavy alkenes, including those branched along multiple bonds [3, 4] and polymers with C=C bonds [5]. Not only petrochemicals but also unsaturated vegetable compounds have been used as substrates [6–8]. Alkoxycarbonylation can be carried out in the presence of Pd, Rh, Ru, Ir, Ir, Pt, Os, Co, Ni, Mo, W, Cr, and Sn catalysts [9]. In recent decades, research has mostly focused on homogeneous palladium–phosphine catalytic systems, which are commonly assumed to be the most catalytically active [1–8, 10–12]. Industrial production of methyl methacrylate over a catalyst of this sort has been implemented using Lucite’s Alpha process [8, 12]. Furthermore, some recent studies have successfully used Co- and Ru-based catalysts. For example, a commercially valuable reaction such as the propoxycarbonylation of ethylene was catalyzed by homogeneous Co2(CO)8-based systems [13], and the methoxycarbonylation of diisobutylene (2,3,4-trimethylpent-1-ene) was carried out over supported cobalt catalysts [14]. Studies have also been published on the methoxycarbonylation of cyclohexene over homogeneous ruthenium catalysts [15, 16] and the methoxycarbonylation of ethylene over heterogeneous ruthenium catalysts [17]. Among the most promising alkoxycarbonylation approaches, special attention should be drawn to the so-called isomerizing alkoxycarbonylation of alkenes with internal multiple bonds over palladium catalysts with some diphosphines [5, 8, 10, 11]. This process consists of two successive steps: migration of the C=C bond to the terminal position; and alkoxycarbonylation that preferentially produces linear esters. Yang et al [18] recently investigated an isomerizing alkoxycarbonylation process as part of the industrially relevant selective synthesis of adipinates from 1,3-butadiene.
Isomerizing alkoxycarbonylation can also occur via the dehydration of secondary alcohols catalyzed by strong protonic acids such as p-toluenesulfonic acid monohydrate (TsOH∙H2O) and the resulting in situ formation of internal alkenes [19]. In particular, when a linear secondary alcohol (e.g., 2-hexanol, 2-heptanol, or 2-nonanol) was used, its dehydration and the alkoxycarbonylation of the resultant internal alkene were arranged in one reactor to preferentially produce a linear carboxylic ester (2-hexyl heptanoate, 2-heptyl octanoate, and 2-nonyl decanoate, respectively). When cyclohexanol was used as a model alcohol in the dehydration combined in one reactor with the subsequent cyclohexene alkoxycarbonylation, the yield of cyclohexyl cyclohexanecarboxylate (CHCHC) reached up to 86% over 4–6 h. In this process, the strong protonic acids played a key role: under their effect, the yield of cyclohexene amounted to 20–34% within the initial 25–30 min [19, 20]. However, the cyclohexene alkoxycarbonylation rates were insufficient [19–22]. In alkoxycarbonylation over homogeneous palladium catalysts, relatively inactive ballast species are known to form and negatively affect the formation rate of target products [3, 23–25]. In-depth kinetic studies on the effects of various reactants on the reaction rate are able to shed light on these patterns and enable researchers to assess the relative stability of the various ballast complexes in the alkoxycarbonylation system [24]. We have previously investigated the effects of different reactants, Pd(PPh3)2Cl2 (as a catalytic precursor), and PPh3 (as a promoter) on the rate of cyclohexene alkoxycarbonylation with cyclohexanol and CO (reaction (1)) [23, 25]. We believe that to gain deeper insight into the mechanism of this reaction, the kinetics of the TsOH·H2O concentration effect on the reaction also need to be investigated in detail. It is worth noting that cyclohexane carboxylic esters are of significant practical interest as intermediates in pharmaceutical synthesis [9], and cyclohexene is a convenient model substrate with an internal C=C bond because its alkoxycarbonylation produces a single ester. Cyclohexanol is an available large-tonnage secondary alcohol. In contrast to methanol, an alcohol most commonly used in alkoxycarbonylation [1–8, 12, 14], other alcohols—including secondary alcohols—have been underexplored as contributors to alkoxycarbonylation.

EXPERIMENTAL
The details of the reactor, as well as the methods employed for the kinetic study of alkoxycarbonylation and for the gas chromatography of the reaction mixture samples using o-xylene as an internal standard, are described in Supplementary Information. The correlation between the cyclohexene alkoxycarbonylation rate and the TsOH∙H2O concentration was investigated in a toluene medium at PCO = 2.1 MPa with the following initial concentrations being used in all experiments (mol/L): c0 (C6H10) = 0.100; c0 (C6H11OH) = 0.400; c0 (Pd(PPh3)2Cl2) = 2.0×10–3; and c0 (PPh3) = 8.0×10–3. The reaction mixture samples were tested on a Tsvet 162 chromatograph equipped with a flame ionization detector and 3000×3 mm glass columns. The separation was carried out using a Chromaton N-AW-DMCS packing (0.125 to 0.160 mm particles) with a 5% XE-60 stationary phase at an argon carrier gas flow rate of 30 mL/min and an injector temperature of 250°C. The temperature was programmed to ramp up from 65 to 205°C at a rate of 12°C/min. Chromatographic peaks were identified by comparing the retention times of the components in the test samples with those in commercial cyclohexene and cyclohexanol samples and in a synthesized CHCHC sample. The CHCHC synthesis procedure is described in [19], and its NMR and IR spectra are provided in that article’s Supplementary Information. The concentrations both of the reactants and the products were measured with an accuracy of ±3%. The confidence intervals (CIs) of the kinetic parameters are provided in the body of that article. The test data on the steel reactor resistance in a TsOH∙H2O environment and the evaluation of the potential diffusion effects on the reaction rate are provided in the Supplementary Information of the present article.
RESULTS AND DISCUSSION
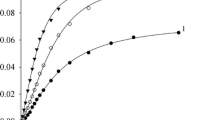
In order to investigate the effects of TsOH∙H2O concentration and process temperature on the rate of cyclohexene alkoxycarbonylation with cyclohexanol and CO, five series of experiments were carried out at 363–383 K, with the TsOH∙H2O concentration varying from 0 to 0.150 mol/L in each series. Like the kinetic curves previously plotted for an identical reaction [23], the CHCHC accumulation curves were roughly S-shaped, with a short induction period of 5–10 min. The initial alkoxycarbonylation rates were determined using the initial sections of the kinetic curves following the induction period. These initial sections, which included 4–6 data points (in accordance with the number of reaction mixture samples), were approximated by straight lines (with a correlation coefficient of at least 0.985). The line slopes were taken as the initial reaction rates. Figure 1 illustrates the initial reaction rates (indicated by the dots on the curves) at varying TsOH∙H2O concentrations and temperatures.

Rate of cyclohexene alkoxycarbonylation with cyclohexanol and CO as a function of TsOH∙H2O concentration at various temperatures (K): (1) 363; (2) 368; (3) 373; (4) 378; and (5) 383. Reaction conditions: PCO = 2.1 MPa; initial concentrations (mol/L): c0 (C6H10) = 0.100; c0 (C6H11OH) = 0.400; c0(Pd(PPh3)2Cl2) = 2.0×10–3; c0 (PPh3)=8.0×10–3.
In recent decades, it has been commonly assumed that the alkoxycarbonylation of unsaturated compounds over palladium–phosphine catalytic systems with strong protonic acids occurs by a hydride mechanism [5–8]. In our previous studies on the effects of different reactants, Pd(PPh3)2Cl2, and PPh3 on the rate of cyclohexene alkoxycarbonylation with cyclohexanol and CO, we suggested that this hydride mechanism additionally involves ligand exchange reactions that produce inactive (ballast) Pd complexes [23, 24]. In particular, the following reaction involving one TsOH molecule was formulated for the formation of a hydride complex (which is the key intermediate in the hydride mechanism for alkoxycarbonylation):
In more recent research we used a Pd(OAc)2–PPh3–TsOH∙H2O catalytic system with TsOH∙H2O concentrations up to 6.4×10–2 M, and the cyclohexene methoxycarbonylation rate was found to vary as a parabolic function of the TsOH∙H2O concentration and as an extremal function of the concentration of the added water [25]. This extremal dependence reflected an accelerating effect of minor amounts of water on methoxycarbonylation and a slowdown of this reaction at higher water concentrations in the reaction mixture. These patterns were explained in the context of an updated version of the hydride mechanism, according to which the formation of the hydride palladium complex involves one TsOH molecule and one H2O molecule. The experimental data were found to be consistent with a new kinetic equation derived for cyclohexene methoxycarbonylation [25]. In the present study, for cyclohexene alkoxycarbonylation with cyclohexanol and CO, we made a similar update to the hydride mechanism previously proposed in [23, 24] by adding ligand exchange reactions (17) and (18). These reactions produce ballast Pd complexes under the effect of water (Scheme 1, reactions (10)–(18), where Sol denotes the toluene solvent molecules).

Hydride mechanism for cyclohexene alkoxycarbonylation with cyclohexanol and CO [reactions (2)–(9)], with additional formation of ballast Pd complexes [reactions (10)–(18)].
Taking into account this modification as well as the negligible contribution of reactions (12) and (13) to the total balance of reactions in the system under study (as found in [24]), the previously proposed kinetic equation for cyclohexene alkoxycarbonylation with cyclohexanol and CO (Eq. (10) in [24]) takes the following form:
where r0 is the initial alkoxycarbonylation rate;
cPd is the total concentration of all Pd forms: cPd = [X0] + [X1] + ... + [X8] + [X11] + ... + [X14], matching the initial concentration of Pd(PPh3)2Cl2.
Given that TsOH∙H2O is the only water source in the system, Eq. (19) transforms into:
Under the single-factor experimental conditions with respect to TsOH∙H2O, Eq. (20) can be simplified:
where keff = kcPdc0(C6H10)c0(C6H11OH)[CO]; and A = 1 + dc02(C6H11OH) + e[CO]2 + hc02(PPh3) + ncPd.
Table 1 presents the parameters of Eq. (21) derived using the Sigma Plot 11.0 package.
The data of Table 1 clearly show that (l/A) is statistically negligible. In all probability, Eq. (21) does not optimally describe the experimental data obtained in the study, especially given that no strong correlations were observed in the variations of (keff/A) and (j/A) with a temperature increase. With (l/A) being statistically negligible, Eq. (21) was converted to Eq. (22), the parameters of which are presented in Table 2:
Using the parameters of Table 2, the r0 variation as a function of c0 (TsOH∙H2O) is graphically illustrated in Fig. 1. At all the tested temperatures, the experimental data (indicated by the dots) were well fitted by Eq. (22). Most probably, reaction (18) does not make a major contribution to the overall reaction balance in this system.
Using the values of the effective constants (keff/A) and (j/A) indicated in Table 2, the effective constants (j/keff) were derived. For these constants, the following equation holds true:
where HCO is Henry’s equilibrium constant for the CO–toluene system.
With a new parameter being further introduced:
we arrived at the following equation:
The values of (j′/k′eff) are also presented in Table 2. Special attention should be drawn to a significant rise in (keff/A) and (j/A) between 373 and 378 K. This rise can likely be attributed to overall errors (both upward and downward) in the calculation of individual (keff/A) and (j/A) constants within the tested range of 363–383 K. In fact, these parameters represent combinations of a variety of equilibrium constants applicable to different steps of the tested reaction. For a less sophisticated parameter such as (j′/k′eff), we see a smaller difference between the values for 373 and 378 K.
The (j′/k′eff) behavior fitted the Arrhenius function with a correlation coefficient of 0.986 (Eq. (26); Fig. 2):

Arrhenius function for effective constant (j′/k′eff).
In [24], an Arrhenius correlation was obtained for the effective constant (d/keff 4):
According to the findings of [24], the constant (d/keff 4) is described fairly well by the equation:
Within the framework of Eq. (27), the following holds true at 373 K:
A new parameter was further introduced:
Using the initial experimental conditions imposed in the previous study regarding the effect of the cyclohexanol concentration on the cyclohexene alkoxycarbonylation rate [24], this constant was calculated as follows:
The following was derived from Eq. (25) in combination with Eq. (29):
and therefore: ln (j′/k′eff) – ln (d′/k′eff 4) = ln K15 – ln K8.
Based on the effective activation energies found above, we arrived at:
and, consequently:
The differential between the two effective activation energies equals to:
which resulted in:
The following was reasonably derived from Eq. (31):
Using Eq. (26), (j′/k′eff) was evaluated at 373 K:
Based on Eq. (33), and using the effective constants evaluated above in (30) and (34), we obtained:
The values of ΔH = ΔH15 – ΔH8 and ΔS = ΔS15 – ΔS8 reflect the enthalpy and entropy changes during the ligand exchange between the two complexes:

At 373 K, the Gibbs free energy change for this reaction amounted to:
Thus, the ΔH value shows the weakly endothermic nature of reaction (36). We see that the binding energy between one cyclohexanol molecule and the Pd site in the complex X7 was about 4 kJ/mol higher than the binding energy of the water molecule in the complex X13. Therefore, under the conditions of reaction (1), X7 was more stable and less reactive than X13. This is consistent with the common assumption of more pronounced donor–acceptor properties of alcohols than those of water. The positive ΔS (see the above calculations) can likely be explained by the fact that two H2O molecules (small-sized) were bound and two cyclohexanol molecules (medium-sized) were released. On the other hand, the ΔG value indicates that, under the conditions of cyclohexene alkoxycarbonylation with cyclohexanol in the presence of TsOH∙H2O, reaction (36) is near equilibrium.
CONCLUSIONS
Within the temperature range of 363–383 K, the dependence between the rate of cyclohexene alkoxycarbonylation with cyclohexanol and CO and the concentration of TsOH∙H2O was found to be similar to an S-shaped curve. Based on the kinetic data obtained in this study, as well as our previous findings with regard to the effects of water and TsOH∙H2O concentrations on the cyclohexene methoxycarbonylation rate, the hydride mechanism for the alkoxycarbonylation process was updated by adding relevant ligand exchange reactions between ballast palladium complexes, specifically reactions that produce a palladium aqua complex. The accordingly-modified kinetic equation for cyclohexene alkoxycarbonylation with cyclohexanol and CO was found to be consistent with the experimental data. Effective constants were evaluated for the modified kinetic equation over the studied temperature range. The new data on the effects of the TsOH∙H2O concentration, in combination with the previous findings on the cyclohexanol concentration effects on the cyclohexene alkoxycarbonylation rate between 368 and 388 K, enabled us to further evaluate, in light of the activated complex theory, a number of relevant parameters: the effective activation energy and the changes in enthalpy, entropy, and Gibbs free energy during the ligand exchange between the complexes Pd(PPh3)2(C6H11OH)2 and Pd(PPh3)2(H2O)2. This reaction was found to be near equilibrium at 373 K.
REFERENCES
Kalck, Ph. and Urrutigoïty, M., Inorg. Chim. Acta , 2015, vol. 431, pp. 110–121. https://doi.org/10.1016/j.ica.2015.02.007
García-Suárez, E.J., Khokarale, S.G., van Buu, O.N., Fehrmann, R., and Riisager, A., Green Chem., 2014, vol. 16, no. 1, pp. 161–166. https://doi.org/10.1039/C3GC41380B
Nifant’ev, I.E., Sevostyanova, N.T., Batashev, S.A., Vinogradov, A.A., Vinogradov, A.A., Churakov, A.V., and Ivchenko, P.V., Appl. Catal. A: Gen., 2019, vol. 581, pp. 123–132. https://doi.org/10.1016/j.apcata.2019.05.030
Nobbs, J.D., Low, C.H., Stubbs, L.P., Wang, C., Drent, E., and van Meurs, M., Organometallics, 2017, vol. 36, no. 2, pp. 391–398. https://doi.org/10.1021/acs.organomet.6b00813
Liu, Y., Dong, K., Beller, M., and Mecking, S., ACS Catal., 2018, vol. 8, no. 10, pp. 9232–9237. https://doi.org/10.1021/acscatal.8b03117
Biermann, U., Bornscheuer, U.T., Feussner, I., Meier, M.A.R., and Metzger, J.O., Ang. Chem. Int. Ed., 2021, vol. 60, no. 37, pp. 20144–20165. https://doi.org/10.1002/anie.202100778
Nomura, K. and Awang, N.W.B., ACS Sust. Chem. Eng., 2021, vol. 9, no. 16, pp. 5486–5505. https://doi.org/10.1021/acssuschemeng.1c00493
Sevostyanova, N.T. and Batashev, S.A., Catal. Ind., 2023, vol. 15, no. 4, pp. 333–349. https://doi.org/10.1134/S2070050423040104
Lapidus, A.L. and Pirozhkov, S.D., Russ. Chem. Rev., 1989, vol. 58, no. 2, pp. 117–137. https://doi.org/10.1070/RC1989v058n02ABEH003430
Neubert, P., Steffen, M., and Behr, A., J. Mol. Catal. A: Chem., 2015, vol. 407, pp. 122–127. https://doi.org/10.1016/j.molcata.2015.06.019
Goldbach, V., Krumova, M., and Mecking, S., ACS Catal., 2018, vol. 8, no. 6, pp. 5515–5525. https://doi.org/10.1021/acscatal.8b00981
Mahboub, M.J.D., Dubois, J.-L., Cavani, F., Rostamizadeh, M., and Patience, G.S., Chem. Soc. Rev., 2018, vol. 47, no. 20, pp. 7703–7738. https://doi.org/10.1039/c8cs00117k
Gorbunov, D.N., Nenasheva, M.V., and Kardashev, S.V. Russ. J. Appl. Chem., 2019, vol. 92, no. 8, pp. 1069–1076. https://doi.org/10.1134/S1070427219080032
Song, H., Lei, S., Jin, F., and Liu, H., Mol. Catal., 2022, vol. 527, Art. 112408. https://doi.org/10.1016/j.mcat.2022.112408
Wu, L., Liu, Q., Jackstell, R., and Beller, M., Org. Chem. Front., 2015, vol. 2, no. 7, pp. 771–774. https://doi.org/10.1039/C5QO00071H
Sevostyanova, N.T. and Batashev, S.A., Russ. J. Phys. Chem. B, 2018, vol. 12, no. 3, pp. 593–594. https://doi.org/10.1134/S1990793118030296
An, J., Wang, Y., Lu, J., Zhang, J., Zhang, Z., Xu, S., Liu, X., Zhang, T., Gocyla, M., Heggen, M., Dunin-Borkowski, R.E., Fornasiero, P., and Wang, F., J. Am. Chem. Soc., 2018, vol. 140, no. 11, pp. 4172–4181. https://doi.org/10.1021/jacs.8b01742
Yang, J., Liu, J., Ge, Y., Huang, W., Ferretti, F., Neumann, H., Jiao, H., Franke, R., Jackstell, R., and Beller, M., Angew. Chem. Int. Ed., 2021, vol. 60, no. 17, pp. 9527–9533. https://doi.org/10.1002/anie.202015329
Sevostyanova, N.T., Batashev, S.A., Rodionova, A.S., and Kozlenko, D.K., Tetrahedron, 2023, vol. 146, Art. 133653. https://doi.org/10.1016/j.tet.2023.133653
Sevostyanova, N.T., Batashev, S.A., and Rodionova, A.S., Russ. Chem. Bull., 2023, vol. 72, no. 8, pp. 1936–1939. https://doi.org/10.1007/s11172-023-3980-1
Eliseev, O.I., Bondarenko, T.N., Stepin, N.N., and Lapidus, A.L., Mendeleev Commun., 2006, vol. 16, no. 2, pp. 107–109. https://doi.org/10.1070/MC2006v016n02ABEH002232
Dong, K., Sang, R., Liu, J., Razzaq, R., Franke, R., Jackstell, R., and Beller, M., Angew. Chem. Int. Ed., 2017, vol. 56, no. 22, pp. 6203–6207. https://doi.org/10.1002/anie.201701950
Aver’yanov, V.A., Sevost’yanova, N.T., Batashev, S.A., and Nesolenaya, S.V., Petrol. Chem., 2006, vol. 46, no. 6, pp. 405–414. https://doi.org/10.1134/S0965544106060053
Aver’yanov, V.A., Sevost’yanova, N.T., and Batashev, S.A., Petrol. Chem., 2008, vol. 48, no. 4, pp. 287–295. https://doi.org/10.1134/S0965544108040063
Averyanov, V.A., Sevostyanova, N.T., Batashev, S.A., Vorobiev, A.A., and Rodionova, A.S., Russ. J. Phys. Chem. B, 2014, vol. 8, no. 2, pp. 140–147. https://doi.org/10.1134/S1990793114020031
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct thisparticular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest requiring disclosure in this article.
Additional information
Publisher's Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sevostyanova, N.T., Batashev, S.A. Investigation of the Dependence of the Rate of Cyclohexene Alkoxycarbonylation with Cyclohexanol and CO on the p-Toluenesulfonic Acid Monohydrate Concentration and Temperature. Pet. Chem. (2024). https://doi.org/10.1134/S0965544124040042
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S0965544124040042