
Abstract
Two novel metal-organic coordination polymers [Sr(ttdc)(dma)2] (1) and [Zn(ttdc)(bpy)]·DMA·4H2O (2) (H2ttdc = thieno[3,2-b]thiophene-2,5-dicarboxylic acid, bpy = 2,2′-bipyridyl, DMA = N,N-dimethylacetamide) are synthesized under solvothermal conditions. Structures of the compounds are established by the single crystal X-ray diffraction (XRD) analysis. The coordination polymers are characterized by powder XRD, elemental and thermogravimetric analyses, and IR spectroscopy. Compound 1 is a 2D coordination polymer while compound 2 is composed of zigzag chains linked by π–π stacking between bpy molecules into a porous supramolecular framework with a free volume of 42%.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Interest in the chemistry of metal-organic framework structures (MOFs) has been constantly high in the recent decades owing to the rich structural diversity and intriguing properties of this class of materials [1-4]. The application of organic ligands whose structures contain electron-enriched heteroatoms affects a number of significant functional features of respective compounds. Thus, it is shown that a replacement of the “traditional” aromatic bridging ligand (terephthalate) by heterocyclic 2,5-thiophenedicarboxylate or 2,5-selenophenedicarboxylate substantially improves the sorption properties of porous MOFs due to additional induction dipole-dipole interactions between the framework and substrate molecules [5-8]. Moreover, electron-rich heterocyclic thiophene-based fragments are responsible for interesting luminescent and magnetic properties of corresponding MOFs [9-17]. Thieno[3,2-b]thiophene-2,5-dicarboxylic acid (H2ttdc) is a close structural analogue of 2,6-naphthalenedicarboxylic acid, one of the most often used bridging ligands in the chemistry of MOFs [18]. At present, the Cambridge Crystallography Data Center ([19, CSD v. 5.42, May 2021) contains almost 500 unique coordination compounds with the respective anion and only eight MOFs with the ttdc2– anion [20, 21]. Apparently, the elaboration of methods to synthesize MOFSs with bridging ttdc2– is important not only for the development of this insufficiently studied field but also for the preparation of new porous materials with improved functional properties. In this work, we synthesized two novel coordination polymers based on the bridging ttdc2–anion with double charged Sr(II) and Zn(II) cations, determined their crystal structures, and investigated their thermal stability.
EXPERIMENTAL
The synthesis of thieno[3,2-b]thiophene-2,5-dicarboxylic acid (H2ttdc) was performed by oxidizing thieno[3,2-b]thiophene-2,5-dicarbaldehyde with the Tollens reagent according to the procedure described by A. M. Samsonova (Journal of Structural Chemistry, 2019). The other precursors and solvents were used as commercially available reagents without additional purification.
IR spectra were measured in the range of 4000-400 cm–1 from pellets with KBr on a Scimitar FTS 2000 Fourier spectrometer. The thermogravimetric analysis (TGA) was conducted on a NETZSCH TG 209 F1 thermoanalyzer upon linear heating of the samples in the He atmosphere with a rate of 10 deg/min. The elemental analysis was carried out on a VarioMICROcube CHN-analyzer. Powder XRD patterns were recorded on a Shimadzu XRD 7000S diffractometer (CuKα radiation, λ = 1.54178 Å).
Synthesis of [Sr(ttdc)(dma)2] (1). Single crystals of compound 1 were obtained by heating a mixture of strontium(II) nitrate tetrahydrate (28.4 mg, 0.1 mmol), thieno[3,2-b]thiophene-2,5-dicarboxylic acid (H2ttdc) (22.8 mg, 0.1 mmol), and 2.5 mL of N,N-dimethylacetamide (DMA) in a glass vial with a tightly screwed lid, preventing the evaporation of the solvent, at 130 °C for 48 h. The colorless crystalline precipitate obtained was washed with DMA (3×5 mL) and dried in air. Yield: 6.9 mg (14%). Product composition and structure were determined by the single crystal XRD analysis. IR spectrum, ν, cm–1: 420 w, 509 w, 598 w, 684 m, 768 m, 792 w, 847 w, 905 w, 1102 w, 1182 w, 1230 w, 1329 s, 1383 s, 1411 s, 1485 s, 1558 s, 3068 w, 3087 w, 3447 w, sh. Found (%): C 38.9, H 4.6, N 6.2, S 12.1. Calculated for [Sr(ttdc)(dma)2]·0.1DMA·0.8H2O (C16.4H22.5N2.1O6.9S2Sr) (%): C 38.5, H 4.4, N 5.8, S 12.5.
Synthesis of [Zn(ttdc)(bpy)]·DMA·4H2O (2). Compound 2 was obtained by heating a mixture of zinc(II) nitrate hexahydrate (29.7 mg, 0.1 mmol), thieno[3,2-b]thiophene-2,5-dicarboxylic acid (H2ttdc) (22.8 mg, 0.1 mmol), 2,2′-bipyridyl (15.6 mg, 0.1 mmol), and 5 mL of N,N-dimethylacetamide in a glass vial with a tightly screwed lid, preventing the evaporation of the solvent, at 120 °C for 48 h. The colorless crystalline precipitate obtained was washed with DMA (3×5 mL) and dried in air. Yield: 52.2 mg (86%). Product composition and structure were determined by the single crystal XRD analysis. IR spectrum, ν, cm–1: 414 m, 424 m, 495 w, 517 w, 638 w, 652 w, 660 w, 669 w, 690 w, 732 m, 769 s, 789 s, 868 w, 901 w, 970 w, 1019 m, 1031 m, 1063 w, 1095 m, 1158 w, 1189 w, 1252 w, 1328 s, 1381 s, 1445 s, 1482 s, 1582 s, 1600 s, 3034 w, 3061 w, 3074 w, 3099 m, 3415 w, sh. Found (%): C 49.2, H 4.4, N 8.4, S 10.8. Calculated for [Zn(ttdc)(bpy)]·1.5DMA·0.5H2O (C24H24.5N3.5O6S2Zn) (%): C 49.1, H 4.2, N 8.3, S 10.9.
Single crystal XRD analysis. XRD data for 1 and 2 single crystals were collected at 100 K on the BELOK beamline [22, 23] in the Kurchatov National Research Center, using an area Rayonix SX165 CCD detector (λ = 0.74500 Å, ω-scanning with a step of 1.0°). Integration, absorption correction, and determination of unit cell parameters were carried out using the XDS program package [24]. Structures were solved using the SHELXT software [25] and refined by full-matrix LSM in the anisotropic (except hydrogen atoms) approximation using the SHELXL program [26]. Positions of hydrogen atoms of organic ligands were calculated geometrically and refined in the riding model. The crystal structure of 2 was solved in the chiral symmetry group, the refinement being performed with regard to racemic twining (the fraction of the second component was 0.477(19)). Attempts to pass to a centrosymmetric space group failed. A part of guest molecules in the structure of 2 is strongly disordered and cannot refined as a set of discrete positions. Therefore the composition of the guest subsystem in 2 was determined based on SQUEEZE/PLATON [27] (300 e– in 1000 Å3). Crystallographic data and details of XRD experiments are listed in Table 1. Full tables of interatomic distances and bond angles, atomic coordinates and displacement parameters have been deposited with the Cambridge Crystallography Data Center (CCDC 2102550-2102551).
RESULTS AND DISCUSSION
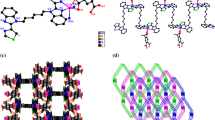
Compound [Sr(ttdc)(dma)2] (1) was synthesized by heating an equimolar mixture of Sr(NO3)2·4H2O and H2ttdc in N,N-dimethylacetamide at 130 °C. Despite a low yield (14%), it is formed as single crystals in reproducible amounts without any impurities. According to the single crystal XRD data, compound 1 crystallizes in the monoclinic crystal system and the symmetry space group I2/a with Z = 4. The asymmetric unit of the 1 structure contains one Sr2+ cation, one ttdc2– anion, and two N,N-dimethylacetamide molecules. The environment of the Sr2+ ion consists of four oxygen atoms of four bridging carboxyl groups and two oxygen atoms of cis-coordinated DMA molecules. Thus, the coordination number of the Sr atom is 6, and the coordination environment is a slightly distorted octahedron (Fig. 1). The Sr–O bond lengths are within 2.456(2)-2.513(3) Å, which indicates their mainly ionic character. The Sr2+ cations and carboxylate groups of ttdc2– ligands are organized in {Sr(RCOO)(DMA)2}1∞ chains along the а crystallographic axis (Fig. 1). Then by means of bridging ttdc2– ligands these fragments are linked into layers parallel to the aс crystallographic plane (Fig. 2) that alternate along the b axis as ABAB (Fig. 3), forming a 2D crystal packing of 1. The DMA ligands are coordinated to strontium ions so that they are alternatively above and below the coordination layer plane.
Colorless crystals of [Zn(ttdc)(bpy)]·DMA·4H2O compound (2) were obtained by heating an equimolar mixture of Zn(NO3)2·6H2O, H2ttdc, and bpy in N,N-dimethylacetamide at 120 °C. According to the single crystal XRD data, compound 2 crystallizes in the monoclinic crystal system and the chiral space group P21 with Z = 8. The asymmetric unit of the coordination structure of 2 contains four Zn(ttdc)(bpy) formula units, with the structures of the fragments being practically identical. Despite the similarity of different parts of the structure, our attempts to reveal the hidden symmetry elements failed.

Structure of the chain fragment in 1; for organic ligands only carboxylate groups (ttdc) and oxygen atoms (DMA) are shown.

Projection of the layer of coordination polymer 1 in the ac crystallographic plane; for N,N-dimethylacetamide molecules only oxygen atoms are shown; hydrogen atoms are omitted.

Layer packing in compound 1 (perspective view along the layers); for N,N-dimethylacetamide molecules only one of two disordered positions is shown; hydrogen atoms are omitted for clarity.
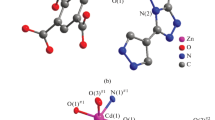
The Zn(II) atoms are surrounded by four oxygen atoms of two chelating carboxylate groups of ttdc2– ligands and two nitrogen atoms of bpy molecules (Fig. 4). The Zn–N bond lengths are within a narrow range of 2.060-2.103 Å whereas the Zn–O bond lengths are significantly different. Each carboxylate group coordinates to Zn2+ ions via one short Zn–O bond (1.985(7)-2.041(7) Å) and one long contact (2.323(7)-2.460(7) Å). The Zn(II) coordination number (CN) is 6, or if we take into account non-equal Zn–O interactions, CN = 4 + 2, with the shape of the coordination polyhedron being a distorted octahedron. The Zn(II) atoms are linked by ttdc2– ligands into zigzag chains {Zn(ttdc)(bpy)} whose periphery is decorated with chelating bpy ligands. The polymeric chains are packed into layers parallel to the ac plane, and the layers in turn alternate along the с axis. The neighboring layers are shifted relative to each other by half of the translation along the a axis. Interestingly that side bpy ligands of the neighboring chains are located in parallel above each other with a minor shift (Fig. 5a), forming short π–π contacts with average distances between the aromatic planes of 3.55 Å, which slightly exceeds the sum of carbon Van der Waals radii (1.7 Å). Due to these specific stacking contacts the zigzag chains are “glued” into a 3D supramolecular structure of 2, in which there are hourglass-shaped channels with a characteristic size of 13×6 Å and a narrow neck of 3 Å, which extend along the а axis (Fig. 5b). The calculated volume available for the accommodating guest molecules is 42% [27]. This space contains solvent molecules (1 DMA and 4 H2O per formula unit). The positions of guest N,N-dimethylacetamide molecules are found directly from the single crystal XRD data, and the water amount was determined with regard to the TGA data and the results of the SQUEEZE/PLATON procedure.

Fragment of the {Zn(ttdc)(bpy)} chain in compound 2; weak Zn⋯O interactions are shown by dashed lines.
The phase and chemical purity of compounds 1 and 2 is confirmed by chemical analysis, TGA, powder XRD data, and IR spectroscopy. Positions of reflections in the XRD diffraction pattern of the powder sample of 1 are well consistent with the theoretical ones calculated from the single crystal XRD analysis (Fig. 6). In the XRD pattern of compound 2 an appreciable small angle shift of reflections relative to theoretically expected ones is observed. We indexed the experimental data using the “Match!” program [28] and determined the monoclinic Р-cell with the following parameters: a = 7.6987(13) Å, b = 24.5374(13) Å, c = 28.9346(4) Å, β = 93.385(8)°, V = 5456.4 Å3. A small increase in the unit cell volume by ~4.5%, as compared to the single crystal XRD data, agrees with a general shift of reflections to the small angle range in the experimental powder XRD curve. The most remarkable relative change is concerned with а (+2.0%) and с (+3.2%) crystallographic parameters, which corresponds to an extension of zigzag chain fragments {Zn(ttdc)(bpy)} that, as we remember, are perpendicular to the b axis. Results of the chemical analysis (C, H, N, S) for 1 and 2 are consistent with the proposed composition of the compounds, which was determined from the single crystal XRD data with correction for a partial change in the composition of guest molecules during the contact of crystalline samples with the atmosphere. The IR spectrum of compound 1 exhibits bands corresponding to out-of-plane bending vibrations of the C–H bond at 768 cm–1. Low-intensity bands at 1102 cm–1 and 1182 cm–1 correspond to stretching vibrations of the C–N bond in DMA molecules. Characteristic intense bands at 1329 cm–1 and 1383 cm–1 belong to symmetric stretching vibrations of the С–О bond in carboxylate groups, and those at 1411 cm–1 and 1485 cm–1 belong to asymmetric stretching vibrations of the same bond. There is a band at 1558 cm–1 that corresponds to stretching vibrations of the С=O bond in the DMA molecule. Stretching vibrations of the C–H bond in methyl groups of the DMA molecule are shown by bands at 3068 cm–1 and 3087 cm–1, while a broad band at 3445 cm–1 corresponds to stretching vibrations of the О–Н bond in the water molecule. The IR spectrum of compound 2 also contains the bands of out-of-plane vibrations of the C–H band (769 cm–1), of stretching vibrations of the C–N bond in the DMA molecule (1095 cm–1 and 1158 cm–1), of symmetric (1328 cm–1 and 1381 cm–1) and asymmetric (1445 cm–1 and 1482 cm–1) stretching vibrations of the С–О bond in carboxylate groups and stretching vibrations of the С=O bond (1600 cm–1) in the DMA molecule. At 3061 cm–1 and 3074 cm–1 one can observe bands corresponding to stretching vibrations of the C–H bond in methyl groups of the DMA molecule, and a broad moderate-intensity band at 3415 cm–1 corresponds to stretching vibrations of the О–Н bond in the water molecule.

View of stacking interactions of bpy ligands from the neighboring chains (а), projection of the crystal packing in compound 2 (view in the bc crystallographic plane) (b); guest molecules and hydrogen atoms are omitted.
According to the TGA data (Fig. 7), compound 1 does not lose weight during heating to Т ~ 170 °C, and then there is an abrupt weight loss (Δm = 30%) caused by the removal of coordinated N,N-dimethylacetamide molecules. The coordination framework irreversibly destructs at Т ≥ 430 °С. Compound 2 has three steps of decomposition. The first step of the weight loss Δm = 12% in the range from the room temperature to Т ~ 130 °С may be explained by the removal of guest water molecules (calculation for 4Н2О: 12%). The second extended step in the range of 130-250 °С is characterized by the weight loss Δm = 14% and is associated with the removal of guest DMA molecules (calculation for 1DMA: 14%). The complete decomposition of the coordination polymer is observed at Т ≥ 280 °С. A substantially lower thermal stability of compound 2 in comparison with that of 1 is most likely to be due to the presence of coordinated bpy molecules in the latter. Their removal initiates the irreversible destruction of chains and pyrolysis of bridging organic ligands.

Comparison of experimental (black) and theoretical (gray) powder XRD patterns for compounds 1 and 2; for compound 2 the dark-gray powder XRD pattern is derived from the single crystal XRD data; the light-gray one was constructed after the refinement of parameters using the “Match!” program.

TGA curves for compounds 1 and 2.
CONCLUSIONS
Two new MOFs based on bridging thieno[3,2-b]thiophene-2,5-dicarboxylate ligands are synthesized and characterized. The compound with Sr(II) has a layered structure; the compound with Zn(II) is composed of coordination chains organized into a 3D supramolecular framework by specific π–π stacking interactions, and this framework contains channels filled with guest molecules.
REFERENCES
S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li, and H.-C. Zhou. Adv. Mater., 2018, 30, 1704303. https://doi.org/10.1002/adma.201704303
H.-C. Zhou and S. Kitagawa. Chem. Soc. Rev., 2014, 43, 5415-5418. https://doi.org/10.1039/c4cs90059f
Q. Wang and D. Astruc. Chem. Rev., 2020, 120, 1438-1511. https://doi.org/10.1021/acs.chemrev.9b00223
A. Kirchon, L. Feng, H. F. Drake, E. A. Josepha, and H.-C. Zhou. Chem. Soc. Rev., 2018, 47, 8611-8638. https://doi.org/10.1039/c8cs00688a
V. A. Bolotov, K. A. Kovalenko, D. G. Samsonenko, X. Han, X. Zhang, G. L. Smith, L. J. McCormick, S. J. Teat, S. Yang, M. J. Lennox, A. Henley, E. Besley, V. P. Fedin, D. N. Dybtsev, and M. Schröder. Inorg. Chem., 2018, 57, 5074-5082. https://doi.org/10.1021/acs.inorgchem.8b00138
P. A. Demakov, S. S. Volynkin, D. G. Samsonenko, V. P. Fedin, and D. N. Dybtsev. Molecules, 2020, 25, 4396. https://doi.org/10.3390/molecules25194396
M. Yoon and D. Moon. Microporous Mesoporous Mater., 2015, 215, 116-122. http://dx.doi.org/10.1016/j.micromeso.2015.05.030
J. Hu, Y. Liu, J. Liu, C. Gu, and D. Wu. Fuel, 2018, 226, 591-597. https://doi.org/10.1016/j.fuel.2018.04.067
J. Zhao, X.-L. Wang, X. Shi, Q.-H. Yang, and C. Li. Inorg. Chem., 2011, 50, 3198-3205. http://dx.doi.org/10.1021/ic101112b
V. A. Dubskikh, A. A. Lysova, D. G. Samsonenko, A. N. Lavrov, K. A. Kovalenko, D. N. Dybtsev, and V. P. Fedin. Molecules, 2021, 26, 1269. https://doi.org/10.3390/molecules26051269
R.-X. Yang, H.-M. Lan, P.-Y. Zhu, L.-Z. Yang, Y.-M. Yu, L.-L. Wang, and D.-Z. Wang. Inorg. Chim. Acta, 2020, 506, 119410. https://doi.org/10.1016/j.ica.2019.119410
L. Li, J.-Y. Zou, S.-Y. You, K.-H. Chen, J.-Z. Cui, and W.-M. Wang. Polyhedron, 2018, 141, 262-266. https://doi.org/10.1016/j.poly.2017.11.049.
S. Goswami, G. Leitus, and I. Goldberg. ChemistrySelect, 2017, 2, 2322-2329. http://dx.doi.org/10.1002/slct.201700136
S. Zhang, N.-X. Sun, L. Li, Z.-B. Han, and Y.-Z. Zheng. RSC Adv., 2014, 4, 5740-5745. https://doi.org/10.1039/c3ra44559c
V. A. Dubskikh, A. A. Lysova, D. G. Samsonenko, А. N. Lavrov, D. N. Dybtsev, and V. P. Fedin. Coord. Chem., 2021, 47, 598-603. https://doi.org/10.31857/S0132344X21100029
X. Chen and A. M. Plonka. Cryst. Growth Des., 2013, 13, 326-332. https://doi.org/10.1021/cg301471r
A. R. Balendra. J. Mol. Struc., 2017, 1131, 171-180. http://dx.doi.org/10.1016/j.molstruc.2016.11.029
J. B. Baruah. Coord. Chem. Rev., 2021, 437, 213862. https://doi.org/10.1016/j.ccr.2021.213862
C. R. Groom, I. J. Bruno, M. P. Lightfoot, and S. C. Ward. Acta Crystallogr., Sect. B, 2016, 72, 171-179. https://doi.org/10.1107/S2052520616003954
J. L. C. Rowsell and O. M. Yaghi. J. Am. Chem. Soc., 2006, 128, 1304-1315. https://doi.org/10.1021/ja056639q
K. Koh, A. G. Wong-Foy, and A. J. Matzger. J. Am. Chem. Soc., 2009, 131, 4184/4185. https://doi.org/10.1021/ja809985t
R. D. Svetogorov, P. V. Dorovatovskii, and V. A. Lazarenko. Cryst. Res. Technol., 2020, 55, 1900184. https://doi.org/10.1002/crat.201900184
V. A. Lazarenko, P. V. Dorovatovskii, Y. V. Zubavichus, A. S. Burlov, Yu. V. Koshchienko, V. G. Vlasenko, and V. N. Khrustalev. Crystals, 2017, 7, 325. https://doi.org/10.3390/cryst7110325
W. Kabsch. Acta Crystallogr., Sect. D, 2010, 66, 125-132. https://doi.org/10.1107/S0907444909047337
G. M. Sheldrick. Acta Crystallogr., Sect. A, 2015, 71, 3-8. https://doi.org/10.1107/S2053273314026370
G. M. Sheldrick. Acta Crystallogr., Sect. C, 2015, 71, 3-8. https://doi.org/10.1107/S2053229614024218
A. L. Speck. Acta Crystallogr., Sect. C, 2015, 71, 9-15. https://doi.org/10.1107/S2053229614024929
Match! Phase Analysis using Powder Diffraction. https://www.crystalimpact.com/match/Default.htm
Funding
The work was supported by the Russian Science Foundation (grant No. 18-13-00203, https://rscf.ru/project/18-13-00203/) and the Ministry of Science and Higher Education of the Russian Federation (project No. 121031700321-3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Russian Text © The Author(s), 2022, published in Zhurnal Strukturnoi Khimii, 2022, Vol. 63, No. 2, pp. 163-170.https://doi.org/10.26902/JSC_id87114
Rights and permissions
About this article

Cite this article
Dubskikh, V.A., Lysova, A.A., Samsonenko, D.G. et al. SYNTHESIS AND STRUCTURES OF COORDINATION POLYMERS BASED ON A BRIDGING LIGAND WITH THE THIENOTHIOPHENE BACKBONE. J Struct Chem 63, 227–234 (2022). https://doi.org/10.1134/S0022476622020032
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476622020032