
Abstract
Thermodynamic models of phases in the Ag–Sb–Sn system are constructed on the basis of the available experimental information. Polythermal sections in the phase diagram of this system are calculated for compositions xAg/xSb = 1, xAg/xSn = 1, xSb/xSn = 1, and xSn = 0.5, along with an isothermal section at 473 K. The coordinates of the invariant points of this system and the projection of its liquidus surface are determined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Alloys belonging to the Ag–Sb–Sn system are promising materials for use as high-temperature lead-free solders [1, 2] and anode materials for lithium-ion batteries [3]. The creation and operation of such materials requires good knowledge of phase equilibria in the Ag–Sb–Sn system. However, experimental studies of this system have revealed uncertainty associated with the most low-temperature invariant equilibrium in the Ag–Sb–Sn system. Three possible variants of this equilibrium have been proposed in the literature: L + (SbSn) ↔ ε + (Sn) at 508 K [4] (information on the solid phases in the Ag–Sb–Sn system is given in Table 1), L + (Sb2Sn3) ↔ ε + (Sn) at 502 K [2], and L + Sb3Sn4 ↔ ε + (Sn) at 506 K [5] (in this work, the Sb3Sn4 phase is denoted as Sb2Sn3, though the composition found by the authors for this phase (57 at % of Sn [6]) corresponds to the Sb3Sn4 phase (57.14 at % of Sn) instead of the Sb2Sn3 phase (60 at % of Sn)).
To solve this problem, we must thermodynamically model the Ag–Sb–Sn system with allowance for the existence of both Sb2Sn3 and Sb3Sn4 phases [7]. Earlier thermodynamic calculations of phase equilibria in the Ag–Sb–Sn system considered either the high-temperature Sb2Sn3 phase [8, 9] or the low-temperature Sb3Sn4 phase [10]. In addition, the liquid models used in these calculations were characterized by considerable deviation from the recent data on the integral enthalpy of mixing of ternary liquid alloys [2] (459 experimental points; the error of measuring is estimated by the authors as 150 J/mol). The numbers of points whose deviation from the data [2] is more than 300 J/mol for the liquid models used in [8–10] are 71, 52, and 58, respectively.
It is also noteworthy that some results of [8] are erroneous. When modeling the Ag–Sb–Sn system, the authors of [8] used thermodynamic models of phases in the Sb–Sn system taken from [11] to determine that the lowest temperature stable invariant equilibrium exists in this system at 505 K and can be described by the reaction L + Sb2Sn3 ↔ ε + (Sn). This means the Sb2Sn3 phase metastable (with respect to (SbSn) and (Sn)) below 515 K in the binary Sb–Sn system [11] becomes stable at 505 K in the ternary Ag–Sb–Sn system. This can happen only when Ag is dissolvable in the Sb2Sn3 phase, but such solubility was taken to be zero in [8]. Hence, the unexpected stability of the Sb2Sn3 phase at 505 K [8] is apparently explained by artificial metastability that can emerge in the (SbSn) phase due to flaws in the used software. As a result, all of the computational results in [8] for equilibria with participation of the (SbSn) phase cannot be reproduced by minimizing the Gibbs energy of this system. Possible errors in the thermodynamic description of the (SbSn) phase were discussed in [7].
Another shortcoming of [8] is that a simplified model of the ε ((Ag)0.75(Sb,Sn)0.25) phase was used to calculate invariant equilibria in the Ag–Sb–Sn system (according to Table 3 in [8] \(x_{{{\text{Ag}}}}^{\varepsilon }\) = 0.75 in all the calculated equilibria). The composition of the ε phase in the binary Ag–Sb system varies in the range \(x_{{{\text{Ag}}}}^{\varepsilon }\) = 0.728–0.785 [12], so approximation \(x_{{{\text{Ag}}}}^{\varepsilon }\) ≡ 0.75 [8] contradicts these data and prevents correct calculation of the composition of the ε phase in equilibria.
In [9], a simplified ε phase model [8] was also used to calculate invariant equilibria in the Ag–Sb–Sn system (the calculation results given in Table 4 in [9] are reproduced only if approximation \(x_{{{\text{Ag}}}}^{\varepsilon }\) ≡ 0.75 is used). In addition, the temperature calculated in [9] for invariant equilibrium L + (Sb) ↔ ε + (SbSn) (661.2 K) differs appreciably from the corresponding experimental values (648 [2], 649.5 [13], 651.5 [4], and 652 K [5]).
In [10], the composition of the Sb3Sn4 phase was assumed to be 58 at % of Sn. This value deviates from both the stoichiometric composition of this phase (57.14 at % of Sn) and the value of 57 at % of Sn, which was used for the composition of this phase in other works by the same authors [6, 14, 15].
Our modeling of the Ag–Sb–Sn system is based on recent thermodynamic data for ternary liquid alloys and the modified Sb–Sn phase diagram in [7], which considers the existence of both Sb2Sn3 and Sb3Sn4. Different sections of the Ag–Sb–Sn phase diagram and the liquidus surface projection were calculated after thermodynamic models of the ternary liquid phase and solid (Ag), ζ, ε, and (Sb2Sn3) solution were constructed.
Ag–Sb, Ag–Sn, AND Sb–Sn SYSTEMS
The Ag–Sb system, a binary subsystem of the Ag–Sb–Sn system, contains solid solutions based on pure components and intermediate ζ and ε solid solutions in addition to a liquid phase. Information about the solid phases of this system is given in Table 1. The available experimental data on the thermodynamic properties of phases and the phase equilibria in the Ag–Sb system were reviewed in [8, 16]. This system was modeled thermodynamically in [8, 10, 12, 16]. In this work, the phases in the Ag–Sb system were described thermodynamically using the parameters in [12].
The set of phases in the Ag–Sn system is the same as in the Ag–Sb system. The available experimental information about the thermodynamic properties of phases and the phase equilibria in the Ag–Sn system was reviewed in [8, 17, 18]. This system was modeled thermodynamically in [8, 10, 17–20]. In this work, the phases in the Ag–Sn system was described thermodynamically using parameters from the COST MP0602 thermodynamic database [21, 22].
In addition to the liquid phase, the Sb–Sn system contains solid solutions based on pure components, an intermediate (SbSn) solid solution, Sb2Sn3, and Sb3Sn4 [7, 23]. The available experimental data on the thermodynamic properties of phases and the phase equilibria in the Sb–Sn system were reviewed in [6, 23, 24]. This system was modeled thermodynamically in [6–8, 11, 24, 25]. In this work, the phases in the Sb–Sn system was described thermodynamically using the parameters in [7, 25].
THERMODYNAMIC MODELS OF PHASES IN THE Ag–Sb–Sn SYSTEM
According to [1, 5, 26], the Ag–Sb–Sn system contains no ternary compounds and has two continuous intermediate solid solutions (ζ and ε) that begin in the Ag–Sb system and terminate in the Ag–Sn system. In addition, the solubility of Ag in the (Sb) and (SbSn) phases is very low [1, 4, 5].
The temperatures of the liquidus and other phase transitions in this system were determined in [2, 4, 5, 9, 13]. The integral enthalpy of mixing of liquid Ag–Sb–Sn alloys was studied in [2, 27, 28]. The activity of tin in ternary liquid alloys was also investigated in [28].
In this work, the molar Gibbs energy of the liquid phase, the intermediate ζ solid solution, and the solid solutions based on pure components were described with the formula
where φ is the physical state of the solution; \(G_{k}^{{ \circ \varphi }}\) represents the Gibbs energies of pure components (for function \(G_{k}^{{ \circ \varphi }}\), we used expressions from the SGTE database of version 4.4 for pure elements [29]); xk denotes the molar fractions of components in the solution (k = 1, 2, 3 corresponds to Ag, Sb, and Sn); R is the universal gas constant; Т is the absolute temperature; \(L_{{ij}}^{{n,\varphi }}\) represents the parameters describing the excess Gibbs energy of the φ solution in the binary subsystems of the Ag–Sb–Sn system; and \(L_{{ijk}}^{\varphi }\) denotes the parameters describing the excess Gibbs energy of ternary solutions (the functions (x1− x2)i(x1 − x3)j(x2 − x3)k corresponding to these parameters were used to extend the Redlich–Kister formalism [30] to ternary solutions; i.e., functions (xi − xj)n were used to describe the excess Gibbs energy of binary solutions).
The molar formation Gibbs energy of Sb3/7Sn4/7 (Sb3/7Sn4/7 = 1/7 Sb3Sn4) was described by the expression [7]
The thermodynamic properties of the Sb2Sn3 – xAgx solid solution (denoted below as (Sb2Sn3)) were described by the two-sublattice model of (Sb)0.4(Sn,Ag)0.6. The molar formation Gibbs energy of this solution is characterized by the expression
where \({{\Delta }_{{\text{f}}}}G_{{{\text{Sb:Sn}}}}^{{{\text{S}}{{{\text{b}}}_{{\text{2}}}}{\text{S}}{{{\text{n}}}_{{\text{3}}}}}}\) is the molar formation Gibbs energy of Sb0.4Sn0.6, yi is the molar fraction of the ith element in the second sublattice of the (Sb2Sn3) phase, and \({{\Delta }_{{\text{f}}}}G_{{{\text{Sb:Ag}}}}^{{{\text{S}}{{{\text{b}}}_{{\text{2}}}}{\text{S}}{{{\text{n}}}_{{\text{3}}}}}}\) is a fitted parameter.
The ε solid solution was described thermodynamically using a two-sublattice model of (Ag,Sb)0.75(Ag,Sb,Sn)0.25 [8]. The molar Gibbs energy of the formation of this phase is characterized by the expression
where \(y_{i}^{{(1)}}\) is the molar fraction of the ith element (i = Ag, Sb) in the first sublattice; \(y_{j}^{{(2)}}\) is the molar fraction of the jth element (j = Ag, Sb, Sn) in the second sublattice; and \({{\Delta }_{{\text{f}}}}G_{{i:j}}^{\varepsilon }\), \(L_{{i,k:j}}^{{{\text{0}}{\text{,}}\varepsilon }}\), \(L_{{i:j,k}}^{{{\text{0}}{\text{,}}\varepsilon }}\), \(L_{{i:{\text{Ag}}{\text{,Sb}}{\text{,Sn}}}}^{{1,\varepsilon }}\) are the thermodynamic parameters.
The (SbSn) solid solution was described using a two-sublattice model of (Sb,Sn)0.5(Sb,Sn)0.5 [25]. The molar formation Gibbs energy of this phase is characterized by the expression
where \(y_{i}^{{(s)}}\) is the molar fraction of the ith element (i = Sb, Sn) in the sth sublattice (s = 1, 2) of the (SbSn) phase, and \({{\Delta }_{{\text{f}}}}G_{{i:j}}^{{{\text{SbSn}}}}\), \(L_{{i,k:j}}^{{{\text{0}}{\text{,SbSn}}}}\), \(L_{{i:j,k}}^{{{\text{0}}{\text{,SbSn}}}}\) are the thermodynamic parameters.
Parameters \(L_{{ij}}^{{n,\varphi }}\), \({{\Delta }_{f}}G_{{i:j}}^{\varepsilon }\), \(L_{{i,k:j}}^{{{\text{0}}{\text{,}}\varepsilon }}\), \(L_{{i:j,k}}^{{{\text{0}}{\text{,}}\varepsilon }}\), \({{\Delta }_{{\text{f}}}}G_{{i:j}}^{{{\text{SbSn}}}}\), \(L_{{i,k:j}}^{{{\text{0}}{\text{,SbSn}}}}\), and \(L_{{i:j,k}}^{{{\text{0}}{\text{,SbSn}}}}\) were taken from the thermodynamic descriptions of the binary subsystems of the Ag–Sb–Sn system in [7, 12, 22, 25], and parameters \(L_{{ijk}}^{\varphi }\), \({{\Delta }_{{\text{f}}}}G_{{{\text{Sb:Ag}}}}^{{{\text{S}}{{{\text{b}}}_{{\text{2}}}}{\text{S}}{{{\text{n}}}_{{\text{3}}}}}}\), and \(L_{{i:{\text{Ag}}{\text{,Sb}}{\text{,Sn}}}}^{{1,\varepsilon }}\) were determined by minimizing the objective function
where P is the sought set of parameters; \(T_{i}^{ * }\) and \(Z_{j}^{ * }\) are the experimental values for the temperatures of phase transitions and the thermodynamic properties of phases; Ti(P) and Zj(P) are the calculated values corresponding to \(T_{i}^{ * }\) and \(Z_{j}^{ * }\); and ωi represents the weight multipliers taken to be equal to the reciprocal error of estimating parameters \(T_{i}^{ * }\) and \(Z_{j}^{ * }\). Zj(P) was found using the analytical expressions for the Gibbs energies of phases and the well-known relationships of thermodynamics. Ti(P) was found by solving the system of nonlinear equations that follow from the condition of equilibrium between phases [31, 32].
The experimental information used to find the parameters consisted of
• the temperatures of the liquidus and secondary crystallization determined in [2, 4, 5, 9, 13];
• the temperatures of invariant equilibria in [2, 4, 5, 13];
• the integral mixing enthalpies of ternary liquid alloys at 803, 873, and 903 K (sections with xAg/xSn = 1 : 3, 1 : 1 and xSb/xSn = 3 : 7, 1 : 1, 7 : 3) [2], at 912 and 1075 K (section with xSb/xSn = 1 : 1) [28], at 1224 K (SbxSn1−x–Ag0.9Sn0.1 sections with x = 0.2, 0.4, 0.6, 0.8), and at 1253 K (section with xSb/xSn = 4 : 1) [27];
• the activities of tin in the Ag–Sb–Sn liquid alloys at 1073 and 1223 K (sections with xAg/xSb = 1 : 3, 1 : 1, 3 : 1) [28].
Function (6) was minimized by the Marquard method [33]. The set of parameters found as a result of optimization was (in J/mol):
The obtained set of parameters perfectly describes the experimental temperatures of phase transitions with average absolute deviations (AADs) of 8.9 K (liquidus), 6.7 K (secondary crystallization), and 1.7 K (invariant equilibria). The deviation of calculation results on the enthalpies of mixing of liquid alloys from the experimental data [2] is AAD = 107 J/mol; 455 of the 459 experimental points [2] are in this case described within 300 J/mol, and the deviation for the other four points [2] is less than 350 J/mol. The difference between the calculated and experimental values of the integral enthalpy of mixing of the Ag–Sb–Sn liquid alloys is characterized by AADs of 211 J/mol [28] and 241 J/mol [27]. The description of experimental data on the activity of tin in the liquid alloys [28] is characterized by AAD = 0.044.
CALCULATING PHASE EQUILIBRIA IN THE Ag–Sb–Sn SYSTEM
Thermodynamic models obtained for all phases of the Ag–Sb–Sn system by finding parameters (7) were used to calculate phase equilibria by minimizing the Gibbs energy of the system. The calculated polythermal sections of the Ag–Sb–Sn phase diagram for the compositions xAg/xSb = 1, xAg/xSn = 1 : 1, and xSb/xSn = 1 : 1 are shown in Figs. 1–3. The obtained phase diagrams are characterized by the existence of several large regions of primary crystallization belonging to the phases ε (all sections), ζ, (Ag) (the section with xSb/xSn = 1 : 1), and (Sb) (sections with xAg/xSn = 1 : 1 and xAg/xSb = 1 : 1). The calculated temperatures of phase transitions are in good agreement with the available experimental data.


Polythermal section with xAg/xSn = 1 in the Ag–Sb–Sn phase diagram: calculations (lines), experimental data [13] (points); (1) L + ε + (SbSn), (2) L + ε + (Sb2Sn3), (3) ε + (Sb2Sn3), (4) ε + (Sb2Sn3) + (SbSn), (5) L + ε + (Sn), (6) ε + (Sn), (7) L + ε + Sb3Sn4, (8) ε + Sb3Sn4 + (Sb2Sn3), (9) ε + Sb3Sn4, (10) regions of ε + Sb3Sn4 + (SbSn) phase coexistence.

Polythermal section with xSb/xSn = 1 in the Ag–Sb–Sn phase diagram: calculations (lines), experimental data [4] (points); (1) L + (Sb), (2) L + (Sb) + (SbSn), (3) L + ε + (Sb), (4) L + ζ + ε, (5) L + ζ + (Ag), (6) ζ + (Ag), (7) (SbSn) + Sb3Sn4 + ε.
The calculated polythermal section of the Ag–Sb–Sn system for compositions xSn = 0.5 is shown in Fig. 4. Two-phase L + ε and (SbSn) + ε regions and three-phase L + ε + (Sb2Sn3) region predominate in this phase diagram.

Polythermal section with xSn = 0.5 in the Ag–Sb–Sn phase diagram: calculations (lines), experimental data [13] (points); (1) L + ε + (Sn), (2) L + ε + Sb3Sn4, (3) ε + (Sb2Sn3), (4) ε + (Sb2Sn3) + Sb3Sn4, (5) Sb3Sn4 + ε.
The isothermal section of the Ag–Sb–Sn system at 473 K is shown in Fig. 5. The (Sb) + ε, (Sb) + (SbSn) + ε, (SbSn) + ε, and Sb3Sn4 + (Sn) + ε phase fields predominate in this phase diagram.

Calculated isothermal section in the Ag–Sb–Sn phase diagram at 473 K: (1) (SbSn) + Sb3Sn4 + ε, (2) Sb3Sn4 + ε, (3) regions of Sb3Sn4 + (Sn) phase coexistence.
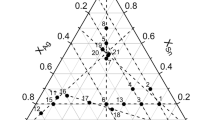
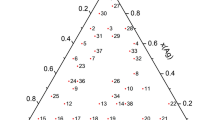
The calculated projection of the liquidus surface of the Ag–Sb–Sn system is shown in Fig. 6, where the solid line represents monovariant equilibria, and the fine lines are the liquidus isotherms at 573–1173 K. The points of intersection between the solid lines correspond to invariant equilibria, and their coordinates are given in Table 2. Of these equilibria, six are transitional, and one is of the eutectoid type. In earlier thermodynamic calculations of the Ag–Sb–Sn system [8–10] that considered only one of the (Sb2Sn3) and Sb3Sn4 phases, only three non-variant equilibria were found.

Calculated projection of the liquidus surface in the Ag–Sb–Sn system and isotherms at (1) 1173, (2) 1073, (3) 973, (4) 873, (5) 773, (6) 673, and (7) 573 K.
According to our calculations, invariant equilibrium L + Sb3Sn4 ↔ ε + (Sn) has the lowest temperature (501.8 K) of the invariant equilibria of the Ag–Sb–Sn system (Table 2). However, the temperatures of metastable equilibria L + (Sb2Sn3) ↔ ε + (Sn) and L + (SbSn) ↔ ε + (Sn) are very close to it, enhancing the role of kinetic factors in predetermining which of these equilibria is attained.
The calculated solubility of Ag in the (Sb2Sn3) phase, which participates in several invariant equilibria, is 2.7 at % at 582 K (L + (SbSn) ↔ ε + (Sb2Sn3)) and 1.3 at % at 507 K ((Sb2Sn3) ↔ ε + Sb3Sn4 + L).
CONCLUSIONS
Thermodynamic modeling of the Ag–Sb–Sn system was performed using a modified Sb–Sn phase diagram [7] and recent thermodynamic data for ternary liquid alloys [2, 28]. Four polythermal sections of the Ag–Sb–Sn phase diagram were calculated along with an isothermal section at 473 K. The coordinates of invariant equilibria in this system and the projection of the liquidus surface were determined.
REFERENCES
C.-Y. Lin, C. Lee, X. Liu, and Y.-W. Yen, Intermetallics 16, 230 (2008).
D. Li, S. Delsante, A. Watson, and G. Borzone, J. Electron. Mater. 41, 67 (2012).
J. Yin, M. Wada, S. Tanase, and T. Sakai, J. Electrochem. Soc. 151, A867 (2004).
D. B. Masson and B. K. Kirkpatrick, J. Electron. Mater. 15, 349 (1986).
S.-W. Chen, P.-Y. Chen, C.-N. Chiu, et al., Metall. Mater. Trans. A 39, 3191 (2008).
S. W. Chen, C. C. Chen, W. Gierlotka, et al., J. Electron. Mater. 37, 992 (2008).
V. A. Lysenko, J. Alloys Compd. 776, 850 (2019).
C.-S. Oh, J.-H. Shim, B.-J. Lee, and D. N. Lee, J. Alloys Compd. 238, 155 (1996).
Z. Moser, W. Gasior, J. Pstrus, et al., Mater. Trans. JIM 45, 652 (2004).
W. Gierlotka, Y.-C. Huang, and S.-W. Chen, Metall. Mater. Trans. A 39, 3199 (2008).
B. Jönsson and J. Ågren, Mater. Sci. Technol. 2, 913 (1986).
E. Zoro, C. Servant, and B. Legendre, J. Phase Equilib. Dif. 28, 250 (2007).
J. Lapsa and B. Onderka, J. Phase Equilib. Dif. 40, 53 (2019).
S.-W. Chen, A.-R. Zi, W. Gierlotka, et al., Mater. Chem. Phys. 132, 703 (2012).
W. Gierlotka, J. Electron. Mater. 45, 2216 (2016).
B. Z. Lee, C. S. Oh, and D. N. Lee, J. Alloys Compd. 215, 293 (1994).
I. Karakaya and W. T. Thompson, Bull. Alloy Phase Diagrams 8, 340 (1987).
P. -Y. Chevalier, Thermochim. Acta 136, 45 (1988).
U. Kattner and W. J. Boettinger, J. Electron. Mater. 23, 603 (1994).
Y. Xie and Z. Qiao, J. Phase Equilib. 17, 208 (1996).
A. Kroupa, A. Dinsdale, A. Watson, et al., J. Min. Metall. B 48, 339 (2012).
G. P. Vassilev, V. Gandova, N. Milcheva, and G. Wnuk, CALPHAD 43, 133 (2013).
C. Schmetterer, J. Polt, and H. Flandorfer, J. Alloys Compd. 728, 497 (2017).
H. Ohtani, K. Okuda, and K. Ishida, J. Phase Equilib. 16, 416 (1995).
A. Kroupa and J. Vizdal, Def. Dif. Forum 263, 99 (2007).
C. Lee, C.-Y. Lin, and Y.-W. Yen, J. Alloys Compd. 458, 436 (2008).
B. Gather, P. Schröter, and R. Blachnik, Z. Metallkd. 78, 280 (1987).
J. Łapsa and B. Onderka, J. Electron. Mater. 45, 4441 (2016).
A. T. Dinsdale, CALPHAD 15, 317 (1991).
O. Redlich and A. T. Kister, Ind. Eng. Chem. 40, 345 (1948).
V. A. Lysenko, Russ. J. Phys. Chem. A 82, 1252 (2008).
V. P. Vassiliev and V. A. Lysenko, J. Alloys Compd. 629, 326 (2015).
G. Reklaitis, A. Ravindran, and K. Ragsdell, Engineering Optimization (Wiley, New York, 1983), Vol. 1.
M. Ellner and E. J. Mittemeije, Acta Crystallogr., Sect. A 58, C333 (2002).
R. C. Sharma, T. L. Ngai, and Y. A. Chang, Bull. Alloy Phase Diagrams 10, 657 (1989).
Funding
This work was supported by the Russian Foundation for Basic Research, project no. 17-08-01723.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Glushachenkova
Rights and permissions
About this article

Cite this article
Lysenko, V.A. Thermodynamic Modeling of the Ag–Sb–Sn System. Russ. J. Phys. Chem. 94, 1747–1755 (2020). https://doi.org/10.1134/S0036024420090174
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024420090174