
Abstract
Two types of nanosilicas with different pore structures are synthesized and decorated via impregnation with cobalt and cerium oxides. Meso–microporous spherical silica particles with thin walls of SiO2 nanochannels having specific surface area and pore volume of up to 1400 m2/g and 0.8 cm3/g, respectively, are used. Macroporous three-dimensionally ordered structures based on SiO2 (so-called synthetic opals) consisting of close-packed submicron spherical silica particles with respective porosity characteristics of 11 m2/g and 0.2 cm3/g are also used. The synthesized materials are characterized via low-temperature nitrogen adsorption, X-ray diffraction, SEM, XPS, and Fourier transform IR spectroscopy, and tested as catalysts for the selective oxidation of CO in excess H2 (CO-PROX). The effect of silica, the ratio of introduced oxides, and the order of their introduction on the structure and catalytic properties of Co–Ce/SiO2 are revealed. The catalytic behavior of the synthesized materials is determined from the specificity of interactions among the metal oxides and with the silica surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Silicon dioxide (or silica) is commonly used in the composition of various materials, both in mineral form and as a synthesized product consisting of colloidal particles or a dry powder. The characteristics and quality of silica are strongly affected by the grain size and pore structure of SiO2. The problem of revealing and analyzing the relationship between the structure and properties of silica particles with a controlled size and a given porosity is of both scientific and practical importance [1, 2]. There are numerous applications of amorphous nanosilicas in the production of cosmetic, biomedical, pharmaceutical, polymeric, building, and other materials [3]. Porous silica particles are used in filters for purifying water of heavy metals and different organic compounds [4, 5], in biomedicine for diagnostics and drug delivery [6, 7], in biocatalysis [8, 9], and as adsorbents [10, 11]. Of particular interest is using them in heterogeneous catalysis as supports or templates for synthesizing catalysts [12–15]. The greatest advantage of using nanosilicas in catalysis is their highly developed surface, which contributes to the best dispersion of the active phase and its stabilization due to interaction with the silanol groups of the support. In addition to metal–support interaction, the chemical composition of the surface, the pore size, and the thickness of support walls are of fundamental importance [16]. Varying the conditions of synthesis allows us to control these parameters, making nanosilicas attractive for use in catalysis.
There are two main ways of synthesizing nanosilicas: sol–gel and deposition using tetraethoxysilane (TEOS), natural raw materials, or silicon-containing industrial waste as sources of silicon [17]. However, TEOS-based sol–gel synthesis remains the preferred way of synthesizing nanosilica, due to its simplicity and the high yield of the final product. The size and morphology of the synthesized particles are determined by the conditions of synthesis, so the resulting materials have different properties [18].
A way of improving the porosity of silica particles by making the walls thinner, increasing the number of mesopores, and forming an additional system of micropores was developed in [19]. The use of two organosilanes in the synthesis of meso–microporous silica (diethoxy(3-glycidyloxypropyl)methylsilane (DEGPMS) and methacryloxypropyltrimethoxysilane (MPTMOS)) ensured the formation of silica particles with specific surface area and pore volume of up to 1400 m2/g and 0.8 cm3/g, respectively.
Macroporous three-dimensionally ordered structures based on SiO2 (so-called synthetic opals) consisting of close-packed submicron spherical silica particles are also of great interest. Opal has two types of pores between close-packed SiO2 particles: octahedral and tetrahedral, with sizes of ~0.41D and ~0.23D (where D is the diameter of a spherical SiO2 particle), respectively [20]. Filling the opal pores with target substances ensures the synthesis of various nanocomposites with desired properties; varying the parameters of filling allows us to control the size and distribution of nanoparticles in the pores. Ways of synthesizing bulk synthetic opals and using them as matrices for stabilizing ensembles of metal oxide clusters were described in [20, 21].
Eurov et al. [22, 23] proposed that opals and nanosilicas decorated with cobalt oxide can be used as catalysts for the total (TOX) and preferential oxidation (PROX) of CO in an inert or hydrogen atmosphere. Against expectations, it was found that among silicas with different pore structures, the highest activity in catalysis was exhibited by Co3O4 aggregates occupying the gaps between the spheres of amorphous SiO2 in the structure of synthetic opal with the least developed specific surface area.
The oxidation of carbon monoxide was chosen as a model for determining the relationship between the structure and properties of CO-decorated silicas, since this reaction is of great fundamental importance when analyzing the nature of active sites and the main stages of heterogeneous catalytic processes [24, 25]. It is also important in terms of protecting the environment from harmful emissions. Interest has risen lately in designing selective catalysts for the oxidation of CO in the presence of hydrogen, which are required for purifying the hydrogen used in fuel cells of CO impurities [26]. Promising systems capable of competing with catalysts based on noble metals are metal oxides, where the joint use of cobalt and cerium oxides, and composites and supported catalysts based on them, is of considerable interest [27–32].
The aim of this work was to synthesize nanosilicas decorated with cobalt and cerium oxides that would have different pore structures, and reveal the relationship between their structure and catalytic behavior during CO-PROX in excess hydrogen. Two silicas with particle sizes of 500–800 nm were chosen as matrices: (a) meso-microporous silica with a specific surface area and pore volume of 1400 m2/g and 0.8 cm3/g, respectively, and (b) synthetic opal with a low specific surface area of around 10 m2/g and a pore volume of 0.2 cm3/g. The content of cobalt and cerium oxides, their ratio, and the order in which they were introduced were varied.
EXPERIMENTAL
The substances and reagents used in this work were cetyltrimethylammonium bromide C16H33N(CH3)3Br (CTAB), 99+% (Acros); aqueous ammonia NH3, 24 wt %, ≥99.99%; ethanol C2H5OH, 95 wt %; deionized water H2O, 10 MΩ; TEOS Si(OC2H5)4, 99+% (Acros); DEGPMS C11H24O4Si, 97% (Aldrich); MPTMOS H2C=C(CH3)CO2(CH2)3Si(OCH3)3, 98% (Aldrich); hydrochloric acid HCl, 37 wt %, ≥99.99%; cobalt nitrate hexahydrate Co(NO3)2·6H2O, 98% (Aldrich); and cerium acetate sesquihydrate Ce(CH3COO)3·1.5H2O, 99.9% (Aldrich).
SiO2 particles with diameters of 820 ± 30 nm were synthesized by hydrolysis of TEOS in an NH3–H2O–C2H5OH mixture [21]. The synthesized particles were calcined in air at 900°C and then redispersed in deionized water. Opals (Siop) were grown from an aqueous suspension of SiO2 particles via sedimentation. The resulting precipitate was dried at a temperature of 100°С and calcined at 800°С. The volume of pores accessible to filling was as high as 26% of the total volume of the opal, and the specific surface area (determined using the Brunauer–Emmett–Teller (BET) model) was 11 m2/g.
Meso-microporous spherical silica particles (Simm) with diameters of 550 ± 40 nm were synthesized according to a procedure developed in [19] for the basic hydrolysis of silicon-containing precursors (TEOS, DEGPMS, MPTMOS) in an NH3–H2O–C2H5OH–CTAB mixture. The molar ratio of reagents (TEOS + DEGPMS + MPTMOS) : NH3 : H2O : C2H5OH : CTAB was 1 : 60 : 370 : 230 : 0.2, respectively, and the weight of the solution was 1 kg. The temperature of the reaction mixture was 40°С, and the synthesis duration was 2 h. To remove organics, the synthesized particles were washed in an alcoholic solution of HCl (0.01 M) and then calcined for 5 h in a flow of O2 at a temperature of 400°C. The specific surface area and pore volume of the synthesized particles were 1400 m2/g and 0.8 cm3/g, respectively.
Cobalt oxide was introduced into the Siop and Simm silicas via capillary impregnation with a 2 M aqueous solution of Co(NO3)2·6H2O at room temperature. Each sample was dried at 70°C. The temperature was then raised to 120°C, and heat treatment was done under normal conditions for 8 h with the temperature being raised again to 400°C (at a rate of 10°C/min). Heat treatment at 400°C was carried out for 2 h.
Cerium and cobalt oxides were introduced in sequence or simultaneously. When cerium oxide was introduced first, a weighed portion of silica was impregnated with a 1 M aqueous solution of Ce(CH3COO)3·1.5H2O at room temperature and then dried at 70°C. The temperature was then raised to 120°C, and heat treatment was done for 8 h under normal conditions. At the next stage, a 2 M aqueous solution of Co(NO3)2·6H2O was introduced. The sample was then dried at 70°С and calcined for 8 h at 120°С under normal conditions. In the final stage, the temperature was raised to 400°C (at a rate of 10°C/min), and heat treatment was done for 2 h. The samples into which cobalt oxide was introduced first were prepared according to the same procedure with the oxides introduced in the opposite order. The compositions and denotations of the synthesized samples are given in Table 1. The contents of cobalt and cerium oxides were converted to the contents of metal.
Microphotographs of the surface of the samples were recorded via scanning electron microscopy (SEM) on a JSM 6000 NeoScope instrument (JEOL, Japan), with the samples on opal preliminarily ground.
The phase composition of the nanocomposites was determined via X-ray diffraction on a Bruker D2 Phaser system (CuKα radiation). Our interpretation of the diffraction patterns was based on data from the ICDD database (Release 2014, PDF2). The average sizes of the coherent scattering regions (CSRs) of individual phases were calculated by means of full profile analysis according to Rietveld, using the Bruker Topas 5 software package and the Scherrer formula
where Lvol is the average size of a CSR, λ is the wavelength, θ is the Bragg angle, and βIB is the integral width. The accuracy of calculation was as high as 1%.
The pore structure of the particles was studied on a Micromeritics ASAP 2020 analyzer at a temperature of 77 K using nitrogen as the adsorbate. Pore size was calculated using the embedded software in terms of the nonlocal density functional theory (NLDFT). Specific surface area was calculated according to Brunauer–Emmett–Teller (BET) method, and the specific pore volume was determined at relative pressure P/P0 = 0.994. In opal consisting of spheres with an average diameter of 820 nm, the sizes of pores between the particles were 100–300 nm [20]. These pores could not be detected by nitrogen adsorption porosimetry because capillary condensation in pores of the given size occurs at P/P0 ~ 0.99. The pore volume in opal was therefore determined gravimetrically, according to the difference between the apparent densities of an opal sample calcined at 500°С (with pores containing air) and a sample impregnated with water (assuming the pores were filled completely).
X-ray photoelectron (XP) spectra were recorded on an Axis Ultra DLD spectrometer (Kratos Analytical, United Kingdom) using a source of monochromatic AlKα radiation (hν = 1486.7 eV, 150 W). The pass energy of the analyzer was set at 160 and 40 eV. The energy scale of the spectrometer was calibrated against the bonding energy of the Au 4f5/2 (83.96 eV), Ag 3d5/2 (368.21 eV), and Cu 2p3/2 (932.62 eV) internal levels of pure metallic gold, silver, and copper. Samples in the form of powder were fixed on a holder using nonconductive double-sided adhesive tape. A charge neutralization system was used with the energy position of the spectra calibrated against the Si 2p line’s bonding energy of 103.6 eV, which is typical of silica. In view of the reduction of cerium on the surface Ce 3d of the samples under the action of radiation, the spectra were recorded at short and long periods of signal acquisition. The inelastic loss background and the decomposition of the spectra into components were calculated using the CasaXPS software (version 2.3.24) as described in [33] for cerium-containing systems. The bonding energies of the Ce3+ and Ce4+ components of the Ce 3d5/2 spectrum varied in the ranges of 880.9–881.2 and 882.7–882.9 eV, respectively. The component of the CeLLM line of the Auger spectrum, which overlapped with the spectrum of Co 2p, was also considered in decomposition. The Co 2p spectra were interpreted using data from [34–36]. The components with bonding energies in the ranges of 781.2–781.7 and 779.7–780.2 eV of the Co 2p3/2 spectrum were attributed to the Co2+ and Co3+ states, respectively.
Infrared spectra of the samples were recorded on an Infralum FT-801 Fourier transform spectrometer (Lyumeks-Sibir, Russia) in the range of 2000–450 cm−1 at a resolution of 2 cm−1. Our studies were performed using tablets prepared by mixing the samples with a KBr powder at different ratios of dilution.
Catalytic testing was done using a reaction mixture composed of 1 vol % CO, 1 vol % O2, 49 vol % H2, and 49 vol % He in a flow quartz reactor with heating–cooling cycles, as described in [23]. The cyclic tests were repeated many times. A mixture of a catalyst (250 mg) with quartz sand was used, with the samples being preheated for 1 h at 350°C in a flow of He. The composition of the gas mixture at the outlet of the reactor was analyzed using a Kristall 2000 chromatograph equipped with a thermal conductivity detector. The flow rate was 10 cm3/min. Under our conditions, the process resulted in the formation of CO2 and H2O exclusively, and no CH4 was observed. The catalytic properties of the samples were compared using the temperature dependences of CO conversion, and parameters whose values were determined from them: the maximum conversion of CO, the temperatures at which 50% and the maximum conversion of carbon monoxide (T50 and Tmax) were achieved.
RESULTS AND DISCUSSION
Morphology of Original and Decorated Silicas
Typical SEM images of samples on silica of two types (Siop and Simm) are shown in Fig. 1. The 2Ce/3Co/Siop image (Fig. 1a) clearly shows the (111) planes of the face-centred cubic structure of opal—i.e., the opal-like structure that formed during the slow sedimentation of silica particles was preserved after introducing the cobalt and cerium oxides. This structure did not have time to form during the drying of the synthesized meso–microporous particles, and there are no fragments of a regular structure in the 9Ce/15Co/Simm image (Fig. 1b).

Scanning electron microscopy images of (a) 2Ce/3Co/Siop and (b) 9Ce/15Co/Simm samples.
Figure 2a shows nitrogen adsorption isotherms at 77 K for both types of nanosilicas before and after decorating them with cobalt and cerium. It is clear that the isotherms for the meso–microporous particles (Fig. 2a, curves 1, 2) have a step-like form (type IV [37]) that can be attributed to the capillary condensation of nitrogen in the mesopores. The specific surface area and pore volume calculated for the original Simm particles were 1400 m2/g (in terms of the BET model) and 0.8 cm3/g, respectively.

(a) Nitrogen adsorption–desorption isotherms at 77 K for the original and decorated silicas: (1) Simm, (2) 15Co/Simm, (3) Siop, and (4) 2Ce/3Co/Siop. (b) Pore size distribution for (1) Simm and (2) 15Co/Simm.
The textural characteristics of the original and decorated silicas, along with structural data determined via X-ray diffraction, are presented in Table 2. The pore size distribution (Fig. 2b, curve 1) calculated via NLDFT has two pronounced maxima, at ~1.3 and ~2.6 nm. The specific surface area and pore volume of the particles fell considerably after decorating the particles with cobalt and cerium, and the calculated values were 970 m2/g and 0.5 cm3/g. It would seem the introduced oxides not only occupied a certain number of pores inside the particles but could also block access to other pores and make them inaccessible to the adsorbate. The cobalt and cerium oxides also increased the apparent density of the particles (by ~30%), reducing the specific surface area and pore volume. A shift of the two maxima was also observed that was attributed to a reduction in both micro- and mesopores (Fig. 2b, curve 2). The reduction in the pore size of particles after decorating their inner surfaces with Co and Ce could be due to several factors. One is the chemisorption of Co2+ and Ce3+ ions from solutions by active silanol groups on the silica surface during the capillary impregnation of the particles [38]. Another is cobalt and cerium oxide crystallites being formed on the inner surface of particle pores after the decomposition of the respective salts. Yet another is the pore diameter being reduced during heat treatment, due to the low temperature stability of meso–microporous particles [19, 39].
The isotherms of the opals (Fig. 2a, curves 3, 4) are of type III [37], which is characteristic of nonporous materials. As noted above, however, opal contains large macropores (100–300 nm) that cannot be detected via low-temperature nitrogen adsorption. The specific surface area of Siop, calculated according to BET before and after decoration with cobalt and cerium, were 11 and 8 m2/g, respectively. The pore volume calculated from the apparent densities (determined gravimetrically) of a dry sample and a sample impregnated with water was almost identical for the original and filled opals (~0.2 cm3/g).
The structure and phase composition of nanosilicas containing cobalt and cerium oxides were studied via X-ray powder diffraction. The diffraction patterns of the samples (Fig. 3) displayed a set of reflections corresponding to Co3O4 and CeO2. No impurity crystalline phases were detected. The patterns also exhibited broad haloes with maxima in the region of 2θ ~ 22°, which were attributed to diffuse reflection from amorphous SiO2 (the template material). The diffraction peaks of Co3O4 and CeO2 were broadened, suggesting that the oxides in the nanosilica pores were nanocrystalline. The degree of crystallinity of the fillers in the meso–microporous particles, was also lower than in the opal. The average sizes of CSRs in the opal and silica particles were 2.4 ± 0.1 and 1.5 ± 0.1 nm, respectively, for CeO2. They were 31.8 ± 0.7 and 5.2 ± 0.1 nm, respectively, for Co3O4 (Table 2). Note that the sizes of the CSRs for cerium oxide were approximately 2 nm in two of our samples (2Ce/3Co/Siop and 9Ce/15Co/Simm), and the degree of cobalt oxide dispersion correlated with the pore sizes of the silica. The Co3O4 particles were much smaller in the meso-microporous sample than in the macroporous opal.

Diffraction patterns of (1) 2Ce/3Co/Siop, (2) 9Ce/15Co/Simm, (3) CeO2 (PDF2 01-075-9470), and (4) Co3O4 (PDF2 00-042-1467).
Electronic State of Metals According to X-ray Photoelectron Spectroscopy (XPS)
Our XP spectra of the samples exhibited lines of cobalt, cerium, oxygen, silicon, and carbon. Typical examples of the high-resolution Co 2p and Ce 3d spectra and their decomposition into components are shown in Fig. 4.

(a) Co 2p and (b) Ce 3d XPS spectra of the 2Ce/3Co/Siop and 9Ce/15Co/Simm samples. The Ce 3d spectra were recorded at short periods of signal acquisition, and E is the energy of bonding.
The atomic fractions of Co and Ce in different electronic states and the ratios of elements on the surface of the samples, calculated from the recorded spectra, are summarized in Table 3. It is clear that all the composites contained cobalt in the Сo3+ and Co2+ states, and the fraction of the higher state of oxidation predominated in almost all the samples, as is characteristic of Co3O4 oxides. Introducing a small amount of cerium (Co/Ce = 3) on an opal surface raised its fraction of Co3+, while a large increase in the amount of cerium in the 3Co/14Ce/Siop composite was accompanied by a drop in the fraction of Co3+. Cerium was in this case mostly in the form of CeO2 and, unlike the 2Ce/3Co/Siop and 3Co–2Ce/Siop composites with low contents of Ce, varied only slightly under the conditions of recording the spectra.
The state of the metals in the 9Ce/15Co/Simm bimetallic sample on meso–microporous silica differed from that in the other composites. The fraction of cobalt in the lower state of oxidation (Co2+) was in this case higher than in the sample’s monometallic 15Co/Simm counterpart. It is also clear from Table 3 that the ability of cerium in this sample to be reduced under the action of X-ray radiation was enhanced, relative to that in the composites on opal. This resulted in a larger increase in the fraction of Ce3+.
The observed differences are likely attributable to the structural features of the silicas and the different patterns of interaction between their surface groups and cobalt and cerium oxides. The penetration of oxides into the porous structure of silicas can be assessed by comparing quantitative data on the composition of elements on the surfaces of the modified supports and in their bulk, while remembering that XPS allows us to analyze only the surface layer of each sample (about 5 nm). The data in Table 3 suggest that the relative amounts of Co and Ce on the surface were lowest in the 15Co/Simm and 9Ce/15Co/Simm samples on meso–microporous silica, despite their high content in bulk. This finding is consistent with a drop in the pore volumes (Table 2) of the Simm samples after decoration and indicates stronger interaction between the introduced components and the surfaces of meso–microporous silica.
On opal, the fraction of cobalt on the surface of the bimetallic systems depended on the order in which the components were introduced. When cobalt was introduced second or along with cerium, the amounts of Co on the surfaces were higher than on their 3Co/Siop monometallic counterpart (the Co/Si ratio rose from 0.09 to 0.15–0.21 when cerium was used). At the same time, the fraction of Co on the surfaces fell considerably when the order of introducing the components was reversed. The Co/Ce atomic ratio on the 2Ce/3Co/Siop surface was almost 6 times less in comparison with the 3Co–2Ce/Siop sample of the same composition (0.26 and 1.6, respectively). The Co/Ce ratios on the surfaces of all composites were lower than volume ratios of 3 : 1 and 4 : 1. The closest values of ~2 were obtained by introducing two components into opal Siop simultaneously, and on the 9Ce/15Co/Simm sample when introducing the components into Simm in sequence.
Infrared Spectroscopy of the Prepared Samples
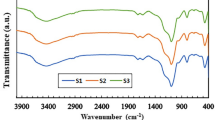
The interaction among the introduced cobalt and cerium oxides and the silica surfaces was studied via transmission Fourier transform IR spectroscopy. Figure 5 compares spectra of the original and decorated silicas in the region of intense bands of vibrations of the supports, recorded at a high ratio of KBr dilution. The absorption bands in the region of 1100–1200 and 805–807 cm−1 were attributed to the asymmetric and symmetric stretching vibrations of Si–O–Si bonds, while the bands at 940 and 476 cm−1 correspond to the bending vibrations of Si–OH and Si–O–Si bonds [40]. It is clear from the figure that the introduction of metal oxides had almost no effect on the position of the bands of Si–O–Si vibrations in macroporous opal Siop, while the bands of the asymmetric stretching vibrations shifted to the region of high frequencies in the meso–microporous Simm supports (from 1086 cm−1 in the original Simm to 1092 and 1096 cm−1 in 3Co/2Ce/Simm and 3Co/Simm, respectively). At the same time, the band of symmetric vibrations shifted toward lower frequencies. The frequency of the Si‒O–Si bending vibrations at 476 cm−1 also grew to 484 or 488 cm−1 upon introducing cobalt oxide or two different oxides into 3Co/2Ce/Simm. The observed spectral changes suggest that the surface groups of silica Simm interact with the modifying additives of cobalt and cerium cations. Note that the effect the interaction between cerium and silicas had on the spectra of the silicon–oxygen supports was greater than the one caused by introducing cobalt.

Infrared spectra of original and Co/Ce-decorated silicas of different compositions.
Figure 6 shows the IR spectra recorded for more concentrated mixtures of samples with KBr. The emergence of two distinct bands at 565–568 (ν1) and 661–662 cm−1 (ν2) attributed to the stretching vibrations of metal–oxygen bonds confirms the formation of a Co3O4 oxide spinel on the surfaces of the modified silicas [23, 41, 42]. The ν1 band can be attributed to the vibrations of Co3+ in the octahedral position on the spinel lattice, while the ν2 band corresponds to the vibrations of Co2+ in the tetrahedral holes [41, 42]. It is clear from Fig. 6a that the bands of vibrations of Co3O4 in the spectra of the bimetallic composites on opal shifted slightly toward higher frequencies, relative to the bands of Co/Siop. The largest shifts (from 566 to 568 cm−1), which were accompanied by band broadening, were observed for the ν1 band of the Co3+–O vibrations in the composites with large amounts of cerium oxide (3Co/14Ce/Siop) or in those synthesized via the co-introduction two oxides (3Co–2Ce/Siop). Similar changes in the spectra of cobalt oxide that testify to interaction between the oxides of the two metals were observed earlier for Co3O4–СеО2 systems [43].

Infrared spectra of Co/Ce-modified (a) Siop and (b) Simm silicas of different compositions.
It is clear from Fig. 6b that the spectra of Co- and Co/Ce-modified silicas Simm containing 3% Co, unlike those of the samples of the same composition on Siop (Fig. 6a), did not exhibit the ν1 and ν2 absorption bands characteristic of the stretching vibrations of oxide Co3O4. Only when the amount of cobalt was raised by 5 times did the spectra of the 15Co/Simm and 9Ce/15Co/Simm composites begin to exhibit bands of cobalt oxide spinel at 662 and 565 cm−1, the positions of which depended only slightly on the presence of cerium. However, cerium had a strong effect on the position of the band of the Si–O–Si bending vibrations of the original Simm support, as was seen with high ratios of dilution at 476 cm−1 (Fig. 5). At lower ratios of dilution, it took the form of a broad peak at 489 cm−1 (Fig. 6b). The frequency of this vibration grew for all samples of the modified silicas. With the composites containing cerium, the frequency rose to 502 and 508 cm−1 for 9Ce/15Co/Simm and 3Co/2Ce/Simm, respectively (Fig. 6b). The observed changes in the spectra of the original support and the lack of cobalt oxide bands in the spectra of the modified 3Co/Simm and 3Co/2Ce/Simm silicas suggest that cobalt and cerium cations interacted strongly with the surface hydroxyl groups of Simm. This agrees with our XPS data (Table 3), which show that the content of cobalt and cerium on surfaces was much lower than in bulk, even in the samples containing 15% Co.
Results from our physicochemical studies of the samples suggests that the distribution of cobalt and cerium oxides, the degree of dispersion of the particles, and the electronic state of the metals were largely determined by the morphology of SiO2 particles and the pattern of the interaction among the introduced oxides and the silica surface. These differences in the structure of the samples affect their behavior in the CO-PROX process.
Catalysis of the Selective Oxidation of CO
Typical temperature dependences of CO conversion are shown in Fig. 7, using the example of 2Ce/3Co/Siop and 3Co/14Ce/Siop samples and data obtained in [23] for a monometallic 3Co/Siop sample. These dependences have the shape typical of CO conversion in excess hydrogen. Conversion began to fall upon reaching a certain temperature (Tmax), due to competition with H2 oxidation—an undesirable process [23, 32, 44]. The curves recorded in the heating and cooling cycles virtually coincide. The greatest conversion of CO was nearly 100% with the most active 2Ce/3Co/Siop bimetallic sample. It fell only slightly when the temperature was raised, in contrast to the 3Co/Siop monometallic composite, for which the conversion of CO was less than 90% at 170°C and then fell abruptly. It is known that introducing cerium oxide enhances the catalytic ability of cobalt oxide, even though cerium oxide is inactive in the oxidation of CO [28–32]. Introducing a larger amount of cerium (Co/Ce = 0.5) reduced the activity of the 3Co/14Ce/Siop catalyst at low temperatures, compared even to that of 3Co/Siop. The activity of this sample grew when the temperature was raised. The maximum conversion of CO reached the values obtained with the monometallic sample, and remained virtually the same when the temperature was raised to 190°C.

Temperature dependences of CO conversion (αCO) in the CO-PROX process with Co/Ce/Siop. The solid lines and colored symbols show heating cycles; the dashed lines and partially colored symbols represent cooling cycles.
The catalytic properties of all studied samples are compared in Table 4. It is clear from a comparison of the data for the samples with the optimum composition on opal, which were synthesized with the sequential (2Ce/3Co/Siop) and simultaneous introduction of the components (3Co–2Ce/Siop), that the order of introduction affected the catalytic properties only slightly. The choice of silica had a strong effect. Both mono- and bimetallic samples on Simm with a Co content of 3% exhibited weak activity in the oxidation of CO (T50 > 200°C). Hydrogen was largely oxidized at high temperatures, therefore, it is not possible to achieve high conversion of CO with these catalysts. Raising the content of Co in the 15Co/Simm and 9Ce/15Co/Simm samples improved their catalytic properties somewhat, but even those samples in which the amount of cobalt was quintupled were inferior to the decorated opals in activity. It is also clear from Table 4 that introducing cerium oxide worsened the catalytic ability of cobalt oxide slightly in the case of Simm, rather than improving it.
These differences can be attributed to different patterns of distribution of the introduced oxides in the structure of the Siop and Simm silicas. In the structure of opal consisting of densely packed particles of nonporous silica, fairly large Co3O4 particles form in the gaps between the spheres. These particles are less prone to interacting with the silanol groups of silica and more readily allow the electronic transitions in Co3+/Co2+ pairs that are required for catalysis [23]. X‑ray diffraction showed that regardless of the silica structure, cerium oxide particles have similar sizes of around 2 nm (Table 2), but their distributions in Siop and Simm are different. In opal, they most likely form on the surfaces of large cobalt oxide particles in the gaps between nonporous SiO2 spheres. This provides the best conditions for the synergistic effect of Co and Ce in catalysis. The reasons for it are associated with the formation of new Co–O–Ce active sites, the emergence of a supplementary source of active oxygen, and an increase in the stability of cobalt oxide with regard to reduction and poisoning by oxidation products such as CO2 and H2O upon interacting with cerium oxide [28–32, 44].
The considerable surface area and developed thin-walled porous structure of Simm (Table 2) result in a large number of reactive silanol groups on its surface, with which the introduced precursors primarily interact [45]. IR spectroscopy showed that cobalt oxides did not form in samples with low contents of cobalt (3%), which displayed weak activity in catalysis.
Raising the content of cobalt to 15% resulted in finely dispersed Co3O4 particles around 5 nm in size, which is comparable to the size of pores in Simm. These particles were most likely localized in the silica pores, as was indicated by the low surface concentration of cobalt (Table 3). When introduced in addition to cobalt oxide, cerium oxide (which is prone to interacting with silica) probably occupies free regions of the surface or partially blocks the silica pores in which cobalt-containing particles active in catalysis are localized. This reduced the catalytic activity of the bimetallic sample somewhat, relative to that of its cobalt-containing counterpart (Table 4). XPS also showed that much of the cobalt in this sample was kept in the less active Co2+ state, as was the case in the original cobalt nitrate (Table 3). This also indicates there was the strong interaction with surface silanol groups, which is unfavorable for catalysis and is likely to result in the formation of cobalt phyllosilicates [46].
CONCLUSIONS
The best conditions for the joint action of Co and Ce were ensured by introducing them into large pores in the gaps between the spheres in the opal structure. Cerium oxide particles formed mostly on the surface of large cobalt oxide particles, due to small area and lack of a system of tiny pores in the opal. This resulted in the formation of new Co–O–Ce active sites in which the electronic transitions in Co3+/Co2+ and Ce3+/Ce4+ pairs that are responsible for catalytic activity readily occured. In terms of activity, these samples are comparable or even superior to catalysts based on bulk cobalt and cerium oxides. With 10% Сo3O4/CeO2, a CO conversion of 94% was achieved at 175°С [29]. The oxidation of CO predominated at temperatures up to 175°C when the order of depositing 10% CeO2 on a Co3O4 surface was reversed. The oxidation of H2 then began and the conversion of CO was reduced [44]. Under similar conditions, supported catalysts based on zeolites modified with Co and Ce ensured 95% conversion of CO at 190°C [32]. Their activity was associated with the formation of new catalytic sites in the zeolite pores. Meso–microporous Simm silicas decorated with cobalt and cerium oxides displayed weak activity, since strong interaction between the active components in these samples and the support hindered their joint action in catalysis. A similar effect of weakening the joint action of Cu and Ce in the CO-PROX process upon raising the specific surface area of SiO2 from 20 to 650 m2/g was observed in [47]. A comparison of these data and the results from this study suggests that the low specific surface of silica contributed to better contact between the active components and raised the efficiency of their joint action as catalysts. In this context, synthetic opal could be a promising support in creating bimetallic catalysts for various processes. The regular structure of this material and the system of wide pores in the gaps between the silica spheres, which ensure the formation and stabilization of the active phase, favorable conditions for the diffusion of the reactants and reaction products, and chemical inertness and high thermal stability, allow us to consider synthetic opal as a nanoreactor for the implementation of catalytic processes.
REFERENCES
J. Farrando-Perez, C. Lopez, J. Silvestre-Albero, and F. Gallego-Gómez, J. Phys. Chem. 122, 22008 (2018). https://doi.org/10.1021/acs.jpcc.8b07278
K. P. Meletov, V. S. Efimchenko, M. A. Korotkova, et al., Int. J. Hydrogen Energy (2023). https://doi.org/10.1016/j.ijhydene.2022.12.297
Deepanjali K. Gautam, and A. V. Ullas, Mater. Today: Proc. 74, 713 (2023). https://doi.org/10.1016/j.matpr.2022.10.281
A. Walcarius and L. Mercier, J. Mater. Chem. 20, 4478 (2010). https://doi.org/10.1039/B924316J
T. Qiang and R. Zhu, Sci. Total Environ. 819, 152929 (2022). https://doi.org/10.1016/j.scitotenv.2022.152929
Q. He and J. Shi, Adv. Mater. 26, 91 (2014). https://doi.org/10.1002/adma.201303123
M. Gisbert-Garzarán, D. Lozano, K. Matsumoto, et al., ACS Appl. Mater. Interfaces 13, 9656 (2021).
S. Majeed, R. Nawaz, T. Rasheed, and M. Bilal, in Nanomaterials for Biocatalysis: Micro and Nano Technologies (Elsevier, Amsterdam, 2022), Chap. 6, p. 171.
S. Muñoz-Pina, P. Amorós, J. El Haskouri, et al., Nanomaterials 10, 1927 (2020). https://doi.org/10.3390/nano10101927
A. Kumar, D. G. Madden, M. Lusi, et al., Angew. Chem. Int. Ed. Engl. 54, 14372 (2015). https://doi.org/10.1002/anie.201506952
D. Zhao, Y. Wan, and W. Zhou, Ordered Mesoporous Materials (Wiley, New York, 2013), p. 523.
R. Huirache-Acuña, R. Nava, C. L. Peza-Ledesma, et al., Materials 6, 4139 (2013). https://doi.org/10.3390/ma6094139
P. Verma, Y. Kuwahara, K. Mori, et al., Nanoscale 12, 11333 (2020). https://doi.org/10.1039/D0NR00732C
P. C. Ribeiro, R. H. G. A. Kiminami, and A. C. F. M. Cos-ta, Ceramics 40, 2035 (2014). https://doi.org/10.1016/j.ceramint.2013.07.115
Z. Xantini and E. Erasmus, Polyhedron 193, 114769 (2021). https://doi.org/10.1016/j.poly.2020.114769
C. C. Chong, Y. W. Cheng, M. B. Bahari, et al., Int. J. Hydrogen Energy 46, 24687 (2021). https://doi.org/10.1016/j.ijhydene.2020.01.086
Y. Liu, Z. Wang, W. Zhao, et al., Chem. Eng. J. 455, 140622 (2023). https://doi.org/10.1016/j.cej.2022.140622
X. Jiang, X. Tang, L. Tang, et al., Ceramics 45, 7673 (2019). https://doi.org/10.1016/j.ceramint.2019.01.067
D. A. Eurov, D. A. Kirilenko, M. V. Tomkovich, M. A. Yagovkina, and D. A. Kurdyukov, Inorg. Mater. 58, 1355 (2022). https://doi.org/10.1134/S002016852212007X
D. A. Kurdyukov, A. B. Pevtsov, A. N. Smirnov, M. A. Yagovkina, V. Yu. Grigorev, V. V. Romanov, N. T. Bagraev, and V. G. Golubev, Phys. Solid State 58, 1216 (2016). https://doi.org/10.1134/S1063783416060275
E. Yu. Trofimova, A. E. Aleksenskii, S. A. Grudinkin, I. V. Korkin, D. A. Kurdyukov, and V. G. Golubev, Colloid. J. 73, 546 (2011). https://doi.org/10.1134/S1061933X11040156
D. A. Eurov, D. A. Kirilenko, M. V. Tomkovich, T. N. Rostovshchikova, M. I. Shilina, O. V. Udalova, and D. A. Kurdyukov, Inorg. Mater. 57, 906 (2021). https://doi.org/10.1134/S0020168521090053
D. A. Eurov, T. N. Rostovshchikova, M. I. Shilina, et al., Appl. Surf. Sci. 579, 152121 (2022). https://doi.org/10.1016/j.apsusc.2021.152121
H.-J. Freund, G. Meijer, M. Scheffler, et al., Angew. Chem. Int. Ed. 50, 10064 (2011). https://doi.org/10.1002/anie.201101378
S. Royer and D. Duprez, ChemCatChem 3, 24 (2011). https://doi.org/10.1002/cctc.201000378
P. Jing, X. Gong, B. Liu, and J. Zhang, Catal. Sci. Technol., No. 10, 919 (2020). https://doi.org/10.1039/C9CY02073J
F. Marino, C. Descorme, and D. Duprez, Appl. Catal. B 58, 175 (2005). https://doi.org/10.1016/j.apcatb.2004.12.008
A. Arango-Diaz, J. A. Cecilia, J. Marrero-Jerez, et al., Ceram. 46, 7462 (2016). https://doi.org/10.1016/j.ceramint.2016.01.151
P. Gawade, B. Bayram, A.-M. C. Alexander, and U. S. Ozkan, Appl. Catal. B 128, 21 (2012). https://doi.org/10.1016/j.apcatb.2012.06.032
L. Zhang, L. Zhang, G. Xu, et al., New J. Chem. 41, 13418 (2017). https://doi.org/10.1039/c7nj02542d
M. Shilina, O. Udalova, I. Krotova, et al., ChemCatChem 12, 2556 (2020). https://doi.org/10.1002/cctc.201902063
I. A. Ivanin, I. N. Krotova, O. V. Udalova, et al., Kinet. Catal. 62, 798 (2021). https://doi.org/10.1134/S0023158421060082
I. Y. Kaplin, E. S. Lokteva, K. I. Maslakov, et al., Appl. Surf. Sci. 594, 153473 (2022). https://doi.org/10.1016/j.apsusc.2022.153473
Yu. A. Teterin, A. V. Sobolev, A. A. Belik, Ya. S. Glazkova, K. I. Maslakov, V. G. Yarzhemskii, A. Yu. Teterin, K. E. Ivanov, and I. A. Presnyakov, J. Exp. Theor. Phys. 128, 899 (2019). https://doi.org/10.1134/S1063776119050066
A. Medvedeva, E. Makhonina, L. Pechen, et al., Materials 15, 8225 (2022). https://doi.org/10.3390/ma15228225
L. Qiao, H. Y. Xiao, H. M. Meyer, et al., J. Mater. Chem. C 1, 4628 (2013). https://doi.org/10.1039/C3TC30861H
S. Gregg and K. Sing, Adsorption, Surface Area, and Porosity (Academic, London, 1982).
D. A. Kurdyukov, E. N. Chernova, Y. V. Russkikh, et al., J. Chromatogr. A 1513, 140 (2017). https://doi.org/10.1016/j.chroma.2017.07.043
D. A. Kurdyukov, D. A. Eurov, D. A. Kirilenko, et al., Microporous Mesoporous Mater. 223, 225 (2016).https://doi.org/10.1016/j.micromeso.2015.11.018
A. V. Kiselev and V. I. Lygin, Infrared Spectra of Surface Compounds and Adsorbed Molecules (Nauka, Moscow, 1972), p. 459 [in Russian].
Y.-Z. Wang, Y.-X. Zhao, C.-G. Gao, and D.-S. Liu, Catal. Lett. 116, 136 (2007). https://doi.org/10.1007/s10562-007-9099-4
C.-W. Tang, C.-B. Wang, and S.-H. Chien, Termochim. Acta 473, 68 (2008). https://doi.org/10.1016/j.tca.2008.04.015
X.-D. Hou, Y.-Z. Wang, and Y.-X. Zhao, Catal. Lett. 123, 321 (2008). https://doi.org/10.1007/s10562-008-9426-4
L. Lukashuk, K. Föttinger, E. Kolar, et al., J. Catal. 344, 1 (2016). https://doi.org/10.1016/j.jcat.2016.09.002
Chemistry of Graft Surface Compounds, Ed. by G. V. Lisichkin (Fizmatlit, Moscow, 2003), p. 49 [in Russian].
I. Puskas, T. H. Fleisch, J. B. Hall, et al., J. Catal. 134, 615 (1992). https://doi.org/10.1016/0021-9517(92)90347-K
I. S. Tiscornia, A. M. Lacoste, L. E. Gomez, et al., Int. J. Hydrogen Energy 45, 6636 (2020). https://doi.org/10.1016/j.ijhydene.2019.12.126
Funding
Synthesis and structural analysis of the materials were financed within the framework of the state budget agreement (Grant no. 0040-2019-0012). Spectral studies and catalytic tests were performed as part of a State Task for Lomonosov Moscow State University, topic no. АААА-А21-121011590090-7, and the Semenov Federal Research Center of Chemical Physics, topic no. 122040500058-1 “Physics and Chemistry of New Nanostructured Systems and Composite Materials with Desired Properties.” SEM and XPS data were obtained using equipment purchased under the Program for the Development of Lomonosov Moscow State University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
This work did not include any experiments performed by the authors with human participants or animals.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
Additional information
Translated by M. Timoshinina
Rights and permissions
About this article

Cite this article
Rostovshchikova, T.N., Eurov, D.A., Kurdyukov, D.A. et al. Effect of the Pore Structure of Nanosilicas Decorated with Cobalt and Cerium Oxides on Catalytic Activity in the Selective Oxidation of Carbon Monoxide. Russ. J. Phys. Chem. 97, 1978–1989 (2023). https://doi.org/10.1134/S0036024423090212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024423090212