
Abstract
The effect the structural properties of a support, magnesium aluminum spinel, with different structural characteristics has on the physicochemical properties of Fe–K-containing catalysts for the hydrogenation of CO is studied. It is shown that hematite is the main phase in Fe-containing catalysts. A highly disperse distribution of iron oxide on the surface of the substrate is observed in case of a bimodal distribution of meso-pores. It is shown via IR spectroscopy that Fe2+ cations and metallic iron are the main sites of adsorption. The introduction of potassium reduces the surface concentration of clusters (including Fe2+) in different coordination environments. A rise in the contribution from the subcarbonyl forms of adsorption to the spectrum of the absorption bands and a drop in the amount of oxidized iron on the surfaces of metallic iron particles are observed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Iron-containing catalysts supported on an oxide support are traditionally used in the hydrogenation of CO, or Fischer–Tropsch synthesis (FTS). The promotion of the catalysts by metals (K and Cu) allows us to achieve high activity along with olefin and C5+ hydrocarbon selectivity [1–3]. Carbides of different chemical compositions are considered the active phase of Fe-containing catalysts [1, 4, 5]. Highly disperse iron carbides can form during the activation of iron catalysts in a flow of CO or synthesis gas and remain stable in a wide range of reaction conditions [6]. The role of the active component on the support is important, since adsorption proceeds on it in particular. The catalytic properties of the catalyst depend largely on how the stage of adsorption proceeds. Alumina is a traditional support both for iron and cobalt FTS catalysts. However, the formation of iron– and cobalt–magnesium aluminum spinels with quite small metal crystallites that are subsequently reduced only at high temperatures and are inactive in FTS, proceeds over such catalysts during FTS at a high degree of conversion [7, 8]. To prevent oxide–oxide interaction with the active component, it is modified by metals and other chemical compounds, e.g., magnesium oxide or silica gel [3, 9]. Stabilization of an Al2O3 support by 5% silica gel allows us to achieve rates of around 49 mmol CO \({\text{g}}_{{{\text{cat}}}}^{{ - 1}}\) h−1 under the standard conditions of FTS. They are lower when traditional commercial alumina supports are used: 26–39 mmol CO × \({\text{g}}_{{{\text{cat}}}}^{{ - 1}}\) h−1 [10]. The authors of that work explained this by the formation of a thermally stable bimodal porous structure that allows us to achieve high dispersity of the metal in the catalysts prepared via impregnation [10]. According to [11], adding small amounts of silica gel to alumina can prevent the formation of inactive cobalt aluminates and thus prevent deactivation of the catalyst during FTS. It was noted that the properties of the porous support, its specific surface area, average pore size, and pore size distribution substantially affect the adsorption and catalytic properties of the prepared catalyst.
As was noted above, potassium as a promoter for a Fe-containing catalyst affects the physicochemical properties of the catalysts, and the activity and selectivity in the catalytic hydrogenation reaction of CO (FTS). The authors of [12] showed that adding a small amount of potassium stabilizes the surface of the supported iron oxide and prevents further sintering of the particles upon calcination. At a high concentrations of potassium, the active sites of the catalyst can be closed by potassium cations, reducing the catalytic activity.
The aim of this work was to investigate the effect the structural properties of the support and the means of preparation have on the physicochemical properties and forms of adsorption of carbon monoxide on the surfaces of potassium-promoted iron-containing catalysts for the hydrogenation of CO that are based on magnesium aluminum spinels with different textures. To rule out the influence of processes that occur during activation, untrained (freshly prepared) catalysts were taken as the objects of study.
EXPERIMENTAL
Preparation of the Catalysts
The catalysts were prepared via successive or simultaneous impregnation of the support (magnesium aluminum spinel) with solutions of Fe(NO3)2 · 9H2O and KNO3 with a 2 wt % concentration of potassium and a constant concentration of iron of 15 wt %. Spinels of the AlxMgyOz type (SASOL Germany GmbH) of the Puralox MG 30, Spinel (S), and Spinel Precursor (SP) grades were used as the supports. We denote these spinels as MA-S and MA-SP, respectively; their technical characteristics are presented in Table 1. The catalysts were dried for 1 h at 80°C on a rotary evaporator and then calcinated for 3 h in a nitrogen flow at 450°C in the programmed mode (the volumetric flow rate of nitrogen was 1200 h−1 and an 8 K/min rate of heating). Drying and calcination were repeated after each impregnation stage with the corresponding metal nitrates. The resulting catalysts are denoted below as Fe/MA, Fe/K/MA, K/Fe/MA, and (FeK)/MA. The supports of magnesium aluminum spinel are MA-S with a small specific surface area and MA-SP with a large specific surface area.
PHYSICOCHEMICAL STUDIES
Low-Temperature Adsorption of Nitrogen
The porous structure of the support and iron-containing catalysts based on magnesium aluminum spinel (specific surface area, specific micropore volume, and average pore size in the samples) were studied via the low-temperature adsorption of nitrogen on a Micromeritics ASAP 2000 sorbtometer. Immediately prior to each experiment, samples of 0.2–0.3 g were degassed for 4 h in vacuum (P < 10 mmHg) at 300°C. Measurements were made at intervals of 5 s. The results are presented in Table 2.
X-ray Diffraction Analysis
X-ray diffraction analysis (XRD) was performed on a DRON-3 diffractometer with CuKα radiation and a graphite monochromator. Measurements were made in the 2θ range of 20°–70°. Scanning step was 0.1 deg, with exposure time per point of 3 s. To determine the sizes of the crystallites, recording was done in increments of 0.02° and a time of exposure per point of 10 s. Average particle size DHKL of hematite was estimated using the Scherrer formula [13]:
where λ is a wavelength of 1.54 Å, β is the integral half-width of a line equal to the area of the peak divided by the intensity at the maximum, and θ is the angle of reflection.
IR Spectroscopic Study
Diffuse reflection IR spectra were recorded on an EQUINOX 55/S Fourier-transform IR spectrometer (Bruker). A sieved powder 2θ range of each sample was placed into a quartz ampoule with a window made of CaF2 and calcinated at 400°C (1 h in air and 2 h under vacuum of at least 5 × 10−5 Torr). Gaseous CO was purified by passing it through a liquid nitrogen trap and with prolonged exposure over a calcinated zeolite. The differential spectra of adsorbed CO were obtained by subtracting the background spectrum from the experimental spectrum of a sample containing adsorbed CO, followed by adjustment of the baseline with the OPUS 6.0 software (Bruker). The digital noise subtraction for low-intensity spectra was performed.
RESULTS AND DISCUSSION
Two porous magnesium aluminum spinels that were white powder-like substances of the 0.05–0.1-mm fraction were chosen as the supports for Fe–K supported catalysts. Table 2 presents the values of the main structural parameters of the spinels and catalysts based on them according to data obtained via the low-temperature adsorption of nitrogen. The specific surface area of the MA-SP spinel was an order of magnitude larger than that of MA-S: the average pore volume in MA-SP was 500% larger and the average pore diameter was 300% smaller, compared to MA-S. This pattern was retained in the catalysts based on these spinels. An exception was the sample of Fe/MA-S, where the specific surface area was notably larger than that of the initial support.
Figure 1 presents the dependence of pore volume on diameter for the (FeK)/MA-SP and (FeK)/MA-S catalysts. As is seen from Fig. 1, a bimodal dependence of pore volume on diameter with maxima at 33 and 75 Å is observed for the sample based on the MA-SP spinel, where the pore volume consists mainly of mesopores. For the sample based on the MA-S spinel, this dependence is of an almost unimodal character with a maximum at 620 Å, and the contributions from mesopores and macropores are comparable. For the other samples based on MA-SP and MA-S spinels, the pore size distribution is similar to that of the (FeK)/MA-SP and (FeK)/MA-S systems, respectively.

Dependence of pore volume V on pore diameter d for the (a) (FeK)/MA-SP and (b) (FeK)/MA-S catalysts.
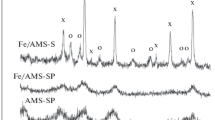
The data from X-ray diffraction analysis of the catalysts showed that the main phases were magnesium aluminum spinel and hematite. As examples, Fig. 2 presents the X-ray diffraction patterns for samples of Fe/MA-S and Fe/MA-SP. The X-ray diffraction pattern of the Fe/MA-S sample (Fig. 2a) is formed by relatively narrow reflections of the crystalline phase that correspond to magnesium aluminum spinel (denoted as S) and hematite (denoted as O). The X-ray diffraction pattern of Fe/MA-SP corresponds to a system with a fairly large contribution from the amorphous phase (Fig. 2b). It is interesting that despite the identical procedure for introducing the active phase into the catalyst, no reflections corresponding to hematite are in this case observed. The X-ray diffraction pattern has quite broadened peaks that correspond to magnesium aluminum spinel against the background of the amorphous phase. This effect is due to the effect the texture of the support had on the state of the supported phase: a highly disperse distribution of iron oxide formed over the surface with the bimodal distribution of the support’s mesopores. This result agrees with the data obtained for silica gel-modified alumina in [10]. The particles of iron oxide were quite small, so the reflections from them were strongly broadened. The X-ray diffraction patterns of catalysts MA-SP and MA-S based on the same spinel virtually coincide with those presented in Fig. 2. X-ray diffraction analysis of the synthesized samples showed they contained no potassium-containing phases, due to the weak concentration of potassium in the catalyst and its low degree of crystallinity.

XRD diffraction patterns of (a) Fe/MA-S and (b) Fe/MA-SP: (s) reflections of spinel and (o) reflections of iron oxide (hematite).
Along with determining the phase composition of the catalysts using the results from XRD (Fig. 2a), we used the Scherrer equation [13] to estimate the average size of the coherent scattering region (CSR) for hematite in the catalysts based on MA-S spinel (Table 2). By assuming the particles were of uniform size, the dimensions of the CSR allowed us to estimate the average particle size. According to our estimates, the average particle size of hematite for the Fe/MA-S catalyst was 8 nm and fell to 6.5 nm when there was potassium in the K/Fe/MA-S catalyst.
Carbon monoxide is an effective spectral probe for determining the nature of adsorption sites. The use of CO as a probe molecule allows us to differentiate cations in different states of oxidation, determine their electron-accepting properties, and estimate their coordination environment. In addition, CO is a reagent in the reaction of FTS. It is therefore of considerable interest to investigate the forms of CO adsorption on the surfaces of our systems.
The adsorption of carbon monoxide on the surfaces of our samples was investigated via diffuse reflection IR spectroscopy. Figures 3 and 4 present the difference IR spectra of CO adsorbed at room temperature on the catalysts based on MA-S and MA-SP, respectively. The spectra were formed by the superpositioning of several absorption bands (ABs), and two groups of overlapping absorption bands are easily distinguished. According to our XRD data, the main iron-containing phase was hematite Fe2O3. However, the literature data [14–16] indicate that Fe3+ cations do not form carbonyl complexes, since they are in the coordinatively saturated state and are not capable of forming complexes that are stable at room temperature and contain molecules with weak electron-donating properties. The absorption bands observed in the high-frequency region of the spectrum are therefore attributed to carbonyl complexes with Fe2+ cations. According to the data of [17], refined with DFT quantum-chemical calculations [18], the absorption bands at 2185–2188 and 2175–2180 cm−1 (Figs. 3a–3d) are due to the adsorption of CO in linear form on sites of the [FeOFe]2+ and [FeO]2+ types, respectively. The authors of [14] believed that the AB at 2175 cm−1 in this case corresponds to the dicarbonyl form of adsorption, while those of [19] assumed the tricarbonyl form of adsorption was involved. The contribution from the absorption band corresponding to the complexes of CO with isolated Fe2+ cations (2205–2218 cm−1) is negligible, and this band can be distinguished only when a surface is sparsely occupied by CO molecules (Figs. 3c, 3d, spectrum 1; Fig. 3b, spectra 2, 3).

Difference IR spectra of CO adsorbed at room temperature on (a) Fe/MA-S, (b) (KFe)/MA-S, (c) Fe/K/MA-S, and (d) K/Fe/MA-S catalysts. The pressure of CO is (1) 5, (2) 20, and (3) 50 Torr; F(R) is the Kubelka–Munk function.

Difference IR spectra of CO adsorbed at room temperature on the (a) Fe/MA-SP, (b) (KFe)/MA-SP, (c) Fe/K/MA-SP, and (d) K/Fe/MA-SP catalysts. The pressure of CO is (1) 5, (2) 20, and (3) 50 Torr.
The absorption band at 2150–2160 cm−1 (Figs. 3c, 3d; Figs. 4a–4d) appears in the spectrum when there is interaction between iron and the aluminum-containing support. It corresponds to the adsorption of CO on the Fe2+ cations that form on the surface of iron aluminate at the points of contact between iron and spinel [17, 20]. The isotope shift effect showed that this form of adsorption corresponded to tricarbonyls Fe2+(CO)3 [19].
The most intense ABs in the low-frequency group of bands at 2090–2115 and 2070–2075 cm−1 (Figs. 3a–3d, spectra 2, 3; Figs. 4a–4d) can be attributed to the carbonyl complexes with Fe2+ cations. The authors of [20] assumed that these two structures differ by the immediate environment of the cation. If the AB at 2070 cm−1 corresponds to the carbonyl complex with a Fe2+ cation surrounded by Fe0 metallic iron atoms on the surface of metallic iron, the AB at 2100 cm−1 corresponds to the complex with a Fe2+ cation, the coordination sphere of which includes oxygen or carbon atoms [20]. Such adsorption sites emerge on the surface of iron oxide or carbide as a result of the surface of metallic iron being oxidized or carbidized.
According to the data of [19], the low-intensity AB at 2123–2137 cm−1 (Fig. 3a; Figs. 3b–3d, spectra 2, 3; Figs. 4a, 4c, 4d) corresponds to polycarbonyls. The authors assume that this AB corresponds to the tetracarbonyls on Fe+ cations. This state of oxidation is not typical of iron, but it can be stabilized by the carbonyl environment. Quantum chemical calculations do not rule out the possible existence of carbonyl complexes with Fe+ [18].
The low-frequency ABs at 2050–2065 and 2000–2015 cm−1 (Figs. 3b, 3c, 3d; Figs. 4a, 4d) correspond to the linear form of adsorption of CO on Fe0 metallic iron atoms that differ by the coordination environment. If the AB at 2015 cm−1 corresponds to the carbonyl complexes on Fe0 atoms surrounded by the same Fe0 atoms, the AB at 2050 cm−1 corresponds to the complex with Fe0, the coordination sphere of which includes oxygen or carbon atoms [20]. The bands with frequencies of 1995 cm−1 or less correspond to the subcarbonyl forms of adsorption Fe\(_{n}^{0}\)(CO)m [14].
We assume that small (several atomic layers) islets of iron oxide on surfaces of spinel are the predecessors of particles of metallic iron. Particles of metallic iron form as a result of the reduction of iron oxide during vacuum heat treatment, or directly in a CO atmosphere. The ABs of carbonyl complexes with Fe2+ cations, the coordination sphere of which includes oxygen atoms (2100 cm−1), in the spectrum allows us to consider the incomplete reduction of iron oxide in these islets.
Comparing the spectra for a sample of Fe/MA-S (Fig. 3a) and those of potassium-promoted samples, we can that introducing potassium into the composition of the catalyst leads to a substantial (5–7 times) drop in the intensity of all absorption bands for the samples on the MA-S spinel, and the ratio of their intensities is substantially altered. While we attribute the AB at 2185 cm−1 to the carbonyl complexes with [FeO]2+ that predominate in the sample of Fe/MA-S (Fig. 3a), the AB at 2175–2180 cm−1 that we attribute to the complexes [FeOFe]2+ is the central band for the potassium-containing samples on the same spinel (Figs. 3b–3d). The relative contribution from the ABs at 2100 and 2072 cm−1 of the carbonyl complexes with Fe2+ cations on surfaces of the oxide or metallic phase is thus reduced, and the relative contribution from the ABs at 2050 and 2015 cm−1 that correspond to the complexes with metallic iron increases. The relative intensity of the ABs below 2000 cm−1 that correspond to subcarbonyl complexes of the Fe\(_{n}^{0}\)(CO)m type simultaneously grows substantially. Such forms are characteristic of metallic iron particles. The much higher intensity of the absorption bands at 2020 cm−1 and lower, which correspond to the carbonyl complexes on Fe0 and the ABs of subcarbonyl complexes, can be noted for the sample of (KFe)/MA-S (Fig. 3b).
In the group of samples based on MA-SP spinel, the introduction of potassium also leads to suppression of the high-frequency bands in the spectrum. In this case, however, it is not as great (2–4 times) as with the previous group. For potassium-containing samples, the contribution from the absorption band at 2200–2215 cm−1 (Figs. 4b–4d), which corresponds to complexes with isolated Fe2+ cations, becomes noticeable. Like the effect observed for the samples on MA-S, the introduction of potassium suppresses the adsorption sites bound to Fe2+ cations on the surface of the oxide or metallic phase. However, the intensity of the ABs at 2050 and 2020 cm−1, which correspond to the carbonyl complexes with Fe0 on surfaces of the metallic phase and the ABs of subcarbonyl complexes for the samples of (KFe)/MA-SP, K/Fe/MA-SP, and Fe/K/MA-SP (Figs. 4b–4d), grows in relation to the sample of Fe/MA-SP (Fig. 4a). The substantially greater (4 times) suppression of most adsorption sites of CO can be noted for the sample of K/Fe/MA-SP, but the intensity of the ABs of subcarbonyl complexes is in this case even higher than with Fe/MA-SP (Fig. 4d).
Systems obtained via simultaneous impregnation of components (KFe)/MA-SP and (KFe)/MA-S stand out against this background. When using this impregnation method, the relative contribution from the adsorption sites bound to metallic iron grows maximally against the background of a drop in the concentration of the adsorption sites bound to Fe2+ (Figs. 3b, 4b).
Impregnation in the order Fe → K results in the maximum suppression of adsorption sites (Figs. 3d, 4d). Impregnation in the reverse order produces a differently directed effect. While the suppression of adsorption sites is similar to the type observed in the K/Fe/MA-S system (Fig. 3d) can be noted in the Fe/K/MA-S system (Fig. 3c), the composition and relative concentrations of the adsorption sites for the Fe/K/MA-SP system (Fig. 4c) are close to those of the (KFe)/MA-SP system (Fig. 4b). We may assume the local concentration of potassium is in this case important, as it is 10 times lower on the surface of MA-SP.
Along with the emergence of the absorption band of the carbonyl complexes, new ABs in the region below 1700 cm−1 appear upon the adsorption of CO. These ABs characterize the vibrations of the bonds in the surfaces of carbonate–carboxylate compounds formed as a result of the reaction with CO adsorbed on the surface at room temperature (Fig. 5). According to the literature [17], there are several ways that carbonate-like structures can form on a surface: through the dissociation of CO on small Fe0 particles, through the reaction of CO with hydroxyl groups with the formation of CO2 [CO + OHads → CO2 + (1/2)H2] or through the formation of formate [CO + OHads → HCOOads] [21]. The formation of carbonates generally does not affect the spectra of carbonyl complexes [17].

Difference IR spectra of the carbonate-like structures appearing in the CO atmosphere: (a) Fe/MA-S, (b) K/Fe/MA-S, (c) (FeK)/MA-S, and (d) Fe/K/MA-S. The pressure of CO is 50 Torr.
When CO interacts with the surface of Fe/MA-S (Fig. 5a), the ABs at 1563 and 1328 cm−1, which can be attributed to bidentate carbonates, appear in the spectrum (Table 3). Introducing potassium into the composition of the samples based on the MA-S spinel greatly enhances the formation of carbonates in a CO atmosphere at room temperature. The highest concentration of carbonates is observed on the sample of (KFe)/MA-S (Fig. 5c). We assume the main path of carbonate formation is the emergence of carboxylate ions on particles of metallic iron, followed by their migration to the surface of the support. The formation of carbonate-like structures proceeds with the participation of basic oxygen on the surface of magnesium aluminum spinel (Table 3). The following carbonates form as a result: monodentate Δν ∼ 150 cm−1 and bidentate carbonates of three types that are characterized by splitting between the components: Δν ∼ 290, 370, and 420 cm−1 (Table 3). Note that the number of the types of carbonate-like structures and the distances between the components of the doublet in the spectrum are close to those of the carbonates on the surface of MgO [22], indicating that the observed carbonates formed with the participation of oxygen introduced into the coordination sphere of Mg2+ cations. The introduction of potassium enhances the oxidizing capacity of the catalysts.
The concentration of carbonates on the samples of the MA-SP spinel was much lower. A characteristic feature of these difference spectra is the number of intense negative peaks in the region of 1200–1500 cm−1 (Fig. 6). This testifies to the decomposition or evolution of some carbonate-like structures on the surfaces after vacuum heat treatment in a CO atmosphere. The amount of predominantly monodentate carbonates with Δν ∼ 120–170 cm−1 and symmetric carbonates (free ions) falls. Since these structures were stable in a vacuum at 400°C but changed at room temperature in a CO atmosphere, we assume these structures did not decompose but evolved into bidentate carbonates with considerable splitting of the doublet, due likely to the drop in the basicity of oxygen on the surface upon the adsorption of CO.

Difference IR spectra of the carbonate-like structures appearing in the CO atmosphere: (a) Fe/MA-SP, (b) K/Fe/MA-SP, (c) (FeK)/MA-SP, and (d) Fe/K/MA-SP. The pressure of CO is 50 Torr.
CONCLUSIONS
Hematite is the main phase in iron-containing catalysts. but since there is no adsorption of CO on Fe3+, the main adsorption sites are Fe2+ cations and metallic iron. The effect promotion by potassium has on the adsorption properties of a surface were studied, along with the possible types of adsorption sites on the catalysts, which differ by the method of the promoter introduction. Addition of potassium lowers the surface concentration of clusters, including Fe2+ in different coordination environments, and from our point of view promotes the enlargement of metallic iron particles on the surface. It is assumed there is a change in the distribution of iron, from the state of mixed clusters and particles to predominantly metallic iron particles. This explains the rise in the contribution from the absorption bands of the subcarbonyl forms of adsorption to the spectrum and the drop in the amount of oxidized iron on the surfaces of these metallic particles. The introduction of potassium enhances the oxidizing capacity of the catalysts’ surfaces and results in the formation of carbonate-like structures at room temperature.
REFERENCES
M. E. Dry, Studies in Surface Science and Catalysis (Elsevier, Amsterdam, 2004).
P. A. Chernavskii, G. V. Pankina, and V. V. Lunin, Catal. Lett. 66, 121 (2000).
G. V. Pankina, P. A. Chernavskii, Yu. A. Krylova, and V. V. Lunin, Kinet. Catal. 48, 567 (2007).
P. A. Chernavskii, G. V. Pankina, R. V. Kazantsev, and O. L. Eliseev, ChemCatChem 10, 1 (2018).
J. M. Gracia, F. F. Prinsloo, and J. W. Niemantsverdriet, Catal. Lett. 133, 257 (2009).
E. de Smit, F. Cinquini, A. M. Beale, et al., J. Am. Chem. Soc. 132, 14928 (2010).
J.-S. Girardon, A. S. Lermontov, L. Gengembre, et al., J. Catal. 230, 339 (2005).
K. Jalama, N. J. Coville, H. Xiong, et al., Appl. Catal., A 395, 1 (2011).
G. V. Pankina, P. A. Chernavskii, A. S. Lermontov, and V. V. Lunin, Pet. Chem. 42, 217 (2002).
M. Rahmati, B. Huang, M. K. Mortensen, et al., J. Catal. 359, 92 (2018).
S. Barradas, E. A. Caricato, P. J. van Berge, and J. van de Loosdrecht, Stud. Surf. Sci. Catal. 143, 55 (2000).
H. Kolbel and M. Ralek, Catal. Rev. Eng. 21, 225 (1980).
A. Taylor, An Introduction to X-ray Metallography (Springer, New York, 1945).
A. Davydov, Molecular Spectroscopy of Oxide Catalyst Surfaces (Wiley, New York, 2003).
C. Angell and P. C. Schaffer, J. Phys. Chem. 70, 1413 (1966).
D. Ballivet-Tkatchenko and G. Coudurier, Inorg. Chem. 18, 558 (1979).
J. Couble and D. Bianchi, Appl. Catal. A 409–410, 28 (2011).
M. F. Fellah, J. Phys. Chem. C 115, 1940 (2011).
M. Mihaylov, E. Ivanova, K. Chakarova, et al., Appl. Catal. A 391, 3 (2011).
A. F. H. Wielers, A. J. H. M. Kock, C. E. C. A. Hop, et al., J. Catal. 117, 1 (1989).
D. Bianchi, T. Chafik, M. Khalfallah, and S. J. Teichner, Appl. Catal. A 105, 223 (1993).
A. A. Davydov, M. L. Shepot’ko, and A. A. Budieva, Kinet. Katal. 35, 299 (1994).
ACKNOWLEDGMENTS
The authors are grateful to Prof. P.A. Chernavsky for his continuing interest in our work and his valuable comments, and to A.A. Malyshev (Hamburg) for kindly providing magnesium aluminum spinel samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Boltukhina
Rights and permissions
About this article

Cite this article
Kharlanov, A.N., Pankina, G.V. & Lunin, V.V. Physicochemical Properties of Potassium-Promoted Fe-Containing Catalysts for the Hydrogenation of CO over Magnesium Aluminum Spinels: IR Spectroscopy. Russ. J. Phys. Chem. 93, 2356–2364 (2019). https://doi.org/10.1134/S0036024419120136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024419120136