
Abstract
Borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) are synthesized by the Pechini method followed by annealing of the intermediates and examined by X-ray diffraction (XRD) and differential scanning calorimetry (DSC). The crystallographic parameters of the synthesized LnBWO6 are refined by powder X-ray diffraction in two crystal systems: monoclinic (space group P21) and orthorhombic (space group P222). Reversible first-order phase transitions in the synthesized LnBWO6 are detected by the DSC method, and the temperatures and enthalpies of phase transformations are determined. The doping of LnBWO6 with Nd3+ and Gd3+ ions is shown to decrease the temperature of the LT (low-temperature) → HT (high-temperature) phase transition of LaBWO6. Two independent positions of Gd in the La0.99Gd0.01BWO6 structure are determined from the experimental electron paramagnetic resonance (EPR) data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Tungstates and borate tungstates containing rare-earth elements (REE) are being actively studied as new materials for practical application [1–16]. Borate tungstate LaBWO6 with the structural formula La(BO2)(WO4) is chemically and thermally stable heteroanionic compound that evokes interest due to the nonlinear optical properties and a possibility of using as a basis for the preparation of functional materials of various design, including luminophores, which are used in the manufacturing technology of solid-state white light emitting diodes (WLED) [6–11, 14–16].
Researchers have no single opinion concerning the LaBWO6 structure. The pioneers of the synthesis of this compound in the course of studying the phase equilibria in the subsolidus range of the La2O3–B2O3–WO3 system [17–19] believe that LaBWO6 belongs to the family of LnBWO6 (Ln = La, Pr, and Nd) and crystallizes in the monoclinic crystal system (space group P21) with the parameters a = 6.019(2) Å, b = 4.106(1) Å, c = 9.468(4) Å, β = 99.71(3)°, and V = 230.6 Å3 [18]. The results were obtained for the single crystals grown by the spontaneous crystallization of the LaBWO6 melt preliminarily formed by the solid-phase interaction of lanthanum oxide La2O3, tungsten oxide WO3, and boric acid H3BO3 in the ratio La2O3 : B2O3 : WO3 = 1 : 1 : 2.
The crystals of LaBWO6 prepared by glass crystallization in a 50WO3–25La2O3–25B2O3 (mol %) system and mechanochemical activation of the amorphous product corresponded to monoclinic LaBWO6 (JCPDF 057-1075), i.e., crystallized in the space group P21 [12, 13, 20]. Optical crystals of LaBWO6 prepared [6] by solution−melt crystallization in a LaBWO6−(Li2WO4/LiF)−B2O3 system were indexed in the orthorhombic crystal system (space group P222) with the parameters a = 4.1 Å, b = 10.34 Å, and c = 21.71 Å. The authors confirmed the absence of an inversion center in the LaBWO6 structure by the determination of the second harmonic generation equal to 0.3 relative to KDP (potassium dihydrophosphate, crystal known in nonlinear optics). Nonlinearity of the second and third orders were found for monoclinic crystals of LаBWO6 [7], although the title of the article indicates that the studied LaBWO6 crystal is monoclinic, but the crystallographic parameters presented in the text refer to an orthorhombic crystal (space group P222): a = 4.1 Å, b = 10.34 Å, c = 21.71 Å (cited data [6]).
The α (low-temperature) and β (high-temperature) polymorphic modifications were found for monoclinic borate molybdate LaBMoO6 [21, 22]. At the same time, monoclinic tungstate LaBWO6 and molybdate LaBMoO6 are known to be isostructural [18, 19]. Published data on polymorphism of monoclinic borate tungstate LaBWO6 are lacking.
An important characteristic of the structures of crystalline borate tungstates LnBWO6 is the number of nonequivalent crystallographic positions of rare-earth (RE) ions, which can be substituted by optically active RE ions exhibiting diverse luminescence properties. The La3+ ion in LaBWO6 has one crystallographic position and is localized at the center of the distorted polyhedron LaO10 (nine La−O distances range from 2.27 to 2.80 Å, and one La−O distance is 3.01 Å) [18]. If assuming that the family of borate tungstates LnBWO6 is formed only with ions Ln3+ = La3+, Pr3+, and Nd3+, then a solid substitution solution is formed at a low level of alloying/doping of LaBWO6 with Pr3+ or Nd3+ ions and ion-dopants Pr3+ (Nd3+), the structures of these LaBWO6 cannot have more than one crystallographic position.
We have previously shown [9] that the luminescence decay curve of the 4F3/2 level of the Nd3+ ions in polycrystalline La0.999Nd0.001BWO6 (Nd concentration is 0.1 at % relative to La3+ ions) is approximated by the monoexponential function, indicating the luminescence of one impurity optical center of the Nd3+ ions, which substitute the base ions (La3+). The doping of the LaBWO6 matrix with Eu3+ ions resulted in the appearance of two luminescence centers in monoclinic La1–xEuxBWO6 (x = 0.01, 0.05, 0.1, 0.15, 0.2, and 0.25) isostructural to borate tungstate LaBWO6 (no. 35-0261, ICDD) synthesized in [17]. The luminescence decay kinetics of the Eu3+ ions in La1–xEuxBWO6 was approximated by the sum of two exponential functions with close radiation lifetimes [16]. According to [8], compounds LnBWO6 (Ln = La, Gd, and Y) doped with Sm3+ ions are isostructural to LaBWO6 (no. 057-1075, JCPDS), i.e., crystallize in the monoclinic crystal system (space group P21) like LnBWO6 (Ln = La, Pr, and Nd). Thus, the family of monoclinic LnBWO6 becomes larger due to members of isostructural compounds: increases up to Gd and further along the REE series, which contradicts published data [17, 19].
The purpose of this work is to determine the crystal structures and crystallographic parameters of borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01), as well as the number of crystallographic positions of gadolinium in the La0.99Gd0.01BWO6 structure. The synthesis of LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) was conducted by the Pechini sol−gel method, whose advantage for the preparation of LnBWO6 was shown by an analysis of the spectral properties of La1–x–yYbxEryBWO6 [9].
It seemed important to study the thermal properties of the synthesized borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) to establish possible polymorphic phase transformations.
EXPERIMENTAL
Synthesis of borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) was carried out by the Pechini method with citric acid and mannitol followed by the annealing of the intermediates. The initial reagents were lanthanum nitrate La(NO3)3·6H2O (reagent grade), gadolinium acetate Gd(CH3COO)3· 4H2O (reagent grade), Nd2O3 (high-purity grade), HNO3 (reagent grade), boric acid H3BO3 (reagent grade), ammonium tungstate (NH4)4W5O17·2.5H2O (analytical grade), citric acid C6H8O7 (high-purity grade), and mannitol С6H14O6 (analytical grade). A 10% H3BO3 excess was added to compensate boron losses during the high-temperature annealing. Oxide Nd2O3 was dissolved on heating in a minimum amount of HNO3 (∼1 : 1), lanthanum nitrate and gadolinium acetate were dissolved in water on heating with magnetic stirring, and citric acid was added to the resulting solution. Ammonium tungstate was dissolved in another beaker and added with boric acid and the calculated amount of citric acid. The solutions were poured together, and mannitol was added to the formed mixture to make it plastic. The molar ratios of the metal to citric acid and of citric acid to mannite were similar to those used in the sol−gel synthesis of Ln3BWO9 [5]. Thus prepared mixture was evaporated with stirring at 80°С for 2 h. Then the solution was placed into a porcelaneous cup and hold at 140°С for 2 h. A brown foamy gel was formed. The temperature was increased to 300°С and maintained for 3 h to form a black plastic mass. After the end of polymerization, the precursor temperature was increased to 700°С. Thus prepared powder was triturated and annealed at 700, 800, and 950°С holding at each temperature for 3 h.
X-ray powder diffraction studies of the intermediates and final reaction products was carried out on a Bruker D8 Advance diffractometer (CuKα radiation, Ni filter, LYNXEYE detector, reflection geometry, angle range 2θ = 10°–60°, scan increment 0.0133°) using the ICDD PDF2 crystallographic database.
Refinement of crystallographic parameters of LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) was performed by the powder XRD method at the Belok/XSA beamline of the Kurchatov specialized source of synchrotron radiation “KISI-Kurchatov” [23]. Monochromatic radiation with the wavelength λ = 0.75 Å focused on the sample with the size to 400 × 400 μm2 was used for measuring diffraction patterns. The measurements were conducted in the Debye–Scherrer (transmission) geometry. A sample was placed in a cryoloop 300 μm in size and rotated about the horizontal axis during measurement, which made it possible to average the diffraction patterns over orientations of the sample. Diffraction patterns were detected with a Rayonix SX165 2D detector arranged at a distance of 150 mm from the sample at an angle of 29.5° to the synchrotron radiation beam. The exposure time was 5 min. The obtained 2D diffraction patterns were integrated to the one-dimensional form I (2θ) using the Dionis program [24]. The angular scale of the detector was calibrated and the apparatus broadening of diffraction reflections was determined by measuring the polycrystalline standard LaB6 (NIST SRM 660a). The crystallographic parameters of the samples were refined analyzing the diffraction patterns by the Rietveld method using the Jana2006 program [25].
Thermal analysis of the synthesized LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) was carried out in a temperature range of 30–1000°С on a DSC 404 F1 Pegasus® differential scanning calorimeter (Netzsch). Temperature and sensitivity calibrations were performed by measuring the melting points of the standard metal samples in 85-μL PtRh20 crucibles with caps and Al2O3 inserts in an argon medium (trade mark 5.5) with a rate of 10 deg/min. According to the calibration results, the temperature inaccuracy was ±0.5 K and that for an enthalpy change was at most 2.8%. An empty PtRh20 crucible with a cap served as the reference during measurements.
Electron paramagnetic resonance (EPR) spectra of La0.99Gd0.01BWO6 were recorded on an Elexsys E-680X radio spectrometer (Bruker) in the X range (working frequency ~9.8 GHz) at room temperature with a modulation amplitude of 5 G and a microwave radiation power of 2 mW.
RESULTS AND DISCUSSION
XRD and Refinement of Crystallographic Parameters for LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01)
Since there are two opinions concerning the LaBWO6 structure, the crystallographic parameters of the synthesized LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) were refined in two crystal systems: monoclinic (space group P21 [18]) and orthorhombic (space group P222 [6]).
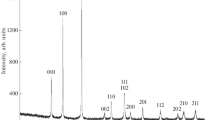
All reflections on the XRD patterns of the synthesized LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) corresponded to monoclinic LaBWO6 (no. 57-1075, JCPDF; space group P21) and, hence, the crystallographic parameters of LnBWO6 were first refined in the space group P21 (no. 2) using the data on monoclinic LaBWO6 [18]. In this case, we failed to describe several reflections by the calculation curve (Fig. 1). The crystallographic parameters and Miller indices published [6] for orthorhombic LaBWO6 (space group P222) made it possible to index all reflections on the XRD patterns of LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) synthesized by the Pechini method (Figs. 2, 3).

Diffraction patterns of LaBWO6: experimental (black points) and calculated (red curve enveloping experimental points, obtained in the monoclinic system, space group P21 [18]) patterns and the difference between the experimental and calculated patterns (blue curve).

Diffraction patterns of LaBWO6: experimental (black points) and calculated (red curve, obtained in the orthorhombic system, space group P222 [6]) patterns and the difference between the experimental and calculated patterns (blue horizontal line).

Description of the experimental diffraction patterns of LnBWO6 in the orthorhombic system (space group P222 [6]): (a) La0.999Nd0.001BWO6 and (b) La0.99Gd0.01BWO6. Diffraction patterns: experimental (black points) and calculated (red lines) patterns and the difference between the experimental and calculated patterns (blue horizontal line).
The refinement results are given in Table 1. An analysis of the obtained data showed that the refinement in the orthorhombic system (space group P222) is preferred. The parameters of the synthesized compounds LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) refined in the space group P222 are listed in Table 2 along with the published data [6] for LaBWO6.
The doping of the LaBWO6 matrix with Nd3+ ions results in a minor decrease in the cell volume. This is due to the fact that Nd3+ ions (that are somewhat smaller, according to [26], than the sizes of La3+ (1.27 Å for La in LaO10)) are incorporated into the structure substituting lanthanum. At the same time, the cell volume of La0.99Gd0.01BWO6 (920.4 Å3) is higher than the volume of undoped LaBWO6 (917.48 Å3), although the Gd3+ ion radius in the coordination polyhedron LnO10 is shorter than that of Nd3+ in the same coordination. It is most likely that a solid insertion solution is formed in the case of doping LaBWO6 with Gd3+ ions.
Thermal Properties of LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01)
The reversible phase transition of the first order was found [21] for crystals of monoclinic borate molybdate LaBO2MoO4 (space group P21), and the transition temperature depends on the thermal prehistory of the sample. According to authors’ opinion, if the sample was stored at room temperature for a long time, the authors failed to detect thermal effects of polymorphic transitions because of a low rate of phase transformations. We tried to detect similar effects and, therefore, repeated the described [21] experiment for the synthesized tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) using cyclic heating and cooling.
Cycle 1: temperature maintenance at 30°С (10 min) → heating to 1000°С with a rate of 10 deg/min → cooling to 200°С with a rate of 10 deg/min. Cycles 2 and 3: heating to 950°С with a rate of 10 deg/min → temperature maintenance at 950°С for 20 min → cooling to 200°С with a rate of 10 deg/min.
The DSC data obtained for LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) differ. For undoped LaBWO6, the temperature of the endothermic effect related to the rearrangement of the low-temperature phase (LT) to the high-temperature phase (HT) and the backward exothermic effect of the phase transition HT → LT on cooling were detected in all the three thermal cycles (Fig. 4). The temperatures of the LT → HT transitions lie in a range of (945.3–935.7) ± 0.5°С, and the temperatures of the backward HT → LT transitions with supercooling lie in a range of (892.2–871.1) ± 0.5°С depending on the thermal prehistory of the sample.

DSC curves for LaBWO6 synthesized by the Pechini sol–gel method followed by the annealing of the intermediates at 950°С.
For neodymium-doped La0.999Nd0.001BWO6, the phase transition temperatures were detected already in the first heating–cooling cycle (Fig. 5, Table 3), whereas the exothermal effect of the HT → LT arrangement of gadolinium-doped La0.99Gd0.01BWO6 was detected only in the second cycle after the temperature of the sample was preliminarily maintained at 950°С for 20 min and with significant supercooling (Тon2 = 553.4 ± 0.5°С) (Fig. 6, Table 3).

DSC curves for La0.999Nd0.001BWO6 synthesized by the Pechini sol–gel method followed by the annealing of the intermediates at 950°С.

DSC curves for La0.99Gd0.01BWO6 synthesized by the Pechini sol–gel method followed by the annealing of the intermediates at 950°С.
The thermal effects observed on the DSC curves of the synthesized LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) indicate that the studied compounds undergo the reversible phase transitions of the first order. Their temperatures and enthalpies are listed in Table 3. The addition of the dopants (Nd and Gd) decreases the temperature of the LT → HT phase transition of borate tungstate LaBWO6. The HT →LT transition occurs with a lower rate than the transition from the low-temperature (LT) to high-temperature (HT) phase. As shown for LaBWO6 as an example, maintenance at a high temperature accelerates the rearrangement of one phase to another.
Determination of the Number of Positions of Gd in the Structure of Borate Tungstate La0.99Gd0.01BWO6
The number of positions of Gd in the structure of La0.99Gd0.01BWO6 synthesized by the Pechini method was determined using the experimental EPR spectra of this compound (Fig. 7).

Experimental EPR spectra of trivalent gadolinium complexes are known to be very diverse. The single method of interpreting these spectra is to simulate the theoretical spectrum with a certain set of spin-Hamiltonian parameters. The spin-Hamiltonian (SH) is the crystal field expansion over multipoles and achieving its agreement with the experimental SH. Just this method was used to determine the number of positions of Gd in La0.99Gd0.01BWO6.
When simulating EPR spectra of coordination compounds of high-spin ions, it is necessary to take into account the influence of the crystal field expansion terms of orders higher than the second order. For instance, fourth-order expansion terms should be included for elements with S ≥ 2 (Mn+2, Fe+3), whereas the terms of the fourth and sixth orders are needed simultaneously for S ≥ 3 (Gd+3).
In the general form, the SH of the high-spin ion in the S state in a nonisotropic medium can be written as follows:
where \(O_{k}^{q}\) are the equivalent Stevens spin operators, and \(B_{k}^{q}\) are the Stevens parameters that describe the crystal field expansion over multipoles.
Since the total number of terms in SH (1) is very high, the EPR spectrum of a single crystal in different orientations is usually studied. If the compound is rather simple (oxide, chloride), the cubic fields with slight distortions of a lower symmetry are the main terms of the expansion. In the case of coordination compounds of a high-spin ion with a more complicated structure, it is usually considered that the second-order Stevens operators (D = 3\(B_{2}^{0}\), E = \(B_{2}^{2}\)) mainly contribute to the crystal field. However, possible contributions of the fourth and sixth orders (even very low) exert a substantial effect on the shape of the EPR spectrum. Therefore, two problems were solved simultaneously: to take into account the fourth- and sixth-order terms and to shorten their number if possible.
Two simplifying assumptions were made for this purpose. First, it was accepted that one of the principal axes of the second-order tensor is simultaneously the axis of the fourth and sixth orders. Second, in the case of the cubic environment of the high-spin ion, the Z axis of the second-order tensor can coincide with either the axis of the fourth-order cube, or the third-order axis coinciding with the long diagonal of the cube. Two SH (2) presented below correspond to the cubic ligand field for different arrangements of the system of coordinates relative to the cube
The upper SH corresponds to the cubic field potential where the Z axis passes through the centers of opposite faces of the cube, and the lower SH is the case where the Z axis passes through the long diagonal of the cube [27].
The direction of the Z axis is significant in the case of contributions of lower symmetry.
Thus, SH (1) is modified to Eq. (3)
The EPR spectrum of La0.09Gd0.01BWO6 is the sum of the spectra of two centers with the spin S = 7/2 in different environments of the described SH (3).
The parameters of the complexes were determined using the best-fit algorithm between the experimental and theoretical spectra by the minimization of the error functional
where \(Y_{i}^{E}\) is the set of experimental intensities of the EPR signal with a constant increment over magnetic field Н, \(Y_{i}^{T}\) are the theoretical values at the same field intensity Н, and N is the number of points.
SH (3) was numerically transformed into the diagonal form. The resonance fields of SH (3) were calculated by the Belford method for the construction of the theoretical spectrum [28]. The crystal field parameters and concentrations of the centers in various environments corresponding to the best fit of the simulated spectrum with the experimental spectrum are given in Table 4.
The results for the number of positions of gadolinium in the structure of La0.99Gd0.01BWO6 are given in Table 5 along with the data of other authors on the number of positions occupied by Pr, Nd, Sm, Eu, Tb, and Dy dopants in the structures of doped LnBWO6. These results were obtained by the luminescence decay kinetics of the active ions in the LaBWO6 matrix.
CONCLUSIONS
Borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) were synthesized by the Pechini sol–gel method followed by the annealing of the intermediates, and their crystallographic parameters were refined in the monoclinic (space group P21) and orthorhombic (space group P222) crystal systems. The obtained results indicate that the synthesized compounds LnBWO6 are orthorhombic. The temperatures and enthalpies of the reversible phase transitions for borate tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) were detected for the first time by the DSC method. Doping with Nd3+ and Gd3+ ions of the LaBWO6 matrix was shown to decrease the temperature of the LT → HT transition. Two independent positions of gadolinium in the structure of La0.99Gd0.01BWO6 were determined from the EPR spectrum of this compound.
REFERENCES
Lu-Ling Li, Xiao-Yu Yue, Wen-Jing Zhang, et al., Chin. Phys. B. 30, 077501 (2021). https://doi.org/10.1088/1674-1056/abf916
S. J. Dai, D. Zhao, R. J. Zhang, et al., J. Alloys Compd. 891, 161973 (2022). https://doi.org/10.1016/j.jallcom.2021.161973
X. Jin, Y. Xie, R. Tang, et al., J. Alloys Compd. 899, 162739 (2022). https://doi.org/10.1016/j.jallcom.2021.162739
D. Flavián, J. Nagl, S. Hayashida, et al., Phys. Rev. B 107, 174406 (2023). https://doi.org/10.1103/PhysRevB.107.174406
V. A. Krut’ko, M. G. Komova, and D. V. Pominova, Inorgan. Mater. 54, 1144 (2018). https://doi.org/10.1134/S0020168518110092
C. X. Sun, Z. B. Lin, L. Z. Zhang, et al., Chin. J. Struct. Chem. 32, 1088 (2013). www.researchgate.net/publication/283843691.
A. A. Kaminskii, Quantum Electron. 49, 377 (2019). https://doi.org/10.1070/QEL16980
F. B. Xiong, H. Chen, H. F. Lin, et al., J. Lumin. 209, 89 (2019). https://doi.org/10.1016/j.jlumin.2019.01.034
V. A. Krut’ko, M. G. Komova, D. V. Pominova, et al., Inorg. Mater. 59, 982 (2023). https://doi.org/10.1134/S002016852309008X
B. Li, X. Huang, H. Guo, et al., Dyes Pigm. 150, 67 (2018). https://doi.org/10.1016/j.dyepig.2017.11.003
F. B. Xiong, H. F. Lin, Z. Ma, et al., Opt. Mater. 66, 474 (2017). https://doi.org/10.1016/j.optmat.2017.03.002
M. Gancheva, L. Aleksandrov, R. Iordanova, et al., J. Chem. Technol. Metall. 50, 467 (2015).
L. Aleksandrov, T. Komatsu, K. Shinozaki, et al., J. Non-Cryst. Solids 429, 171 (2015). https://doi.org/10.1016/j.jnoncrysol.2015.09.004
D. Zhu and Z. Mu, Displays 35, 261 (2014). https://doi.org/10.1016/j.displa.2014.09.005
L. Aleksandrov, R. Iordanova, Y. Dimitriev, et al., Opt. Mater. 36, 1366 (2014). https://doi.org/10.1016/j.optmat.2014.03.031
Y. Huang and H. J. Seo, Mater. Lett. 84, 107 (2012). https://doi.org/10.1016/j.matlet.2012.06.051
B. F. Dzhurinskii, E. M. Reznik, and I. V. Tananaev, Zh. Neorg. Khim. 25, 2981 (1980).
K. Palkina, V. Saifuddinov, V. Kuznetsov, et al., Zh. Neorg. Khim. 24, 1193 (1979).
B. F. Dzhurinskii and G. V. Lysanova, Russ. J. Inorg. Chem. 43, 1931 (1998).
L. Aleksandrov, T. Komatsu, R. Iordanova, et al., Opt. Mater. 34, 201 (2011).
P. Becker, B. van der Wolf, L. Bohat’y, et al., Laser Phys. Lett. 5, 737 (2008). https://doi.org/10.1002/lapl.200810056
W. Zhao, L. Zhang, G. Wang, et al., Opt. Mater. 31, 849 (2009). doihttps://doi.org/10.1016/j.optmat.2008.09.010
R. D. Svetogorov, P. V. Dorovatovskii, and V. A. Lazarenko, Cryst. Res. Technol. 55, 1900184 (2020). https://doi.org/10.1002/crat.201900184
R. D. Svetogorov, Copyright Certificate of State Registration of Rights no. 2018660965.
V. Petricek, M. Dusek, and L. Palatinus, Z. Kristallogr. 229, 345 (2014). https://doi.org/10.1515/zkri-2014-1737
R. D. Shannon, Acta Crystallogr., Sect. A 32, 751 (1976).
A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions (Oxford University Press, New York, 1970).
G. G. Belford, R. L. Belford, and J. F. Burkhaven, J. Magn. Reson. 11, 251 (1973).
ACKNOWLEDGMENTS
The studies were carried out using the equipment of the Center for Collective Use of Physical Methods of Investigation at the Kurnakov Institute of General and Inorganic Chemistry (Russian Academy of Sciences) and the unique scientific installation “Kurchatov Synchrotron Radiation Source.”
Funding
This work was supported by budgetary financing (State registration no. 1021071612854-0-1.4.2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
ABBREVIATIONS AND NOTATION
Ln | lanthanides |
XRD | powder X-ray diffraction |
DSC | differential scanning calorimetry |
LT | low-temperature |
HT | high-temperature |
EPR | electron paramagnetic resonance |
REE | rare-earth elements |
KDP | potassium dihydrophosphate |
RE | rare-earth |
SH | spin-Hamiltonian |
Additional information
Translated by E. Yablonskaya
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Krut’ko, V.A., Komova, M.G., Svetogorov, R.D. et al. Features of the Structure and Thermal Properties of Borate Tungstates LnBWO6 (Ln = La; La0.999Nd0.001 and La0.99Gd0.01) Synthesized by the Sol–Gel Method. Russ. J. Inorg. Chem. (2024). https://doi.org/10.1134/S0036023624600394
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S0036023624600394