
Abstract
Using the cytochrome b gene (1143 bp), species identification and the phylogenetic analysis of voles of the generic group Microtus from the eastern part of the Greater Caucasus, including the Ismayilli, Khizi, and Balakan Districts of Azerbaijan, have been carried out. Three species, the Major’s pine vole (M. majori), the social vole (M. socialis), and the common vole (M. arvalis form obscurus), have been identified, and five new haplotypes have been described for them. Genetic analysis with the inclusion of the new data showed that for each of the species, the physiographic conditions of the Greater Caucasus played a certain role (isolation, migration route or refugium) during the formation of the modern genetic structure. The obtained results indicate that any new data from the Caucasus could be of critical importance for the reconstruction of the evolutionary history of the modern biodiversity both within the region itself and in adjacent territories.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
One of the urgent problems of evolutionary ecology is the study of the role of ecological and historical factors in the process of speciation. Voles of the of the generic group Microtus are a promising model for such studies. Microtus (tribe Arvicolini) is one of the most rapidly developing groups of mammals in the Northern Hemisphere, characterized by high rates of evolutionary transformations. This group is also the most recent radiation of voles, whose expansion was directly related to the dynamics of physical and geographical conditions in the Quaternary. Their modern morphological and genetic differentiation is due to the peculiarities of the geological history of individual regions against the background of global climatic changes [1–3]. Geological events significant for evolutionary transformations include the development of ice sheets, transgression and regression of sea basins and processes of orogenesis for mountainous regions. These areas include the Greater Caucasus, the geographical location (at the junction of Europe and Asia, in the contact zone of large zoogeographic regions) and the originality of the geological history of which determined its importance for the formation of individual taxa as a factor of isolation, migration route or refugium [4, 5].
Generic group Microtus in the territory of the Greater Caucasus is represented by European (M. arvalis Pallas, 1778 and M. rossiaemeridionalis Ognev, 1924), Asian (M. socialis Pallas, 1773) species and Caucasian endemics (M. majori Thomas, 1773, M. dagestanicus Shidlovsky, 1919). Significant information about the genetic variability, geographical distribution and ecological characteristics of the representatives of the group in the region was obtained based on the data of the analysis of karyotypes [6–8]. However, the complexity of species identification, taxonomy, and phylogeny of voles of the generic group Microtus requires genetic research using molecular-genetic approaches. For M. majori and Microtus rossiaemeridionalis, such studies started in the central and western parts of the Greater Caucasus [9–12], but there is practically no information for other regions and species. Therefore, we carried out species identification and an analysis of intraspecific phylogenetic relationships of voles of the generic group Microtus from the territory of Azerbaijan based on the sequences of the of cytochrome b gene (cyt b) of mtDNA.
For genetic analysis, we used samples of muscle tissue of five voles captured in three localities: Ismailli district, the village of Karakaya (40°47′ N, 48°18′ E), Khizi district, region of Sereki mountain (40°56′ N, 49°6′ E), Balakan district, Zagatala State Nature Reserve (41°46′ N, 46°27′ E). DNA isolation, amplification, sequencing, sequence processing, and phylogenetic reconstructions were performed using the methods described earlier [13]. The sequencing results were detected at the Center for Collective Use of the Institute of Plant and Animal Ecology of the Ural Branch of the Russian Academy of Sciences (1986).
Samples from Ismailli district were identified as common voles (M. arvalis) form obscurus, samples from Khizi district were defined as social voles (M. socialis), samples from Balakan district were determined as Major’s pine vole (M. majori). All sequences of cyt b (1143 bp) were new haplotypes (GenBank acc.no. OK376012–OK376016).
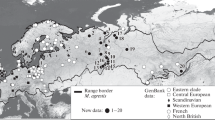
On a phylogenetic tree of M. majori, built on the basis of 14 known cyt b haplotypes (GenBank: AY513814 [1], KM656468–78 [9], DQ841703–04 [14]) and one new, two groups were distinguished (Fig. 1): group I, haplotypes from the territories of Kabardino-Balkaria, Stavropol krai and Krasnodar krai (valley of the Pslukh River); group II, haplotype from Azerbaijan, haplotypes from Turkey and Krasnodar krai (near Krasnaya Polyana).

Localization of the analyzed cyt b sequences (1143 bp) of M. majori within the range, the phylogenetic tree reconstructed using Bayesian analysis based on 15 haplotypes (above the branches, the probabilities BI > 0.70/ML > 50/NJ > 50) and the median network (M–JN) of these haplotypes (the numbers on the branches indicate the number of substitutions).
In group I, a separate subgroup was formed by the M. majori from the Stavropol krai, caught in the island forests of the Strizhament Mountain. In group II, haplotypes from the Krasnaya Polyana region (Krasnodar krai) were significantly differentiated, which, according to the median network, can be considered as an independent phylogenetic line. It should be noted that haplotype from this area, the valley of the Pslukh River, was also included in group I, and within the group demonstrated significant isolation from other haplotypes.
Revealed differentiation of M. majori does not contradict the results obtained earlier by the analysis of both mitochondrial and nuclear markers [9–11], and is most likely associated with the biotopic preferences of the species, the habitats of which are confined to the mountain-forest zone, and with the barrier role of high treeless mountain ranges of the Greater Caucasus. The formation of the modern intraspecific genetic diversity of the M. majori, probably was caused by fluctuations in the boundaries of the forest zone during periods of global climatic changes. The assumption that the differentiation of M. majori from the populations of Strizhament mountain was associated with the displacement of the boundaries of the forest zone in the western part of the Greater Caucasus during the last glaciation and subsequent isolation [9] is probably also true for the significant differentiation of populations from the vicinity of Krasnaya Polyana. Nevertheless, the limited amount of data and significant interpopulation genetic variability indicate the need for further studies of M. majori, including data from both mountainous and foothill regions of the Caucasus.
Haplotypes sequenced by us from the eastern part of the Greater Caucasus and five cyt b sequences of voles from the territory of Georgia, Iran, Crimea, and Kalmykia (GenBank: AY513829-31 [1], GQ352468 [2], KC953626 [15]) were included in the phylogenetic analysis of M. socialis, conducted for the species for the first time. The result showed significant differentiation of the haplotype from the Crimean isolate and the relative proximity of populations from the eastern part of the Greater Caucasus and from the western part of the Transcaucasia (Georgia) (Fig. 2). It is fundamentally important to assess the genetic diversity within the entire species range in the further analysis of the genetic structure of M. socialis and the reconstruction of its evolutionary history.

Localization of the analyzed cyt b sequences (1143 bp) of M. socialis within the range, the phylogenetic tree reconstructed using Bayesian analysis based on 7 haplotypes (above the branches, the probabilities BI > 0.70/ML > 50/NJ > 50) and the median network (M–JN) of these haplotypes (the numbers on the branches indicate the number of substitutions).
Unlike M. majori and M. socialis, a number of studies were dedicated to the analysis of genetic diversity of M. arvalis (the third species, defined in Azerbaijan) [13, 16–19]. The division of M. arvalis form obscurus into the Sino-Russian and South Caucasian clades was demonstrated [17], later the South Caucasian group was included in the Middle East clade together with the new Iranian group [19]. Reconstructions using our data (Fig. 3) indicated a more complex phylogenetic structure of M. arvalis: haplotypes of voles from Azerbaijan formed a separate group, which we called Caspian group, the level of differentiation of which was comparable to the Iranian and South Caucasian. The mutual proximity of the Transcaucasian groups and their isolation from the Sino-Russian clade on the median network suggest that the topology of the phylogenetic tree does not accurately reflect the real structure, probably due to the insufficient amount of data from the studied region. In this regard, we propose to consider the Iranian, South Caucasian, and Caspian groups as independent, the question of the relationship between which cannot be resolved without new data from the Caucasus and the Middle East.

Localization of the analyzed cyt b sequences (1143 bp) of M. arvalis within the range, the phylogenetic tree reconstructed using Bayesian analysis based on 114 haplotypes (above the branches, the probabilities BI > 0.70/ML > 50/NJ > 50; * means no-group for the used method) and the median network (M–JN) of these haplotypes (the numbers on the branches indicate the number of substitutions).
Thus, species identification of voles of generic group Microtus from previously unexplored populations of the eastern part of the Greater Caucasus was determined based on the data of the complete sequences of cyt b. Three species with different ecological and zoogeographic characteristics, which can be conditionally attributed to three different ecologo-faunistic groups were identified [20]: the Caucasian mountain-meadow mesophilic M. majori; the Near Eastern highland-steppe xerophilous M. socialis; and the European forest mesophilic M. arvalis form obscurus. Genetic analysis with the inclusion of new data suggests that, depending on the environmental preferences of the species, physiographic conditions of the Greater Caucasus played a certain role in the course of its evolutionary history. The obtained results indicate the importance of new, even limited, information about the populations of the Greater Caucasus, which may be of fundamental importance for the reconstruction of the pathways of the formation of modern genetic diversity both within the region and in adjacent territories.
REFERENCES
Jaarola, M., Martinkova, N., Gunduz, I., et al., Mol. Phylogen. Evol., 2004, vol. 33, no. 3, pp. 647–663.
Bannikova, A.A., Lebedev, V.S., Lissovsky, A.A., et al., Biol. J. Linn. Soc., 2010, vol. 99, no. 3, pp. 595–613.
Abramson, N.I., Bodrov, S.Y., Bondareva, O.V., et al., Mitochondrial genome phylogeny of voles and lemmings (Rodentia: Arvicolinae): Evolutionary and taxonomic implications. Preprint, 2021. https://doi.org/10.1101/2021.02.23.432437
Tembotova, F.A., Mlekopitayushchie Kavkaza i omyva-yushchikh ego morei. Opredelitel’ (Mammals of the Caucasus and Seas Surrounding It: An Identification Guide), Moscow: KMK, 2015.
Abdurakhmanov, G.M. and Batkhiev, A.M., Yug Ross. Ekol. Razv., 2013, no. 3, pp. 35–52.
Akhverdyan, M.R., Lyapunova, E.A., and Voron-tsov, N.N., Zool. Zh., 1992, vol. 71, no. 3, pp. 96–110.
Kuliev, G.N. and Bickham, J.W., Mus. Texas Tech. Univ., 2010, no. 291, pp. 1–14.
Malygin, V.M., Sablina, S.V., and Meier, M.N., Obyknovennaya polevka: Vidy-dvoiniki (The Common Vole: The Sibling-species), Moscow: Nauka, 1994.
Baskevich, M.I., Potapov, S.G., Khlyap, L.A., et al., Zool. Zh., 2015, vol. 94, no. 8, pp. 963–971.
Baskevich, M.I., Potapov, S.G., and Mironova, T.A., Zh. Obshch. Biol., 2015, vol. 76, no. 4, pp. 319–335.
Bogdanov, A.S., Khlyap, L.A., and Baskevich, M.I., Izv. Ros. Akad. Nauk Ser. Biol., 2020, no. 6, pp. 575–580.
Balakirev, A.E., Mironova, T.A., Khlyap, L.A., et al., Povolzh. Ekol. Zh., 2017, no. 1, pp. 14–23.
Sibiryakov, P.A., Tovpinets, N.N., Dupal, T.A., et al., Genetika, 2018, vol. 54, no. 10, pp. 1162–1176.
Martinkova, N., Zima, J., Jaarola, M., et al., Folia Zool.—Praha, 2007, vol. 56, no. 1, p. 39.
Kryštufek, B., Zorenko, T., and Buzan, E.V., Mammal. Biol., 2012, vol. 77, no. 3, pp. 178–182.
Tougard, C., Renvoise, E., Petitjean, A., et al., PLoS One, 2008, vol. 3, no. 10, e3532.
Tougard, C., Montuire, S., Volobouev, V., et al., Biol. J. Linn. Soc., 2013, vol. 108, no. 2, pp. 434–452.
Haynes, S., Jaarola, M., and Searle, J.B., Mol. Ecol., 2003, vol. 12 , no. 4, pp. 951–956.
Mahmoudi, A., Darvish, J., Aliabadian, M., et al., Mammalia, 2017, vol. 81, no. 6, pp. 583–593.
Batkhiev, A.M., Yug Rossii Ekol. Razv., 2014, no. 1, pp. 61–75.
Funding
This study was supported by the Russian Foundation for Basic Research (grant no. 19-04-00966).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All studies were carried out in accordance with the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (1986).
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
Additional information
Translated by V. Mittova
Rights and permissions
About this article

Cite this article
Yalkovskaya, L.E., Bol’shakov, V.N., Krokhaleva, M.A. et al. Genetic Diversity of Species of Microtus Generic Group (Arvicolinae, Rodentia): New Data from the Greater Caucasus. Dokl Biol Sci 502, 31–35 (2022). https://doi.org/10.1134/S0012496622010100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0012496622010100