
Abstract
The total content (cΣ) of toxic phenols in waters is usually determined by methods involving the introduction of a group reagent, measurement of the generalized signal (AΣ) at a selected wavelength, and estimation of cΣ in terms of C6H5OH. The use of diazotized sulfanilic acid as a group reagent allows one to determine cΣ with errors not exceeding 30 rel %. Further reduction of errors is possible with transition to multiple-wavelength measurements and multivariate calibrations, but for the determination of phenolic toxicants, these techniques have not been used before. To test this possibility, model mixtures (colored aqueous solutions) were prepared, simultaneously containing up to five different phenols with their total concentration from 15 to 70 μmol/L. The generalized signals were measured at m wavelengths in the UV region of the spectrum 10 min after mixing the solutions. Inverted multivariate calibrations were constructed on the basis of the AΣ values of n mixtures of the same type that formed the training set. Under optimized conditions (m = 7, n = 10), systematic errors (δc) in determining the amount of phenols in mixtures from the test sample did not exceed 13 rel %, which is half as much as when recalculating the signal to a standard substance. Obviously, it is advisable to use multivariate calibrations for a generalized assessment of phenolic pollution of water bodies instead of calculating total indices. However, in cases where the samples contained phenols that were not taken into account when constructing the calibration, an increase in systematic errors was observed, reaching as high as 80 rel % (in absolute value). Therefore, to use multivariate calibrations in hydrochemical analysis, a preliminary study of the qualitative composition of phenolic mixtures in waters of different types and taking into account the expected composition of samples when forming a training set are necessary.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Natural waters and wastewaters contain numerous phenolic compounds, many of which are dangerous toxicants. Phenols in water are rarely determined individually; instead, their total content (cΣ) is monitored, expressed as the phenol index (PhI) [1–3]. Unfortunately, determining the PhI often leads to significantly underestimated assessments of the total content [4–7]. This is due to the passivity of certain phenols in the reaction with 4-aminoantipyrine, intragroup signal selectivity, and the improper choice of standard substance. A good alternative to measuring the PhI is a recently developed method [8] involving the conversion of phenols into azo dyes by another reagent: diazotized sulfanilic acid (DSA). After a 10-min or 60‑min exposure, the generalized signal of phenols (AΣ) is measured at 360 nm and expressed relative to a standard substance (Xst) using one-dimensional calibration. This method allows for the assessment of the total content at levels of 10–6–10–4 mol/L; the errors of analysis are systematic and, with the correct choice of Xst, do not exceed 30 rel % in magnitude.
It is known that any assessments of cΣ obtained by recalculating AΣ to a standard substance Xst are metrologically incorrect [9, 10]. Measuring the total content of similar analytes in units of another physical quantity (Xst concentration) not only violates the principle of measurement unity but also increases the uncertainty of the results of analysis. This applies to the method [8] as well. More accurate methods for assessing cΣ are needed that do not require recalculating to Xst. In particular, generalized signals can be measured at multiple wavelengths and then cΣ can be determined using inverted multivariate calibration [11, 12]. In recent years, this approach has been successfully applied to determine the total content of carbohydrates [13], hydrocarbons [14], anthocyanins [15], and other groups of similar organic compounds. Multivariate calibrations (MC) have not been used previously to determine the total content of toxic phenols in natural waters or wastewaters. The possibility of accurately assessing cΣ using this method requires experimental verification, which was the aim of our research. Generalized signals were measured according to the procedure [8], multivariate calibrations were calculated and optimized, and then multicomponent aqueous solutions of phenols with different but known compositions were analyzed. The application of the optimized method in the analysis of different types of natural waters and wastewaters will be discussed in the next article.
EXPERIMENTAL
Objects of research. We used eight individual phenolic compounds containing from one to three hydroxyl groups (Table 1).
Initial aqueous or aqueous alcoholic solutions of phenols were prepared using precise weighed amounts of chemically pure reagents without additional purification. Working solutions were prepared on the day of the experiment, diluting the original ones with distilled water.
Model mixtures (colored multicomponent aqueous solutions) were prepared by mixing the calculated volumes of initial solutions of different phenols and diazotizing reagents. Single mixtures contained from two to five individual phenols, with molar ratios of different phenols in their mixture not exceeding 10 : 1 and total phenolic contents (cΣ) ranging from 15 to 70 μmol/L. Hereinafter, cΣ values are given in final dilution, i.e., after conversion of phenols to azo dyes. In total, more than 60 colored solutions with known cΣ values were prepared. A part of the mixtures (training set) was used for construction of multivariate calibrations. These mixtures contained Ph, N1, G, and R (Table 2). The mixtures of the first test sample had the same qualitative composition, but different ratios of components, which were used to check and compare the efficiency of different calibrations.
The composition of the mixtures for the formation of the second test sample included both the above and other phenols, namely, MC, N2, P, and PG. Mixtures from the third test sample contained only the last four phenols (Table 3). Each test sample contained seven mixtures of known composition. Thus, in contrast to a number of analogous studies, when testing the new methodology of group analysis, we deliberately used mixtures of not only the same but also a different qualitative composition than in the formation of the mathematical model.
Experimental technique. To convert individual phenols into azo dyes, 5.0 cm3 of 0.1 M NaHCO3 solution was introduced into a 50.00 cm3 volumetric flask (to create pH 7.4), along with distilled water to 2/3 of the volume of the flask and V (cm3) of working solution of the phenol under study. Then 1.0 cm3 of DSC solution with a concentration of 5.0 × 10–3 mol/L prepared from reagent of analytical grade according to the method [17] was added; the volume of the solution was brought to the mark with distilled water and mixed. The values of V were chosen so that the optical densities of the photometered solutions (AΣ) in the region of 350–410 nm were in the range from 0.1 to 1.0 units. In τ = 10 min after the addition of DSC, the absorption spectrum of the prepared solutions was recorded using an SF-2000 spectrophotometer in quartz cuvettes (l = 1.00 cm); the blank solution served as a reference solution. The AΣ values were measured at several (m) preselected analytical wavelengths (AWL). Similarly, multicomponent colored solutions were prepared and the generalized signals were measured. The spectra of each colored solution were recorded three times, the AΣ values obtained at the same AWL were averaged. The generalized signals had good precision: when remeasuring the optical density of one solution at any AWL, Sr < 1%, and when repreparing solutions, Sr < 3%. The formation and measurement of generalized signals are described in more detail in [8]. The additivity of generalized signals was checked using the 3S criterion [18].
Construction of multivariate calibrations. Inverted multivariate calibrations were constructed using Microsoft Excel with use of the formula
where cΣ is the total concentration of phenols in the ith mixture, Aij is the optical density of the ith mixture at the jth AWL, and bj is the regression coefficient for the jth AWL. Summation was carried out over all AWLs, the number of which was purposefully varied from three to ten during the experiment. The use of experimental data on n mixtures of the training set made it possible to form an overdetermined system of linear equations, which was solved with respect to coefficients by the least squares method (OLS algorithm [11]) using the Microsoft Excel software package. The determined values of bj were substituted into Eq. (1), obtaining the desired calibration. Thus, with m = 7 and n = 10, we obtained the following regression:
Substituting the values of Aj characterizing the next sample into the resulting equations led to results (\(c_{\Sigma }^{*}\)) close to the total content of phenols in this sample taking into account its dilution during the analysis. Naturally, when changing m and/or n, we obtained slightly different calibrations and slightly different values of \(c_{\Sigma }^{*}\) for the same mixtures.
Estimation of errors. Statistical processing of the results of the analysis of each mixture was carried out using the Student algorithm (n = 3; P = 0.95). The errors in the analysis of single mixtures were found according to the formula
Repeated photometry of the same mixture yielded highly reproducible values of \(c_{\Sigma }^{*}\) (Sr < 2%). The generalized error in the analysis of different mixtures included in a certain test set and analyzed with the help of some calibration was characterized by the RMSEC and RMSEP parameters [11], expressed in μmol/L, as well as in % of the average content of phenols in this set. Both parameters were calculated as follows:
RMSEC characterizes the adequacy of the model when applied to mixtures from the training set, and RMSEP characterizes the adequacy of the mixtures from the test sample.
To optimize the parameters m and n, the RMSEC or RMSEP values obtained using different models for the first test sample were compared. Maximum errors in the analysis of single mixtures were also taken into account.
RESULTS AND DISCUSSION
As previously established and confirmed during this study, phenolic toxicants in neutral aqueous solutions form stable azo dyes under the action of DSA (reagent), which absorb light well in the near UV region of the spectrum. The optical densities of these azo dyes in the concentration range of 10–5–10–4 mol/L are directly proportional to the concentration of the initial phenols (Fig. 1). The molar absorption coefficients of different azo dyes (ε) at the same wavelength vary, but in the range of 350–410 nm, they are of the same order of magnitude. For the set of phenols used, the ratio T = εmax/εmin does not exceed 7 units.

Univariate calibrations for the determination of individual phenols as azo dyes (pH 7.4, τ = 10 min, λ = 380 nm).
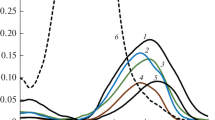
Deviations from additivity of analytical signals at the selected analytical wavelength in the vast majority of cases were found to be statistically insignificant, and in the remaining cases, they did not exceed 5 rel %. The relatively low level of intragroup selectivity and approximate additivity of signals provide the possibility of a correct estimation of cΣ when recalculated to a standard substance [8]. On the other hand, the mentioned characteristics of phenols and their corresponding azo dyes allow for the rapid construction of multidimensional linear models and their application for determining the total content of phenols according to the spectrum of the corresponding mixture. Examples of absorption spectra of mixtures of phenols after conversion of the components into azo dyes are shown in Fig. 2.

Absorption spectra of phenol mixtures after their interaction with DSA (CDSA = 100 μmol/L, pH 7.4, τ = 10 min): (41) N1 + N2 + R + G, 59.8 [mu]mol/L; (42) R + G, 15.3 μmol/L; (43) Ph + N1 + R, 34.9 μmol/L; (44) Ph + R + G, 45.4 μmol/L).
Selecting the optimal mathematical model. To select a mathematical model connecting the total concentrations of phenols and their generalized signals, two series of experiments were conducted. In the first series, absorption spectra of seven model mixtures from the training set were recorded in the range from 350 to 410 nm. Generalized signals were measured at different AWLs, gradually increasing their number from m = 3 to m = 10. The values of Aij for the mixtures from the first test sample were substituted into the obtained regression equations. For each mixture, the values of \(c_{\Sigma }^{*}\) were calculated, the relative errors of group analysis (δcj) were determined, and then the generalized error of analysis of these mixtures (RMSEP) was calculated. As expected, with a fixed volume of the training set, the values of individual errors and RMSEP decreased as the number of AWL increased. Reasonably accurate results (RMSEP ≈ 10% of the average phenol content) were obtained starting from m = 7. Further increasing the number of AWLs did not significantly improve the accuracy of the results, but complicated the procedure; therefore, it was deemed impractical.
In the second series of experiments, the number and set of used AWLs remained unchanged (m = 7), but the parameter n was varied from 5 to 16. Increasing the number of model mixtures in the training set initially led to a reduction in errors of group analysis and then to their increase (presumably due to the accumulation of random errors). The minimum value of RMSEP, equal to 7.8% of the average phenol content in the first test sample, was observed at n = 10. For further application, a multivariate calibration obtained at m = 7 and n = 10 was selected (see Eq. (2)). The total concentrations of phenols in mixtures from test sample no. 1 were determined using this calibration with individual errors not exceeding 13 rel % (in absolute value).
For comparison, the same mixtures were analyzed using the method [8], expressing the results in terms of the best standard substance (1-naphthol), i.e., in the form of an total index (TI). In this case, individual errors reached up to 23 rel %, and the RMSEP parameter was 12.8% of the average phenol content (Table 4, top line). It can be seen that, with the same qualitative composition of the samples and mixtures from the training set, the transition from calculating II to using inverted multivariate calibration reduces the errors in determining the sum of phenols by approximately half. It should be noted that, when determining the sum of hydrocarbons or the sum of aromatics, the transition from II to MC results in even greater gains in accuracy [14, 19].
Obviously, replacing the metrologically incorrect operation (calculating TI) with calculating the total phenol content using inverted MC should increase the accuracy of results in the analysis of natural waters and wastewaters. The development of corresponding methods has already begun. The main problem in this case is the choice of the optimal composition of the training set.
Influence of the qualitative composition of phenolic mixtures on the accuracy of analysis. It is known that the qualitative composition of calibration samples should match the composition of the samples being analyzed. However, it is not always possible to create a training set that includes all components of future samples. This is feasible, for example, in quantitative analysis of synthetic pharmaceuticals containing known sets of components, but impossible in the analysis of heavy petroleum products [11]. In the separate determination of components using multivariate calibration, the presence of foreign substances in the sample belonging to the same group of analytes but not accounted for in the calibration leads to systematic errors [20]. The influence of the same factor on the results of group analysis is difficult to predict and poorly studied [19, 21]. Selecting the composition of the training set for determining the total phenol content in natural waters and waste waters is challenging because the qualitative composition of phenolic mixtures in such waters varies significantly depending on the type of water and the source of phenol contamination [22, 23].
In the course of this study, it was necessary to determine to what extent the results of group analysis would be distorted when the composition of the samples being analyzed does not match the mixtures used to construct the simplified multivariate calibration. A comparison of the results of analysis of different test set (see Table 4) shows that the presence of “foreign” phenols significantly increases the absolute values of systematic errors in group analysis. This occurs both in the calculation of TI and when using simplified MC, and in such cases, the use of MC can lead to even greater (in absolute value) errors than the calculation of TI. A similar conclusion was previously drawn when studying the influence of foreign compounds on the results of determining the sum of arenes [19].
The influence of “foreign” phenols on the results of group analysis is explained by differences in sensitivity coefficients when determining different phenols in the form of azo dyes (see Fig. 1). For example, the simplified multivariate calibration (MC) in the range of 360–410 nm is determined with the same or slightly higher sensitivity than the phenols included in the training set. Therefore, the presence of mC in the mixtures from test sets 2 and 3 led to small, positively biased errors in absolute value. On the contrary, phenols that react slowly with the diazotized sulfanilic acid (H2, PG, and PC) were determined with lower sensitivity after a 10-min exposure compared to the components of the training set. Therefore, the presence of these phenols in the samples should lead to significantly underestimated results, as observed during the conducted experiment.
CONCLUSIONS
On the basis of the results of the experiment, the following conclusions and practical recommendations can be formulated.
(1) The possibility of determining the total content of phenols in the form of azo dyes has been confirmed. It has been established that the analysis of multicomponent aqueous solutions containing phenols at a level of 10–5 mol/L can be carried out in a metrologically correct way based on the construction of inverted multivariate calibrations.
(2) Using inverted multivariate calibrations, it is possible to obtain significantly more correct estimates of the total content of phenols than by calculating the corresponding total index. After optimizing the number of analytical wavelengths and the amount of samples in the training set, the single errors in the group analysis of mixtures from the test sample according to the corresponding multivariate calibration did not exceed 13 rel %, and the generalized error of analysis (RMSEP) was 7.8% of the average phenolic content. At the same time, the sensitivity, precision, and duration of analysis using the new method are close to the characteristics of traditional methods for assessing the total content of phenols.
(3) The main disadvantage of the new technique is the sensitivity of the result of analysis to the individual composition of the samples being studied. If there are phenols in the sample that were not taken into account when constructing a simplified multivariate calibration, the systematic errors of the group analysis increase sharply. In such cases, the use of II may be preferable. Particularly dangerous is the presence of phenols in the sample, which are determined with much greater or much less sensitivity than the phenols used to construct the MC. Therefore, the training set should include the widest possible set of phenols, including compounds determined with particularly high and particularly low sensitivity. This recommendation was confirmed experimentally using multivariate calibration constructed using ten model mixtures, including all eight phenols we used: the total contents of phenols in model mixtures from all three test samples were determined quite accurately (single errors were less than 15 rel %, RMSEP = 8.5%).
(4) The use of multivariate calibrations to determine the total content of phenols in wastewater, as well as in heavily polluted natural waters, is possible and advisable. It is necessary only to form multivariate calibrations in accordance with the expected composition of phenolic mixtures in the corresponding waters, identifying their main components using HPLC, and then include these phenols in the training set. Thus, to analyze waters of different types, different multivariate calibrations will be required. An alternative field of methodological research is the elimination of intragroup selectivity of analytical signals, which can lead to the creation of a unified calibration.
REFERENCES
Vorob’eva, T.V., Terletskaya, A.V., and Kushchevskaya, N.F., Standard and unified methods to determine the phenols in natural and waste water and he basic ways their development, Khim. Tekhnol. Vody, 2007, vol. 29, no. 4, pp. 370–390.
ISO 6439:1990(E): Water Quality. Determination of Phenol Index. 4-aminoantipyrine Spectrometric Methods after Distillation. https://cdn.standards.iteh.ai/samples/12795/21d5440a9f474ce09fd682a4c8084387/ISO-6439-1990.pdf. Accessed June 4, 2023.
ASTM D1783–01(2020): Standard Test Methods for Phenolic Compounds in Water. https://www.astm.org/ d1783-01r20.html. Accessed June 4, 2023.
Goerlitz, D.F. and Brown, E., Methods for Analysis of Organic Substances in Water, Washington: US Geol. Survey, 1984. https://doi.org/10.3133/twri05A3_1972
Khatmullina, R.M., Safarova, V.I., Magasumova, D.T., et al., Determination of some phenols in water by HPLC method with electrochemical detection, Bashkir. Khim. Zh., 2008, vol. 15, no. 3, pp. 48–52.
Tropynina, L.V., Kartashova, A.V., Zhylina, I.V., and Romanov, P.V., Reliability and informatic content of phenolic index, Metody Otsenki Sootv., 2012, no. 12, pp. 27–30.
Vershinin, V.I., Safarova, V.I., Antonova, T.V., and Khatmullina, R.M., Reliability of the assessment for indicator 'phenolic index', Metody Otsenki Sootv., 2013, no. 8, pp. 22–26.
Brilenok, N.S., Bakhareva, M.V., and Vershinin, V.I., UV-spectrometric determination of total phenols using diazotized sulfanilic acid, J. Anal. Chem., 2018, vol. 73, no. 6, pp. 551–559. https://doi.org/10.1134/S1061934818060047
Baena, J.R. and Valcarcel, M., Total indices in analytical sciences, Trends Anal. Chem., 2003, vol. 22, no. 10, pp. 641–649. https://doi.org/10.1016/S0165-9936(03)01101-4
International Vocabulary of Metrology: Basic and General Concepts and Associated Terms (VIM), 3rd ed., JCGM 200:2008, Paris: Int. Org. Legal Metrol., 2008.
Brereton, R.G., Introduction to multivariate calibration in analytical chemistry, Analyst, 2000, vol. 125, no. 11, pp. 2125–2154. https://doi.org/10.1039/B003805I
Vershinin, V.I., Methodological aspects of the group analysis of organic substances, J. Anal. Chem., 2023, vol. 78, no. 2, pp. 162–174. https://doi.org/10.1134/S1061934823020144
Rambla, F.J., Garrigues, S., and de la Guardia, M., PLS-NIR determination of total sugar, glucose, fructose and sucrose in aqueous solutions of fruit juices, Anal. Chim. Acta, 1997, vol. 344, pp. 41–53. https://doi.org/10.1016/S0003-2670(97)00032-9
Vershinin, V.I. and Petrov, S.V., The estimation of total petroleum hydrocarbons content in waste water by IR spectrometry with multivariate calibrations, Talanta, 2016, vol. 148, pp. 163–169. https://doi.org/10.1016/j.talanta.2015.10.076
Janik, L.J., Cozzolino, D., Dambergs, R., et al., The prediction of total anthocyanin concentration in red-grape homogenates using visible-near-infrared spectroscopy and artificial neural networks, Anal. Chim. Act-a, 2007, vol. 594, no. 1, pp. 107–118. https://doi.org/10.1016/j.aca.2007.05.019
GN (Hygienic Guidelines) 2.1.5.1315-03: Maximum Permissible Concentrations (MPC) of Chemical Substances in Waters, 2003.
Whitlock, L.R., Siggia, S., and Smola, J.E., Spectrophotometric analysis of phenols and of sulfonates by formation of an azo dye, Anal. Chem., 1972, vol. 44, no. 3, pp. 532–536. https://doi.org/10.1021/ac60311a021
Vershinin, V.I., Vlasova, I.V., and Tsypko, T.G., Exposure of deviations from additivity in the spectrophotometric analysis of unseparated mixtures, Met. Ob’ekty Khim. Anal., 2010, vol. 5, no. 4, pp. 226–233.
Vershinin, V.I. and Abramova, A.E., Summary determination of similar substances with total indices or multivariate calibrations for strong intragroup selectivity of signals, Zavod. Lab. Diagn. Mater., 2022, vol. 88, no. 10, pp. 5–12. https://doi.org/10.26896/1028-6861-2022-88-10-5-12
Esbensen, K.H., Multivariate Data Analysis – in Practice. An Introduction to Multivariate Data Analysis and Experimental Design, 5th ed., Woodbridge: Camo Process AS, 2004.
Vlasova, I.V. and Vershinin, V.I., Spectrometric determination of the total concentration of single-type analytes using conventional multidimensional calibrations, J. Anal. Chem., 2022, vol. 77, no. 1, pp. 35–42. https://doi.org/10.1134/S1061934822010142
Khatmullina, R.M., Safarova, V.I., and Latypova, V.Z., Reliability of the assessment of water pollution by petroleum hydrocarbons and phenols using some of total indices, J. Anal. Chem., 2018, vol. 73, no. 7, pp. 728–733. https://doi.org/10.1134/S1061934818070080
Bogolitsyn, K.G., Moskalyuk, E.A., Kostogorov, N.M., et al., Application of integral quality indicators of wastewaters in production ecological and analytical control of cellulose production, Khim. Rast. Syr’ya, 2021, no. 2, pp. 343–352. https://doi.org/10.14258/jcprm.2021027871
ACKNOWLEDGMENTS
We are grateful to I.V. Vlasova and N.S. Brilenok for consultations during the experiment and comments when summing up the results of the work.
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors of this work declare that they have no conflicts of interest.
Additional information
Translated by I. Moshkin
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vershinin, V.I., Bazhenova, L.S. Application of Inverted Multivariate Calibrations to Determine the Total Content of Phenols. Inorg Mater (2024). https://doi.org/10.1134/S0020168524700109
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1134/S0020168524700109