
Abstract
Immune system and bone marrow stromal cells play an important role in maintaining normal hematopoiesis. Lymphoid neoplasia disturbs not only development of immune cells, but other immune response mechanisms as well. Multipotent mesenchymal stromal cells (MSCs) of the bone marrow are involved in immune response regulation through both intercellular interactions and secretion of various cytokines. In hematological malignancies, the bone marrow stromal microenvironment, including MSCs, is altered. Aim of this study was to describe the differences of MSCs’ immunological function in the patients with acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL). In ALL, malignant cells arise from the early precursor cells localized in bone marrow, while in DLBCL they arise from more differentiated B-cells. In this study, only the DLBCL patients without bone marrow involvement were included. Growth parameters, surface marker expression, genes of interest expression, and secretion pattern of bone marrow MSCs from the patients with ALL and DLBCL at the onset of the disease and in remission were studied. MSCs from the healthy donors of corresponding ages were used as controls. It has been shown that concentration of MSCs in the bone marrow of the patients with ALL is reduced at the onset of the disease and is restored upon reaching remission; in the patients with DLBCL this parameter does not change. Proliferative capacity of MSCs did not change in the patients with ALL; however, the cells of the DLBCL patients both at the onset and in remission proliferated significantly faster than those from the donors. Expression of the membrane surface markers and expression of the genes important for differentiation, immunological status maintenance, and cytokine secretion differed significantly in the MSCs of the patients from those of the healthy donors and depended on nosology of the disease. Secretomes of the MSCs varied greatly; a number of proteins associated with immune response regulation, differentiation, and maintenance of hematopoietic stem cells were depleted in the secretomes of the cells from the patients. Lymphoid neoplasia leads to dramatic changes in the functional immunological status of MSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Bone marrow is responsible for production and maintenance of blood cell populations, including immune cells, throughout the human life. In addition, it acts as an important lymphoid organ, housing many types of mature lymphocytes including B cells, T cells, natural killer T cells, and innate immune cells [1]. In bone marrow, lymphoid cells interact with the stromal microenvironment and are involved in regulation of hematopoiesis and immune response. Hematopoietic stem cells (HSCs) reside in the specialized niches that maintain them for lifelong blood cell production. Niches also provide homing and survival of HSCs, regulate their dormant state, self-renewal, differentiation, and proliferation. Many cell types are involved in formation and functioning of these niches [2]. Main niche components include mesenchymal stem cells, which differentiate into numerous other stromal cells that make up the niche, keep HSCs in it, and take part in immune responses in the bone marrow [3].
B-cell lymphoproliferative disorders, which include multiple myeloma, Hodgkin’s lymphoma, and chronic lymphocytic leukemia (CLL), as well as precursor conditions such as monoclonal B-cell lymphocytosis, are pathologies characterized by uncontrolled growth of B-lymphocytes [4]. Lymphoproliferative disorders can occur either in the lymphatic tissues (as in lymphoma) or in the bone marrow (as in CLL and multiple myeloma). Course of the disease and treatment vary widely depending on the type of neoplasia and other individual factors; however, even extramedullary tumors affect the bone marrow stromal microenvironment [5]. In addition, the bone marrow stroma can be strongly affected by chronic viral infections. For example, the CXCL12-abundant reticular cells (CAR cells) die in the mice with chronic lymphocytic choriomeningitis due to production of IFN-α and IFN-γ by the virus-specific CD8+ T cells [6]. Chronic viral infections are associated with hematopoiesis suppression, bone marrow failure, and depletion of the HSC pool [7, 8]. Combination of functional analysis with 3D microscopy demonstrated that chronic infection with lymphocytic choriomeningitis virus leads to the death of most mesenchymal CAR cells and pro-inflammatory transcriptional remodeling of the remaining ones. This causes long-term functional defects and reduced competitive repopulation ability of HSCs. Bone marrow immunopathology is caused by the virus-specific activated CD8+ T cells that accumulate in the bone marrow through the interferon-dependent mechanisms. Combined inhibition of the IFN type I and type II pathways by antibodies completely prevents CAR cell degeneration and protects HSCs from chronic dysfunction. Thus, viral infections and subsequent immune response have a lasting effect on the bone marrow homeostasis, permanently reducing repopulation ability of HSCs and disrupting secretion of the key stromal cytokines that support hematopoiesis [9].
Acute lymphoblastic leukemia (ALL) is a neoplasia arising from early progenitors of B-cells (B-ALL) or T-cells (T-ALL). The disease is characterized by uncontrolled proliferation of lymphoid progenitors in the bone marrow and consequent appearance of large numbers of immature lymphocytes, disrupting normal hematopoiesis. B-ALL is considered a genetic disease, but increasing evidence points to the ability of the bone marrow microenvironment to significantly contribute to maintenance, progression, response to treatment, and possibly development of the disease, regardless of the presence of specific genetic abnormalities in hematopoietic cells [10]. There is a large body of evidence suggesting that B-ALL cells can modify the bone marrow microenvironment creating conditions conducive to the survival of malignant cells during chemotherapy, leading to the disease recurrence. Leukemic cells interact with components of the bone marrow microenvironment, including multipotent mesenchymal stromal cells (MSCs) [11]. Studying interactions between the bone marrow microenvironment and ALL cells has led to the discovery of potential therapeutic targets that include cytokines/chemokines and their receptors, adhesion molecules, signal transduction pathways, and hypoxia-associated proteins [12]. Complex interactions between the leukemic cells and components of the bone marrow microenvironment lead to the involvement of MSCs in the suppression of antitumor response, since these cells secrete cytokines such as transforming growth factor-β (TGF-β) and hepatocyte growth factor (HGF), which mediate suppression of T-cells [13].
Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous group of diseases that differ in histological, immunohistochemical, and molecular characteristics [14]. In DLBCL, histological or molecular methods reveal damage to the bone marrow only in 10-25% of patients [15]. It is believed that in the remaining patients bone marrow is not involved in the malignant process. However, bone marrow stromal progenitor cells in the DLBCL patients without bone marrow involvement are altered [16]. These changes cannot be attributed to the contact interaction with tumor cells, as occurs in leukemia [17, 18]. Many tumors secrete cytokines and chemokines [19, 20], moreover, presence of a tumor can be considered an inflammatory process [21, 22]. Inflammation is associated with active release of multiple factors that can activate cells of the bone marrow stromal microenvironment, in particular MSCs [23]. MSCs activated by IL-1β, TNF, and IFN-γ secrete inhibitors and activators of the inflammatory process [24].
The aim of the study was to analyze the effect of lymphoid neoplasia from early progenitor cells in direct contact with the bone marrow stroma (ALL) and more mature cells located exclusively extramedullary (DLBCL) on immunological function of MSCs.
MATERIALS AND METHODS
Patient and donor bone marrow samples. Patient and donor samples were obtained in accordance with the Declaration of Helsinki after the written informed consent. The study was approved by the ethics committee of the Federal State Budgetary Institution National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation, protocol No. 171 dated April 27, 2023.
The work was performed with MSCs isolated from the bone marrow of patients with ALL and DLBCL; MSCs from the bone marrow of healthy donors were used as controls. To account for the age difference, two different donor groups were age-matched with the patients with ALL and DLBCL. As such, the studied parameters were normalized to the value of the corresponding donor group median. Data on the number of samples, patients, and healthy donors are presented in Table 1.
MSCs cultivation. Bone marrow was obtained from the patients during diagnostic punctures and from the hematopoietic stem cells donors during exfusions after informed consent. To prevent clotting, 2-7 ml of bone marrow were placed in sterile tubes with 1 ml of heparin (50 units/ml). The bone marrow samples were diluted 2-fold with an α-MEM (ICN, Canada) containing 0.2% methylcellulose (1500 cP, Sigma-Aldrich, USA) and left for 40 min at room temperature. Supernatant was collected and precipitated by centrifugation at 450g for 10 min. Number of nuclear cells was determined by counting after staining with gentian violet solution (1% solution in 3% acetic acid) in a Goryaev chamber. Cells (3×106) were placed in a flask with a bottom area of 25 cm2 (Corning-Costar, USA) in 5 ml of complete α-MEM nutrient medium (ICN, Canada) supplemented with 10% fetal bovine serum (FBS) (Hyclone, USA), 2 mM L-glutamine (ICN, Canada), 100 U/ml penicillin (Sintez, Russia), and 5 µg/ml streptomycin (BioPharmGarant, Russia). MSCs were cultivated at 37°C and 5% CO2. Culture medium was changed twice a week. After reaching confluency, the cells were passaged. To do this, the cells were washed twice with 5 ml of Versen’s solution and once with 0.25 ml of 0.25% trypsin solution (PanEco, Russia). 0.25 ml of trypsin solution was added, and flasks were left at room temperature until the cells detached from the surface. The cells were resuspended in 1 ml of the medium with FBS, and counted in 0.2% trypan blue solution (Sigma-Aldrich) to determine their number and viability (trypan blue only stains dead cells). During passage, 105 cells were seeded in a flask with a bottom area of 25 cm2 in 5 ml of the medium. Cultures were maintained for 4 passages.
The time to P0 was defined as the number of days from seeding bone marrow to reaching confluence for the first time.
Calculation of cumulative cell production. Cumulative cell production over 3 passages was calculated using the formula (1):
where N0, N1, N2, and N3 are number of the cells removed from 2 culture flasks at passages 0, 1, 2, and 3, respectively.
Surface marker expression analysis by flow cytometry. Surface phenotype of MSCs was studied at the 2nd passage by flow cytometry. After removing MSCs from the flask, they were washed twice with CellWash solution (BD Biosciences, USA) and then 2×104 cells were incubated for 20 min in the dark with antibodies. The antibody panels were as follows: 1) PE-labeled anti-CD90 (5E10, BD Pharmingen, USA), FITC-labeled anti-HLA-ABC (FN50, BioLegend, USA) and APC-labeled anti-HLA-DR (L243, BioLegend); 2) anti-CD105, labeled with FITC (43A3, BioLegend), anti-CD54, labeled with APC (HA58, BioLegend), anti-CD146 PE-labeled (P1H12, BD Pharmingen, USA); 3) PE-labeled anti-CD73 (AD2, BD Pharmingen, USA). The analysis was performed using a CytoFLEX flow cytometer (Beckman Coulter, USA), data were analyzed with Kaluza Analysis 2.1 (Beckman Coulter). MSC population was determined by forward and side light scattering. Mean fluorescence intensity (MFI) was assessed in APC, FITC, and PE channels.
Relative level of gene expression analysis. RNA isolation. To isolate RNA, the cells of the first passage (105-4.5×105 cells) were centrifuged at 300g. The pellet was washed with 1 ml of phosphate buffer and centrifuged at 300g. 400 µl of TriZol (Ambion by Life Technologies, USA) was added to the pellet. Samples with TriZol were frozen at –70°C. After thawing, 120 µl of chloroform was added to the samples, after which they were shaken, incubated for 2 min at room temperature, and centrifuged for 15 min at 13,500g and 4°C in a Centrifuge 5424 R (Eppendorf, Germany). The resulting upper phase was transferred into new tubes. 400 µl of isopropanol was added, the samples were incubated for 10 min at room temperature and centrifuged for 10 min at 13,500g and 4°C. The pellet was washed with 1 ml of 75% ethanol, vortexed, and centrifuged for 5 min at 13,500g at 4°C. The pellet was left to dry for 5 min at room temperature. Next, 100 µl of DEPC-treated water was added to the pellet and left for 30 min on ice for it to dissolve. After vortexing, 1 µl was taken to measure the amount of extracted RNA. The measurement was carried out with a NanoDrop One spectrophotometer (Thermo Fisher Scientific, USA) at a wavelength of 260 nm, RNA purity was determined by the ratio of 260/280 nm (it should be in the range of 1.8-2.0). To the remaining 99 μl of the RNA solution, 10 μl of 3 M sodium acetate and 250 μl of 96% ethanol were added. Samples were stored at –20°C.
cDNA synthesis. RNA in a mixture of ethanol and sodium acetate was centrifuged for 10 min at 13,500g and 4°C. After that, the pellet was washed with 1 ml of 75% ethanol, mixed on a vortex, and centrifuged for 5 min at 13,500g and 4°C. The pellet was left to dry for 5 min at room temperature. 1 µl of DEPC-treated water was added per 1 µg of RNA and the samples were left on ice for 30 min for dissolution. Primers for reverse transcription (T13 primers and random hexamers) were annealed: 2 µl of RNA solution, 1.25 µl of each primer (40 pmol/µl) and 5.5 µl of DEPC-treated water were mixed, incubated in a Tertsik amplifier (DNA-Technology) for 10 min at 70°C and 10 min at 4°C. After that, 15 μl of the reverse transcription mix (5.5 µl milliQ water, 5 µl 5X M-MLV reversease buffer (Promega, USA), 2.5 µl dNTPs mix, 1 µl each RNAsin (Promega) and M-MLV reversease (Promega)) was added, and the samples were incubated in a Tertsik amplifier at 42°C for 1 h. 75 μl of milliQ water were added. The samples were stored at –20°C.
Real-time PCR. Real-time PCR in Taq-man modification was performed with an AbiPrism Real Time PCR System 7500 (Thermo Fisher Scientific) in a 96-well plate; the reaction volume was 25 µl. Each sample was analyzed in triplicate; a positive control (a reference mixture of cDNA from 117 donors) was used to assess the quality of the reaction and correlate the results of different PCRs, and a negative control was included (water was added instead of cDNA). Sequences of primers and probes are presented in Table 2. PCR reagents were mixed into a master mix (12.8 µl milliQ water, 3.5 µl 25 mM MgCl2 (Thermo Fisher Scientific), 2.5 µl 2.5 mM dNTPs mix, 2.5 µl 10X SmarTaq buffer (Promega), 1 µl of forward and reverse primers each (10 pmol/µl), 0.5 µl fluorescent probe (10 pmol/µl), 0.2 µl Taq polymerase (Promega) per reaction). 72 µl of the master mix were placed into wells of a 96-well PCR plate and 3 µl of the cDNA solution was added. The samples were mixed and divided into 3 wells (25 µl per well) to obtain triplicates. PCR started with 10-min incubation at 95°C to activate the polymerase; 40 cycles of PCR were performed for the BACT and GAPDH genes, and 45 cycles for the remaining genes. Cycle parameters: 15 s at 95°C + 40 s at 60°C.
Calculation of relative gene expression level. Relative expression level (REL) of genes was calculated using the modified ΔΔCt method [25]. The mean Ct of three replicates was used for calculations. Ct is the number of cycles required to reach the probe fluorescence threshold in PCR. For each gene, ΔCt was calculated using the formula (2):
The RELs of the housekeeping genes (RELhk) – BACT and GAPDH – were calculated using the formula (3):
Next, the sample normalization factor (NF) was calculated using the formula (4):
RELs for the genes of interest were calculated using the formula (5):
Secretome analysis. Preparation of MSCs conditioned medium. MSCs at the passages 2-3 were seeded at 4×103 cells per cm2 in T175 flasks (Costar, USA). After the cells reached confluence (3-4 days), the flasks were washed 5 times with phosphate buffer without Ca2+/Mg2+ (Invitrogen, USA). The cells were then cultured for 24 h in RPMI 1640 medium without serum and phenol red (HyClone). The conditioned medium was centrifuged at 400g and frozen at –70°C.
Sample preparation for analysis. A protease inhibitor cocktail (Halt Protease Inhibitor Cocktail, Thermo Fisher Scientific) was added to each sample, which were then centrifuged at 1500g for 10 min to remove debris. Supernatants were immediately frozen and lyophilized to reduce volume. The lyophilized samples were resuspended for 30 min in a buffer containing 6 M Gd-HCl, 10 mM Tris-HCl (pH 8) and 2 mM DTT. To precipitate the insoluble fraction, the solutions were centrifuged at 16,000g for 10 min at 4°C. Samples were concentrated using a centrifuge filter (Corning Spin-X UF6, Sigma-Aldrich) to replace the buffer. Buffer (8 M urea, 2 M thiourea, 10 mM Tris-HCl (pH = 8)) was added to the concentrated samples at a ratio of 1 : 3 and incubated at room temperature for 30 min. Disulfide bonds were reduced with 5 mM DTT at room temperature for 40 min and next alkylated with 10 mM iodoacetamide in the dark at room temperature for 20 min. Alkylated samples were diluted by adding 50 mM NH4HCO3 solution at a ratio of 1 : 4 followed by trypsin addition (0.01 μg per 1 μg of protein) and incubation at 37°C for 14 h. The reaction was stopped by adding formic acid to a final concentration of 5%. The peptides were desalted using Discovery DSC-18 tubes (1 ml, 50 mg) (Sigma-Aldrich), dried under vacuum, and stored at –80°C before analysis. Prior to LC-MS/MS, samples were redissolved in 5% acetonitrile with 0.1% trifluoroacetic acid and sonicated.
LC-MS/MS analysis. Analysis was performed using an Orbitrap Q Exactive HF-X mass spectrometer equipped with a nano-ESI source and a high pressure nanoflow chromatograph (UPLC Ultimate 3000) with a C-18 (100 µm) reverse phase column. ×300 mm).
Bioinformatic processing of mass spectrometry data. Raw data from the mass spectrometer were converted into .mgf files using MSConvert (ProteoWizard Software Foundation, USA) with the parameters “--mgf --filter pickPicking true [1, 2]”. To identify proteins, a search was carried out using MASCOT (version 2.5.1, Matrix Science Ltd., UK) and X!Tandem (ALANINE, 2017.02.01, 2017.02.01, The Global Proteome Machine Organization) in the UniProt human protein database with concatenated backtrap dataset. Permissible masses of the precursor and fragment were set to 20 ppm and 0.04 Da, respectively. Database search parameters included the following: tryptic digestion with one possible gap [26], static modification for urea methyl (C), and dynamic modifications for oxidation (M). For X!Tandem, parameters were chosen that allowed to quickly check for the acetylation of the N-terminal residue of the protein, the loss of ammonia from the N-terminal glutamine and water from the N-terminal glutamic acid. The resulting files were processed in Scaffold 5 (version 5.1.0). An algorithm for estimating the local false discovery rate (FDR) with standard grouping of proteins was used. To assess the hits of peptides and proteins, FDR = 0.05 was chosen for both. The samples annotated in the Swiss-Prot database were marked as preferred.
Statistical analysis. Data sets are presented as individual values with indicated median. For each data set, a normality test was performed using the Shapiro–Wilk test (at p < 0.05, the distribution was taken to be different from normal). Significance of differences was analyzed using Mann–Whitney test for non-normal distributions. Differences were considered statistically significant at p < 0.05. Statistical analysis was performed using GraphPad Prism 8.03.
RESULTS
Under the influence of lymphoid neoplasia, the characteristics of MSCs change. Hematopoietic progenitors that initiate tumor development in ALL and DLBCL differ in the stage of differentiation: in ALL, early bone marrow progenitors undergo tumor transformation, while in DLBCL, it happens to more differentiated B cells outside the bone marrow. MSCs only from DLBCL patients without bone marrow involvement were included in this study.
MSCs growth characteristics. Time required for the MSCs of the patients at the onset of ALL to reach P0 increased in comparison to the time required for donor MSCs (p < 0.0001), which may indirectly reflect decrease in the number of stromal progenitor cells in the bone marrow. Upon reaching remission, this parameter returned to normal. MSCs of the patients with DLBCL did not differ from those of the healthy donors at the onset of the disease, and in remission they reached P0 faster than those of the donors (p = 0.0107). As a result, the time to P0 was longer in the cells from the patients with ALL than in the MSCs from the patients with DLBCL at the onset of the disease (Fig. 1a). In remission, the time to P0 did not differ significantly.

Time to passage 0 (P0) (a) and cumulative cell production in 3 passages (b) of MSCs from patients with ALL and DLBCL before treatment and in remission normalized to age-matched donor MSCs’ median value. Data are presented as scatterplots with indicated median. Horizontal red line indicates the median values of healthy donors’ cells.
Cumulative cell production of ALL patients’ MSCs in 3 passages did not differ from that of the healthy donors. When remission was achieved, total cell production of patients with ALL increased significantly compared to the donors (p = 0.0419). Cumulative cell production of the MSCs from the patients with DLBCL increased in comparison to the donors’ MSCs both at the onset (p = 0.0119) and in remission of the disease (p = 0.0011), and at the onset it was also significantly higher than at the onset of ALL (Fig. 1b).
Thus, we have shown that in ALL, the MSC growth is suppressed before treatment and returns to normal in remission, while in DLBCL, on the contrary, the MSCs grow more actively than the cells from the healthy donors. Other studied characteristics of the MSCs from the patients differed from the donors’ cells as well.
Membrane surface marker analysis. MSCs from the bone marrow of the patients with hematological diseases and the healthy donors differ in the expression of surface markers.
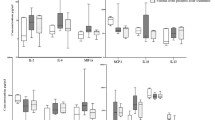
Mean fluorescence intensity (MFI) of the surface markers was studied on the cells of 6 patients before treatment of ALL and 3 in remission of ALL, 9 patients before treatment and in remission of DLBCL, and 10 donors for each of the patient groups. According to the International Society for Cellular Therapy (ISCT) criteria, MSCs express CD90, CD105, and CD73 [27]. These markers were present on all the studied cells, but their MFIs differed between the groups (Fig. 2, a-c). CD73 MFI was significantly increased on the MSCs from the patients with ALL before treatment compared to donors’ (Fig. 2c). This parameter did not differ from the donors neither at the onset nor in the remission of DLBCL. However, in the remission of DLBCL MFI of CD73 on the MSCs became significantly higher than it was before treatment. The MSCs of patients with DLBCL in remission had increased MFI of CD105 (Fig. 2b). Before the treatment of ALL, the observed expression of HLA-ABC on the cells was significantly increased compared to donors; in remission its MFI decreased (Fig. 2d). Before the treatment of DLBCL, HLA-ABC MFI on the cells did not differ from donors’, but in the remission it significantly increased. The same effect was observed for the class II histocompatibility molecules, HLA-DR, in DLBCL (Fig. 2e). Expression of CD146 on the surface of the MSCs in the patients with ALL demonstrated an upward trend compared to the donors (Fig. 2f). In the patients with DLBCL, it did not differ from the donors before treatment, but significantly increased when remission was achieved. There were no significant differences in the CD54 expression between the studied groups except for a significant increase in the patients in remission of DLBCL compared to the donors (Fig. 2g).

Mean fluorescence intensity (MFI) of the surface markers CD 90 (a), CD 105 (b), CD 73 (c), HLA-ABC (d), HLA-DR (e), CD146 (f), and CD54 (g) on the MSCs of patients with ALL and DLBCL, and healthy donors of corresponding ages. Y axis shows MFI fold change relative to the respective median value for the donor cells. Data are normalized to the median values for the donor groups and presented as scatterplots with marked median.
Significant changes in the expression of MSC surface markers were detected.
Changes in gene expression of factors important for immunoregulatory function of MSCs. Patterns of the MSCs gene expression were studied. The genes selected for analysis encode factors directly involved in the regulation of immune response – IL6, IL8, TGFB1; homing and adhesion factors – SDF1, ICAM1; growth factors and their receptors – FGF2, FGFR1, FGFR2, PDGFRB, VEGFA. Expression pattern of the MSC differentiation marker genes BGLAP, SPP1, SOX9, and PPARG was studied as well, since the differentiated descendants of MSCs – osteoblasts, adipocytes, etc. – participate in immune regulation.
In the patients with ALL, expression of the IL6 gene in MSCs was significantly increased in comparison to the donors at the onset of the disease (p < 0.0001) and did not normalize after achieving remission (p < 0.0001), and in DLBCL it increased after chemotherapy compared with the onset of disease (Fig. 3a) and the healthy donors (p = 0.0018). At the same time, at the disease onset, IL6 gene expression was higher in ALL than in DLBCL.

Relative expression levels of genes IL6 (a), IL8 (b), TGFB1 (c), FGF2 (d), FGFR1 (e), FGFR2 (f), PPARG (g), SOX9 (h), SPP1 (i), BGLAP (j), VEGFA (k), SDF1 (l), PDGFRB (m), ICAM1 (n) in the MSCs of the patients with ALL and DLBCL before treatment and in remission of the disease, normalized to the median REL of the group of corresponding healthy donor. Y axis shows mRNA expression level relative to respective median value for the donor groups. The data are presented as scatterplots with indicated median. Horizontal red line indicates the median REL of the donor MSCs.
Additionally, at the onset of DLBCL increase in the IL8 expression level relative to the healthy donors (p = 0.0220) was observed, and after treatment REL of this gene increased even more (Fig. 3b). TGFB1 REL was lowered in the MSCs of patients at the onset of ALL compared to donor cells (p = 0.004) and decreased even more in remission (p = 0.0002). In patients with DLBCL there was also decrease in the REL of this gene after treatment compared with the onset of the disease (Fig. 3c). In the MSCs of the patients with DLBCL at the onset and in the remission, the FGF2 REL was increased compared to donors’ (p = 0.0121, p = 0.0206, respectively), at the onset of the disease it was significantly higher than in the MSCs of ALL patients (Fig. 3d). FGF2 receptors differed in their expression in the patients with ALL and DLBCL (Fig. 3, e, f). At the onset of DLBCL, the MSCs had reduced FGFR1 REL (p = 0.0166) and increased FGFR2 REL (p = 0.0017). In the MSCs of the patients with ALL and DLBCL, FGFR1 REL was insignificantly reduced at the onset and remission of the disease, and FGFR2 expression was significantly elevated in the MSCs of the patients with DLBCL compared with the donors and significantly decreased when remission was achieved; such changes were not observed in the patients with ALL.
RELs of the MSC differentiation markers differed between the studied nosologies. REL of the adipose differentiation marker PPARG was increased in the MSCs of the patients with ALL at the onset of the disease compared to the donors (p < 0.0001) and remained elevated in the remission (p < 0.0001). At both points, PPARG gene expression in the MSCs was higher in ALL than in DLBCL (Fig. 3g). The REL of SOX9, a marker of chondrogenic differentiation, was lower in patients with ALL at the onset of the disease than in donors (p = 0.0049) and lower than in patients with DLBCL at the onset and remission (Fig. 3h).
In DLBCL, expression of some growth factors and receptors was also altered. In DBBCL remission, the expression of PDGFRB in MSCs was reduced in comparison with the donor MSCs (p = 0.0006) and with the onset of the disease (Fig. 3m). SDF1 REL was increased in DLBCL at the onset and remission of the disease compared with donors (p < 0.0001 and p = 0.0028, respectively, Fig. 3i). In ALL, VEGFA expression was reduced at the onset of the disease compared to donors (p = 0.0036), but was restored upon reaching remission (Fig. 3k). ICAM1 expression increased in ALL remission compared to the onset of the disease (Fig. 3n).
Thus, the RELs of growth factors, adhesion factors, and interleukins in the MSCs are altered in ALL and DLBCL. Changes in the MSC differentiation marker genes demonstrate propensity toward adipogenic differentiation in ALL.
Secretome. Secretomes of the MSCs of 2 patients with ALL, 6 patients with DLBCL, and 21 donors were studied. No significant changes corresponding to the changes in the gene expression were observed, which may be due to the small sample number or post-transcriptional modifications. However, there were changes in the secretion levels of the proteins for which gene expression was not studied in this work.
The patients with ALL are typically much younger than the patients with DLBCL, so two groups of donors were used as controls in the secretome analysis. The number of secretome proteins analyzed is shown in Fig. 4.

Diagram of distribution of the studied proteins secreted by MSCs in ALL and DLBCL at the onset and in remission of the disease. Number of proteins not secreted by the given group of MSCs but secreted by other groups is indicated.
938 proteins were commonly secreted by the MSCs of all studied groups. The MSCs from the ALL patients did not secrete 1444 proteins, while donors’ MSCs did not secrete 1140. DLBCL patients’ MSCs did not secrete 433 proteins, of which 31 were found in the secretomes of the MSCs from ALL patients. These include proteins important for regulation of immune response – PDGFA, POSTN, LGALS1, and KIT. The MSCs from both donors and DLBCL patients secreted 800 proteins, while the donor MSCs did not secrete 793 of the studied proteins.
In ALL, secretion of 70 proteins was increased before the treatment compared with donors, the most interesting of which are CXCL12, POSTIN, HLA-DRB, LGALS1. Additionally, secretion of the proteins regulating cell migration and related to cytoskeletal organization was increased (Fig. 5a). Secretion of 15 proteins, including VCAM1, CSF1, CTGF, ADAMTS1, was reduced. Those proteins are involved in vesicular transport and extracellular matrix organization (Fig. 5b).

Enrichment analysis of the signaling pathways with GO database. The most important signaling pathways, components of which were differentially found in the secretomes of MSCs from the patients with ALL and donors, are presented. Only significant changes (p < 0.05) were taken into account. Histograms represent -log10 FDR (false discovery rate) values. a) Signaling pathways components of which were upregulated in the secretomes of MSCs from the ALL patients before treatment compared to donors’ MSCs. b) Signaling pathways components of which were downregulated in the secretomes of MSCs from the ALL patients before treatment compared to donors’ MSCs. c) Signaling pathways components of which were upregulated in the secretomes of MSCs from the ALL patients before the treatment compared to remission. d) Signaling pathways components of which were downregulated in the secretomes of MSCs from the ALL patients before the treatment compared to remission.
After achieving remission of ALL, secretion of the proteins associated with the immune response by the MSCs changes (Fig. 5, c, d). Compared to the onset of the disease, secretion of the proteins involved in the functioning of chemokines and cytokines, vesicular transport, MSC differentiation, etc. increased. At the same time, other proteins involved in the same signaling pathways could decrease. Secretion of 62 proteins was increased relative to the donors, including PTMA, DCD, LIMCH1, and POSTIN, and secretion of 80 proteins was decreased, including VCAM1, LTBP1, C3, ANXA1, IGFBP-1, -3, and -6; LGALS1, ENO1.
The obtained data indicate significant impairment of the MSCs’ functional properties in ALL, both before treatment and in remission.
At the onset of DLBCL, secretion of 77 proteins was increased compared to the donors, including B2M, CD59, HLA-C, PDGFRA, CSF1, SOD1, CAPG, and levels of 44 proteins were reduced, including APOs, ACAN, S100A9, LTBP4. Proteins with the increased secretion are involved in cytokine signaling pathways, including TNF, interferons, and interleukins, and are involved in innate and adaptive immunity (Fig. 6a). Downregulated proteins are also involved in some of these pathways (Fig. 6b). Secretion of the proteins related to the MSC differentiation and complement system was reduced.

Enrichment analysis of the signaling pathways with GO database. The most important signaling pathways components of which were differentially found in the secretomes of MSCs from the patients with DLBCL and donors are presented. Only significant changes (p < 0.05) were taken into account. Histograms represent -log10 FDR (false discovery rate) values. a) Signaling pathways components of which were upregulated in the secretomes of MSCs from the DLBCL patients before treatment compared to donors’ MSCs. b) Signaling pathways components of which were downregulated in the secretomes of MSCs from the DLBCL patients before treatment compared to donors’ MSCs. c) Signaling pathways components of which were upregulated in the secretomes of MSCs from the DLBCL patients before treatment compared to remission. d) Signaling pathways components of which were downregulated in the secretomes of MSCs from the DLBCL patients before treatment compared to remission.
Upon achieving remission, secretion of 15 proteins by the patients’ MSCs increased compared to donors’ MSCs. With the exception of B2M, these proteins differ from those upregulated at the onset of DLBCL. Important signaling pathways altered upon achievement of remission include vesicular transport, immune response, and differentiation (Fig. 6c). Noteworthy is secretion of the proteins associated with antigen presentation by the major histocompatibility complex (MHC) class II. Under the standard cultural conditions, non-activated MSCs express extremely low levels of MHC class II, whereas the MSCs from the DLBCL patients express it much more actively.
At the same time, secretion of 73 proteins decreased, including APOs, ACAN, S100A9, LTBP4, PDGFA, CFHR1, C4BPA, and CRP. Secretion of APOs, ACAN, S100A9, LTBP4 remains reduced in the remission. Secretion of the proteins that are important for immune response, such as PDGFA and various complement components (CFHR1, C4BPA, and CRP) was decreased. Many of the downregulated proteins are involved in the immune system – complement system, activation of platelets and neutrophils (Fig. 6d).
The changes observed in the secretomes of the MSCs from the patients with ALL and DLBCL indicate systemic inflammation affecting bone marrow in both nosologies. In remission, secretion of the inflammation-related proteins decreases, but the secretome does not normalize.
None of the proteins with the altered secretion compared to the donors at the onset and remission of the disease matched between ALL and DLBCL. The same is true for the proteins that differ between the onset and the remission in ALL and DLBCL.
DISCUSSION
Inflammation involves not only the cells directly related to the immune system, but also many others, including MSCs. It has been shown in vitro that MSCs are able to modulate immune response both through direct intercellular interaction and through secretion of various factors. In addition, MSCs, being precursors of the bone marrow stromal microenvironment, regulate hematopoiesis and, as a result, formation of all cells of the immune system. Lymphoid tumors upset the balance of the immune system, not only by impairing lymphopoiesis, but also by altering other cells associated with the immune system.
Lymphoproliferative diseases considered in this work – DLBCL and ALL – differ in cell differentiation stage and localization. In ALL, early lymphoid progenitors are located in the bone marrow, while in DLBCL, a tumor of more mature B cells may not affect the bone marrow, as happened in patients whose MSCs were studied in this work. However, it is known that the properties of MSCs change significantly not only in ALL [28], when they are in close proximity to malignant cells and interact with them, but also in DLBCL, in the absence of direct contact [5].
In the case when the same number of bone marrow mononuclear cells is seeded, the time it takes to achieve confluency depends on the number of plastic-attaching cells capable of proliferating, and thus may indirectly reflect the number of mesenchymal precursors in the bone marrow. The increase in time to P0 only in the MSCs of the patients at the onset of ALL is most likely due to the fact that only in this case the MSCs are in direct contact with tumor cells and are damaged by them. This assumption correlates with the results of other studies indicating damage to the stromal microenvironment in acute leukemia [29-31]. In DLBCL, on the contrary, activation of the stromal microenvironment can be assumed, since increase in the cell production of these patients’ MSCs was observed both at the onset of the disease and in the remission. Moreover, in remission, the time to P0 was also reduced, i.e., concentration of the stromal precursors in the bone marrow increased.
It is believed that leukemia is associated with chronic inflammation [32, 33]. The study of surface phenotype of the MSCs in ALL patients showed increased MFI of the class I major histocompatibility complex (HLA-ABC) and ecto-5′-nucleotidase (CD73) proteins prior to the treatment. In addition, the MSCs of ALL patients before treatment secreted more HLA-DRB than the donor cells. The MSCs of patients at the onset of DLBCL did not differ from the cells of healthy donors in these parameters. However, upon reaching remission, surface expression of HLA of both classes (HLA-ABC and HLA-DR), CD73, CD54, and CD146 significantly increased. In the secretomes of DLBCL patients’ MSCs, content of HLA-C and B2M proteins was increased at the onset of the disease. In remission, B2M remained upregulated. These changes suggest that the MSCs, on which HLAs are typically weakly expressed, are in a pro-inflammatory environment in the bone marrow and remain activated upon transfer to culture. CD73 has an immunosuppressive effect through generation of adenosine. Increase in the surface expression of this marker indicates that the MSCs execute an anti-inflammatory effect. It is known that increased expression of CD73 frequently occurs in the tumor cells and cells of the tumor microenvironment [34-36]. It seems this immune escape mechanism is implemented by MSCs in ALL and DLBCL. CD54, or ICAM-1, is an adhesion molecule that is upregulated when the cells are activated [37, 38]. CD146 is an adhesion molecule considered to be a marker of the MSCs with high proliferative potential and ability to differentiate [39]. Based on this, one could expect faster growth or greater cell production by the MSCs with increased CD146 expression, which was observed in the cells of DLBCL patients after treatment. We studied the MSCs in the DLBCL patients without bone marrow involvement. In the MSCs of DLBCL patients, proliferative activation was not observed before treatment, but occurred in remission, which implies that activation might be associated with the treatment. Chemotherapy activates and severely damages immune system of the patients [40].
In remission of DLBCL, an increased content of CD105, or endoglin, was also found on the MSCs. Endoglin is involved in keeping hematopoietic stem cells in a dormant state along with TGFβ1 [41]. Expression level of the TGFB1 gene in the MSCs was reduced at the onset of ALL, which could contribute to tumor proliferation. After chemotherapy, TGFB1 REL was reduced in both ALL and DLBCL. Perhaps this is necessary to restore hematopoiesis and immunity, which are inevitably damaged during treatment.
Among the factors influencing the immune response, an increase in the REL of the pro-inflammatory cytokine IL6 was observed at the onset of ALL. It did not normalize upon reaching remission. MSCs could contribute to the increase in the IL6 level observed in the blood of the patients [42]. We did not find a significant increase in the IL6 level in the secretome of the MSCs, which could be due to insufficient number of samples and limitations of the method, since other groups observed increased secretion of IL6 by the MSCs from ALL patients using the ELISA method [43]. In the patients with DLBCL, increased IL8 expression was noted before the treatment and increased expression of IL6 and IL8 after the treatment. Upregulated expression of the pro-inflammatory interleukin genes in remission may reflect massive changes in immune processes associated with both chemotherapy and consequent infectious complications, which is consistent with the observations of other authors [9].
SDF-1 (CXCL-12) is important for interaction with HSC. Its expression in the MSCs of the patients did not differ from the healthy donors in either ALL or DLBCL. Previous studies have noted that SDF1 expression does not change in acute myeloid leukemia (AML) [44, 45], but the level of protein on the surface of the MSCs in patients is increased [46]. It has been shown that in ALL, on the contrary, concentration of SDF-1 in the bone marrow decreases [43, 47]. In the patients’ MSCs studied in this work, there was a trend towards increased expression of this factor. In the secretomes of those cells, SDF-1 was significantly upregulated. It can be assumed that regulation of SDF-1 in acute leukemia does not occur at the transcription level.
In both ALL and DLBCL, expression of cytokines involved in formation and regeneration of the stroma was altered. At the onset of ALL expression of VEGFA in the MSCs was downregulated despite the fact that VEGF-A concentration in the blood of the patients is increased [48, 49]. It is possible that the MSCs reduce expression of this growth factor due to a feedback loop, and malignant cells are responsible for its secretion [48]. In DLBCL patients’ MSCs, expression of many growth factors and their receptors was impaired. It is possible that these changes would have contributed to the spread of the tumor to bone marrow if the patients had remained without treatment.
In ALL, change in the relative levels of expression of the gene markers of MSC predisposition to differentiation lineages was observed. Increased expression of PPARG suggests that the MSCs of ALL patients are more prone to adipogenic differentiation than the MSCs of the healthy donors. This is also confirmed by a decrease in expression of SOX9, a chondrogenic differentiation marker, which, according to some studies, is able to inhibit differentiation of MSCs into adipocytes [50]. According to the literature data, the MSCs from ALL patients show an increased tendency to adipose differentiation [28, 43]. Decrease in SOX9 expression has also been noted [43]. It is worth emphasizing that the changes in expression of these genes persisted even after the patients achieved remission. Perhaps more serious changes occurring in ALL are caused by the contact with malignant cells.
In DLBCL, no changes in the RELs of the differentiation markers were noted in the MSCs.
The signaling pathways enrichment analysis showed an indirect relationship between the changes in the expression of IL8, IL6, TGFB1, VEGFA, PDGFRB, PPARG and the secretome.
The secretome analysis revealed decrease in the secretion of proteins involved in regulation of the immune response and vesicular transport both before and after the ALL treatment compared to the donors. Secretion of the vesicles is important for the regulatory and trophic functions of MSCs [51]. Dysregulation of the vesicular transport indicates the MSCs damage. The MSCs also interact differently with the extracellular matrix, which is involved in the regulation of hematopoietic stem cells and B-lymphopoiesis [52].
The proteins downregulated in ALL before treatment include: ADAMTS1 – an angiogenesis-inhibiting metalloprotease associated with VCAM1 (whose secretion is also reduced) [53]; CTGF, connective tissue growth factor, that plays an important role in some forms of cancer, fibrotic diseases, and in many biological processes, including cell adhesion, migration, proliferation, and angiogenesis [54]; CSF1, a macrophage growth factor, stimulates increase in phagocytic and chemotactic activity of macrophages and monocytes, as well as cytotoxicity against tumor cells [55].
Factors with increased secretion in the MSCs of ALL patients before treatment compared to the donors regulate cell migration and are associated with actin cytoskeleton rearrangement. Those can attract both immune cells and circulating tumor cells to the bone marrow.
Presence of a tumor in the body is usually associated with chronic inflammation [21, 22]. Our observations are consistent with this hypothesis. Upon achieving remission of ALL, the MSCs reduced secretion of the factors involved in the response to proinflammatory cytokines, such as IL-1, TNF, and others (Fig. 5b). Secretion of some of these factors was decreased before the treatment and increased when remission was achieved (Fig. 5d). Several signaling pathways have components that were differentially secreted: a part of them was upregulated and a part was downregulated in the same groups, suggesting that some of the changes may be compensatory.
The list of proteins with secretion reduced in ALL remission includes C3 complement component; insulin-like growth factor binding proteins – IGFBP1, IGFBP3, which regulate cell growth, and IGFBP6, which regulates growth and immune response of dendritic cells [56]; LGALS1 or galectin-1, a protein that inhibits cell proliferation and is involved in immunosuppression of CD8+ T cells [57]; ENO1, a glycolytic enzyme that functions as a tumor suppressor and is important for chemoresistance in lymphomas [58]; ANXA1, annexin A1, that inhibits innate immune cells and promotes T cell activation. Activation of T cells results in the release of annexin A1 and expression of its receptor. This pathway appears to finely regulate the strength of T cell receptor (TCR) signaling. Increased expression of annexin A1 under pathological conditions could enhance TCR signaling through the mitogen-activated protein kinase (MAPK) signaling pathway, thereby causing T cell hyperactivation [59]. With the decrease in secretion of these proteins, the tumor more easily escapes immune surveillance, partially provided by the MSCs. It can be concluded that the MSCs of ALL patients undergo a complex of changes that contribute to imbalance of immune system and escape of the tumor cells from immune surveillance.
The MSCs of the DLBCL patients included in the study were immunologically activated, despite the fact that the bone marrow was not directly involved in the tumor process. This is indicated by the increased secretion of the proteins from the signaling pathways that mediate the immune response, including the presentation of antigens by the MHCs and the response to proinflammatory cytokines. After treatment, secretion of the proteins related to inflammatory processes decreased.
In the secretome of MSCs of the patients both before and after DLBCL treatment, secretion of ACAN, S100A9, LTBP4, and a number of APOs (apolipoproteins) was downregulated compared to donors. When remission was achieved, secretion of PDGFA, CFHR1, C4BPA, and CRP decreased as well.
ACAN, or aggrecan, is critical for cartilage skeletal morphogenesis during development and is expressed by chondrocytes [60]. Lack of expression of this protein could be associated with impaired cartilage and bone differentiation in the patients with DLBCL. In addition, aggrecan could be involved in the antigen presentation, as shown for the chondrocytes during inflammation [61].
The S100A9 protein is calcium-binding protein A9, also known as migration inhibitory factor-related protein 14 (MRP14). Intracellular S100A9 is known to reduce the ability of neutrophils to respond to bacterial pathogens [62].
Decrease in the LTBP4 protein, a key regulator of transforming growth factor beta (TGFB1, TGFB2, and TGFB3), which controls activation of TGF-β maintaining it in a latent state during storage in the extracellular space, is associated with the functions of TGF-β. TGF-β is biologically latent after secretion. Thus, LTBP4 is an important regulator of TGF-β signaling and is indirectly associated with the development, immunity, injury recovery, and disease, playing a central role in regulation of inflammation, fibrosis, and cancer progression [63]. CFHR1, C4BPA, CRP are elements of the complement system that are normally secreted by the cells, including MSCs and pericytes, to protect the body from infections [64].
Decrease in all these proteins in the secretome of MSCs confirms deterioration of their physiological functions associated with hematopoiesis and immunity in the DLBCL patients.
Increased secretion of B2M, CD59, HLA-C, PDGFRA, CSF1, SOD1, CAPG at the onset of DLBCL in the MSC secretome indicates MSC activation in the bone marrow of the DLBCL patients without tumor involvement. In the remission, the level of these proteins, with the exception of B2M, normalized, which implies that these changes are partially reversible.
Comparison of MSC secretomes of patients with ALL and DLBCL confirmed the presence of significant functional changes in comparison with the donors and the difference of the nosologies from each other. The MSCs from the DLBCL patients did not secrete many proteins secreted by the MSCs from the donors and the patients with ALL; proteins involved in regulation of the immune response, PDGFA, POSTN, and LGALS1 (reduced in the secretomes of MSCs in ALL) and KIT were not found in their secretomes. POSTN is a secreted extracellular matrix protein that is involved in tissue development and regeneration, binds to integrins to support adhesion and migration, and plays a role in cancer stem cell maintenance and metastasis [65]. Growth factors that support stromal cells and hematopoietic stem cells are also absent in the secretomes of MSCs from the DLBCL patients, which confirms abnormalities in the bone marrow MSCs without direct contact with the tumor B cells.
Thus, regardless of the tumor location, there are significant changes in the functional status of bone marrow MSCs, which contribute to the decrease in antitumor immunity in the lymphoid neoplasia. In addition, in both pathologies, the MSCs acquire features of the senescent cells associated with chronic inflammation. The MSC senescence may be the reason for incomplete restoration of hematopoiesis for a long period after achieving remission.
Analysis of the functional status of MSCs revealed both differences and similarities between the impairments occurring in the MSCs during the transformation of early hematopoietic precursors in the bone marrow (ALL) and more mature ones outside the bone marrow (DLBCL). Some of the differences may be related to the participation of the bone marrow lymphocytes in the MSC differentiation [66]. In ALL, predisposition of the MSCs to bone and fat differentiation changes as evidenced by the gene expression and protein secretion. Some of the differences between the DLBCL and ALL are age related. In DLBCL, signaling pathways of the growth factors IGF and PDGF in the MSCs are altered.
Identification of the specific signs in the bone marrow stroma indicates the possibility of the relapse risk assessment [46]. A more detailed study of the metabolism of MSCs from the patients will allow for identification of the drugs that could modulate reactive oxygen species associated with inflammation and cell aging.
CONCLUSION
Lymphoid neoplasias have a pathological effect on the function of bone marrow MSCs. Concentration of these cells in the bone marrow, their ability to proliferate, immunophenotype, and expression pattern of the genes important for differentiation, maintenance of immunological status, and expression of cytokines change. The functions of MSCs are diverse and form a complex of reactions to the state and demands of the body. Changes in the immunological status of MSCs depend on the nosology and, despite the fundamental differences, generally contribute to optimizing the niche for the needs of the tumor and tumor escape from immunological surveillance. The obtained data demonstrate the importance of MSCs for immunity and imply the possibility to target not only the malignant cells, but their microenvironment as well. Lymphoid tumors are not only the transformation of hematopoietic cells of varying degrees of maturity, but also concomitant changes in the stromal microenvironment.
Abbreviations
- ALL:
-
acute lymphoblastic leukemia
- Ct:
-
number of cycles required to reach the probe fluorescence threshold in PCR
- DLBCL:
-
diffuse large B-cell lymphoma
- HSCs:
-
hematopoietic stem cells
- MFI:
-
mean fluorescence intensity
- MSCs:
-
multipotent mesenchymal stromal cells
- REL:
-
relative expression level
References
Schürch, C. M., Caraccio, C., and Nolte, M. A. (2021) Diversity, localization, and (patho)physiology of mature lymphocyte populations in the bone marrow, Blood, 137, 3015-3026, https://doi.org/10.1182/blood.2020007592.
Kaushansky, K., and Zhan, H. (2018) The regulation of normal and neoplastic hematopoiesis is dependent on microenvironmental cells, Adv. Biol. Regul., 69, 11-15, https://doi.org/10.1016/J.JBIOR.2018.06.003.
Beerman, I., Luis, T. C., Singbrant, S., Lo Celso, C., and Méndez-Ferrer, S. (2017) The evolving view of the hematopoietic stem cell niche, Exp. Hematol., 50, 22-26, https://doi.org/10.1016/j.exphem.2017.01.008.
Luca, D. C. (2021) Update on lymphoblastic leukemia/lymphoma, Clin. Lab. Med., 41, 405-416, https://doi.org/10.1016/J.CLL.2021.04.003.
Petinati, N. A., Bigildeev, A. E., Karpenko, D. S., Sats, N. V., Kapranov, N. M., Davydova, Y. O., Fastova, E. A., Magomedova, A. U., Kravchenko, S. K., Arapidi, G. P., Rusanova, M. I., Lagarkova, M. M., Drize, N. I., and Savchenko, V. G. (2021) Humoral effect of a B-cell tumor on the bone marrow multipotent mesenchymal stromal cells, Biochemistry (Moscow), 86, 207-216, https://doi.org/10.1134/S0006297921020097.
Zhou, X., Ramachandran, S., Mann, M., and Popkin, D. L. (2012) Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: past, present and future, Viruses, 4, 2650-2669, https://doi.org/10.3390/v4112650.
Schürch, C. M., Riether, C., and Ochsenbein, A. F. (2014) Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells, Cell Stem Cell, 14, 460-472, https://doi.org/10.1016/j.stem.2014.01.002.
Demerdash, Y., Kain, B., Essers, M. A. G., and King, K. Y. (2021) Yin and Yang: the dual effects of interferons on hematopoiesism, Exp. Hematol., 96, 1-12, https://doi.org/10.1016/j.exphem.2021.02.002.
Isringhausen, S., Mun, Y., Kovtonyuk, L., Kräutler, N. J., Suessbier, U., Gomariz, A., Spaltro, G., Helbling, P. M., Chyn Wong, H., Nagasawa, T., Manz, M. G., Oxenius, A., and Nombela-Arrieta, C. (2021) Chronic viral infections persistently alter marrow stroma and impair hematopoietic stem cell fitness, J. Exp. Med., 218, e20192070, https://doi.org/10.1084/jem.20192070.
Fallati, A., Di Marzo, N., D’amico, G., and Dander, E. (2022) Mesenchymal stromal cells (MSCs): an ally of B-cell acute lymphoblastic leukemia (B-ALL) cells in disease maintenance and progression within the bone marrow hematopoietic niche, Cancers, 14, 3303, https://doi.org/10.3390/CANCERS14143303.
Kihira, K., Chelakkot, V. S., Kainuma, H., Okumura, Y., Tsuboya, N., Okamura, S., Kurihara, K., Iwamoto, S., Komada, Y., and Hori, H. (2020) Close interaction with bone marrow mesenchymal stromal cells induces the development of cancer stem cell-like immunophenotype in B cell precursor acute lymphoblastic leukemia cells, Int. J. Hematol., 112, 795-806, https://doi.org/10.1007/s12185-020-02981-z.
Chiarini, F., Lonetti, A., Evangelisti, C., Buontempo, F., Orsini, E., Evangelisti, C., Cappellini, A., Neri, L. M., McCubrey, J. A., and Martelli, A. M. (2016) Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: from biology to therapeutic targeting, Biochim. Biophys. Acta, 1863, 449-463, https://doi.org/10.1016/j.bbamcr.2015.08.015.
Zhi-Gang, Z., Wei-Ming, L., Zhi-Chao, C., Yong, Y., and Ping, Z. (2008) Immunosuppressive properties of mesenchymal stem cells derived from bone marrow of patient with hematological malignant diseases, Leuk. Lymphoma, 49, 2187-2195, https://doi.org/10.1080/10428190802455875.
Beltran, B. E., Quiñones, P., Morales, D., Malaga, J. M., Chavez, J. C., Sotomayor, E. M., and Castillo, J. J. (2018) Response and survival benefit with chemoimmunotherapy in Epstein-Barr virus-positive diffuse large B-cell lymphoma, Hematol. Oncol., 36, 93-97, https://doi.org/10.1002/hon.2449.
Swerdlow, S. H. (2017) in WHO Classification of Tumours (Swerdlow, S. H., Campo, E., Harris, N. L., Jaffe, E. S., Pileri, S. A., Stein, H., and Thiele, J., eds) Lyon, International Agency for Research on Cancer, Lyon, pp. 199-215.
Fastova, E. A., Magomedova, A. U., Petinati, N. A., Sats, N. V., Kapranov, N. M., Davydova, Y. O., Drize, N. I., Kravchenko, S. K., and Savchenko, V. G. (2019) Bone marrow multipotent mesenchymal stromal cells in patients with diffuse large B-Cell lymphoma, Bull. Exp. Biol. Med., 167, 150-153, https://doi.org/10.1007/s10517-019-04480-6.
Chen, Q., Yuan, Y., and Chen, T. (2014) Morphology, differentiation and adhesion molecule expression changes of bone marrow mesenchymal stem cells from acute myeloid leukemia patients, Mol. Med. Rep., 9, 293-298, https://doi.org/10.3892/mmr.2013.1789.
Shipounova, I. N., Petrova, T. V., Svinareva, D. A., Momotuk, K. S., Mikhailova, E. A., and Drize, N. I. (2009) Alterations in hematopoietic microenvironment in patients with aplastic anemia, Clin. Transl. Sci., 2, 67-74, https://doi.org/10.1111/j.1752-8062.2008.00074.x.
Kotyza, J. (2017) Chemokines in tumor proximal fluids, Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub., 161, 41-49, https://doi.org/10.5507/bp.2016.062.
Vilgelm, A. E., and Richmond, A. (2019) Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy, Front. Immunol., 10, 333, https://doi.org/10.3389/fimmu.2019.00333.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008) Cancer-related inflammation, Nature, 454, 436-444, https://doi.org/10.1038/nature07205.
Okrój, M., and Potempa, J. (2019) Complement activation as a helping hand for inflammophilic pathogens and cancer, Front. Immunol., 9, 3125, https://doi.org/10.3389/fimmu.2018.03125.
Lund, F. E. (2008) Cytokine-producing B lymphocytes-key regulators of immunity, Curr. Opin. Immunol., 20, 332-338, https://doi.org/10.1016/j.coi.2008.03.003.
Munir, H., Ward, L. S. C., and McGettrick, H. M. (2018) Mesenchymal stem cells as endogenous regulators of inflammation, Adv. Exp. Med. Biol., 1060, 73-98, https://doi.org/10.1007/978-3-319-78127-3_5.
Schmittgen, T. D., and Livak, K. J. (2008) Analyzing real-time PCR data by the comparative CT method, Nat. Protocols, 3, 1101-1108, https://doi.org/10.1038/nprot.2008.73.
Kumar, L. P., Kandoi, S., Misra, R., and Verma, R. S. (2019) The mesenchymal stem cell secretome: a new paradigm towards cell-free therapeutic mode in regenerative medicine, Cytokine Growth Factor Rev., 46, 1-9, https://doi.org/10.1016/j.cytogfr.2019.04.002.
Dominici, M., Le Blanc, K., Mueller, I., Slaper-Cortenbach, I., Marini, F., Krause, D., Deans, R., Keating, A., Prockop, D., and Horwitz, E. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement, Cytotherapy, 8, 315-317, https://doi.org/10.1080/14653240600855905.
Vanegas, N.-D. P., Ruiz-Aparicio, P. F., Uribe, G. I., Linares-Ballesteros, A., and Vernot, J.-P. (2021) Leukemia-Induced cellular senescence and stemness alterations in mesenchymal stem cells are reversible upon withdrawal of B-cell acute lymphoblastic leukemia cells, Int. J. Mol. Sci., 22, 8166, https://doi.org/10.3390/ijms22158166.
Schroeder, T., Geyh, S., Germing, U., and Haas, R. (2016) Mesenchymal stromal cells in myeloid malignancies, Blood Res., 51, 225-232, https://doi.org/10.5045/br.2016.51.4.225.
Passaro, D., Di Tullio, A., Abarrategi, A., Rouault-Pierre, K., Foster, K., Ariza-McNaughton, L., Montaner, B., Chakravarty, P., Bhaw, L., Diana, G., Lassailly, F., Gribben, J., and Bonnet, D. (2017) Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia, Cancer Cell, 32, 324-341.e6, https://doi.org/10.1016/J.CCELL.2017.08.001.
Shipounova, I. N., Petinati, N. A., Bigildeev, A. E., Drize, N. J., Sorokina, T. V., Kuzmina, L. A., Parovichnikova, E. N., and Savchenko, V. G. (2017) Alterations of the bone marrow stromal microenvironment in adult patients with acute myeloid and lymphoblastic leukemias before and after allogeneic hematopoietic stem cell transplantation, Leuk. Lymphoma, 58, 408-417, https://doi.org/10.1080/10428194.2016.1187277.
Ludin, A., Gur-Cohen, S., Golan, K., Kaufmann, K. B., Itkin, T., Medaglia, C., Lu, X.-J., Ledergor, G., Kollet, O., and Lapidot, T. (2014) Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment, Antioxid. Redox Signal., 21, 1605-1619, https://doi.org/10.1089/ars.2014.5941.
Récher, C. (2021) Clinical implications of inflammation in acute myeloid leukemia, Front. Oncol., 11, 623952, https://doi.org/10.3389/fonc.2021.623952.
Beavis, P. A., Stagg, J., Darcy, P. K., and Smyth, M. J. (2012) CD73: a potent suppressor of antitumor immune responses, Trends Immunol., 33, 231-237, https://doi.org/10.1016/j.it.2012.02.009.
Jin, D., Fan, J., Wang, L., Thompson, L. F., Liu, A., Daniel, B. J., Shin, T., Curiel, T. J., and Zhang, B. (2010) CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression, Cancer Res., 70, 2245-2255, https://doi.org/10.1158/0008-5472.CAN-09-3109.
Petruk, N., Tuominen, S., Åkerfelt, M., Mattsson, J., Sandholm, J., Nees, M., Yegutkin, G. G., Jukkola, A., Tuomela, J., and Selander, K. S. (2021) CD73 Facilitates EMT Progression and Promotes Lung Metastases in Triple-Negative Breast Cancer, Sci. Rep., 11, 6035, https://doi.org/10.1038/S41598-021-85379-Z.
Bui, T. M., Wiesolek, H. L., and Sumagin, R. (2020) ICAM-1: a master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis, J. Leukoc. Biol., 108, 787-799, https://doi.org/10.1002/JLB.2MR0220-549R.
Ren, G., Zhao, X., Zhang, L., Zhang, J., L’Huillier, A., Ling, W., Roberts, A. I., Le, A. D., Shi, S., Shao, C., and Shi, Y. (2010) Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression, J. Immunol., 184, 2321-2328, https://doi.org/10.4049/jimmunol.0902023.
Russell, K. C., Phinney, D. G., Lacey, M. R., Barrilleaux, B. L., Meyertholen, K. E., and O’Connor, K. C. (2010) In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment, Stem Cells, 28, 788-798, https://doi.org/10.1002/stem.312.
Schirrmacher, V. (2019) From chemotherapy to biological therapy: a review of novel concepts to reduce the side effects of systemic cancer treatment, Int. J. Oncol., 54, 407-419, https://doi.org/10.3892/ijo.2018.4661.
Borges, L., Oliveira, V. K. P., Baik, J., Bendall, S. C., and Perlingeiro, R. C. R. (2019) Serial transplantation reveals a critical role for endoglin in hematopoietic stem cell quiescence, Blood, 133, 688-696, https://doi.org/10.1182/blood-2018-09-874677.
Portale, F., Cricrì, G., Bresolin, S., Lupi, M., Gaspari, S., Silvestri, D., Russo, B., Marino, N., Ubezio, P., Pagni, F., Vergani, P., Te Kronnie, G., Valsecchi, M. G., Locatelli, F., Rizzari, C., Biondi, A., Dander, E., and D’Amico, G. (2019) ActivinA: a new leukemia-promoting factor conferring migratory advantage to B-cell precursor-acute lymphoblastic leukemic cells, Haematologica, 104, 533-545, https://doi.org/10.3324/haematol.2018.188664.
Balandrán, J. C., Purizaca, J., Enciso, J., Dozal, D., Sandoval, A., Jiménez-Hernández, E., Alemán-Lazarini, L., Perez-Koldenkova, V., Quintela-Núñez del Prado, H., Rios de los Ríos, J., Mayani, H., Ortiz-Navarrete, V., Guzman, M. L., and Pelayo, R. (2017) Pro-inflammatory-related loss of CXCL12 niche promotes acute lymphoblastic leukemic progression at the expense of normal lymphopoiesis, Front. Immunol., 7, 666, https://doi.org/10.3389/fimmu.2016.00666.
Desbourdes, L., Javary, J., Charbonnier, T., Ishac, N., Bourgeais, J., Iltis, A., Chomel, J.-C., Turhan, A., Guilloton, F., Tarte, K., Demattei, M.-V., Ducrocq, E., Rouleux-Bonnin, F., Gyan, E., Hérault, O., and Domenech, J. (2017) Alteration analysis of bone marrow mesenchymal stromal cells from de novo acute myeloid leukemia patients at diagnosis, Stem. Cells Dev., 26, 709-722, https://doi.org/10.1089/scd.2016.0295.
Chandran, P., Le, Y., Li, Y., Sabloff, M., Mehic, J., Rosu-Myles, M., and Allan, D. S. (2015) Mesenchymal stromal cells from patients with acute myeloid leukemia have altered capacity to expand differentiated hematopoietic progenitors, Leuk. Res., 39, 486-493, https://doi.org/10.1016/j.leukres.2015.01.013.
Kim, J.-A., Shim, J.-S., Lee, G.-Y., Yim, H. W., Kim, T.-M., Kim, M., Leem, S.-H., Lee, J.-W., Min, C.-K., and Oh, I.-H. (2015) Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia, Cancer Res., 75, 2222-2231, https://doi.org/10.1158/0008-5472.CAN-14-3379.
Van den Berk, L. C. J., van der Veer, A., Willemse, M. E., Theeuwes, M. J. G. A., Luijendijk, M. W., Tong, W. H., van der Sluis, I. M., Pieters, R., and den Boer, M. L. (2013) Disturbed CXCR4/CXCL12 axis in pediatric precursor B-cell acute lymphoblastic leukemia, Blood, 122, 2643, https://doi.org/10.1182/blood.V122.21.2643.2643.
El-Obeid, A., Sunnuqrut, N., Hussain, A., Al-Hussein, K., Gutiérrez, M. I., and Bhatia, K. (2004) Immature B cell malignancies synthesize VEGF, VEGFR-1 (Flt-1) and VEGFR-2 (KDR), Leuk. Res., 28, 133-137, https://doi.org/10.1016/S0145-2126(03)00188-7.
Mizia-Malarz, A., and Sobol-Milejska, G. (2017) Assessment of angiogenesis in children with acute lymphoblastic leukemia based on serum vascular endothelial growth factor assay, Indian J. Med. Paediatr. Oncol., 38, 321-325, https://doi.org/10.4103/IJMPO.IJMPO_109_17.
Azadniv, M., Myers, J. R., McMurray, H. R., Guo, N., Rock, P., Coppage, M. L., Ashton, J., Becker, M. W., Calvi, L. M., and Liesveld, J. L. (2020) Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with Implications for leukemia cell support, Leukemia, 34, 391-403, https://doi.org/10.1038/s41375-019-0568-8.
Ren, K. (2019) Exosomes in perspective: a potential surrogate for stem cell therapy, Odontology, 107, 271-284, https://doi.org/10.1007/s10266-018-0395-9.
Morales-Hernández, A., and McKinney-Freeman, S. (2021) GABA gets blood on its hands, Blood, 137, 723-724, https://doi.org/10.1182/blood.2020008772.
Luque, A., Carpizo, D. R., and Iruela-Arispe, M. L. (2003) ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165, J. Biol. Chem., 278, 23656-23665, https://doi.org/10.1074/JBC.M212964200.
Hall-Glenn, F., and Lyons, K. M. (2011) Roles for CCN2 in normal physiological processes, Cell. Mol. Life Sci., 68, 3209-3217, https://doi.org/10.1007/s00018-011-0782-7.
Nemunaitis, J. (1993) Macrophage function activating cytokines: potential clinical application, Crit. Rev. Oncol. Hematol., 14, 153-171, https://doi.org/10.1016/1040-8428(93)90022-V.
Liso, A., Venuto, S., Coda, A. R. D., Giallongo, C., Palumbo, G. A., and Tibullo, D. (2022) IGFBP-6: at the crossroads of immunity, tissue repair and fibrosis, Int. J. Mol. Sci., 23, 4358, https://doi.org/10.3390/ijms23084358.
Gandhi, M. K., Moll, G., Smith, C., Dua, U., Lambley, E., Ramuz, O., Gill, D., Marlton, P., Seymour, J. F., and Khanna, R. (2007) Galectin-1 mediated suppression of Epstein-Barr virus specific T-cell immunity in classic hodgkin lymphoma, Blood, 110, 1326-1329, https://doi.org/10.1182/blood-2007-01-066100.
Zhu, X., Miao, X., Wu, Y., Li, C., Guo, Y., Liu, Y., Chen, Y., Lu, X., Wang, Y., and He, S. (2015) ENO1 promotes tumor proliferation and cell adhesion mediated drug resistance (CAM-DR) in non-Hodgkin’s lymphomas, Exp. Cell Res., 335, 216-223, https://doi.org/10.1016/J.YEXCR.2015.05.020.
Perretti, M., and D’Acquisto, F. (2009) Annexin A1 and glucocorticoids as effectors of the resolution of inflammation, Nat. Rev. Immunol., 9, 62-70, https://doi.org/10.1038/nri2470.
Kiani, C., Chen, L., Wu, Y. J., Yee, A. J., and Yang, B. B. (2002) Structure and function of aggrecan, Cell Res., 12, 19-32, https://doi.org/10.1038/sj.cr.7290106.
Sengprasert, P., Leearamwat, N., Ngarmukos, S., Yuktananda, P., Tanavalee, A., and Reantragoon, R. (2022) Upregulation of antigen presentation function and inflammation in chondrocytes by induction of proteoglycan aggrecan peptides (P16-31 and P263-280), Clin. Exp. Rheumatol., 40, 596-607, https://doi.org/10.55563/clinexprheumatol/hjzqfs.
Monteith, A. J., Miller, J. M., Maxwell, C. N., Chazin, W. J., and Skaar, E. P. (2021) Neutrophil extracellular traps enhance macrophage killing of bacterial pathogens, Sci. Adv., 7, eabj2101, https://doi.org/10.1126/sciadv.abj2101.
Su, C.-T., and Urban, Z. (2021) LTBP4 in health and disease, Genes, 12, 795, https://doi.org/10.3390/genes12060795.
Xavier, S., Sahu, R. K., Landes, S. G., Yu, J., Taylor, R. P., Ayyadevara, S., Megyesi, J., Stallcup, W. B., Duffield, J. S., Reis, E. S., Lambris, J. D., and Portilla, D. (2017) Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis, Am. J. Physiol. Renal. Physiol., 312, F516-F532, https://doi.org/10.1152/ajprenal.00604.2016.
Morra, L., and Moch, H. (2011) Periostin Expression and epithelial-mesenchymal transition in cancer: a review and an update, Virchows Arch., 459, 465-475, https://doi.org/10.1007/s00428-011-1151-5.
Ding, L., and Morrison, S. J. (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches, Nature, 495, 231-235, https://doi.org/10.1038/nature11885.
Acknowledgments
The equipment (tablet reader Infinite 200 (Tecan, Austria)) from the collective use center of the Pushchino Scientific Center was used in the work. The authors also express their gratitude to the Center for High-Precision Editing and Genetic Technologies for Biomedicine of the Lopukhin Center for Physical-Chemical Medicine for LC-MS/MS analysis.
Funding
This work was supported by the Russian Foundation for Basic Research (grant no. 17-001-00170) (DLBCL patients and donors’ samples) and by the Russian Science Foundation (grant no. 22-15-00018, https://rscf.ru/project/22-15-00018/ [in Russian]) (all patients and donors’ samples).
Author information
Authors and Affiliations
Contributions
E.N.P. supervised and managed the work with the patients; N.A.P. and N.I.D. conceptualized and managed the study; E.A.F., A.U.M., A.N.V., and O.A.A. worked with the donors and patients; N.A.P., A.V.S., N.V.S., N.M.K., and Yu.O.D. conducted the experiments with MSCs; M.A.L. supervised the secretome analysis and edited the text of the article; G.P.A., V.O.Sh., I.P.S., and O.V.P. performed the secretome study and bioinformatic analysis; N.A.P., A.V.S., and N.I.D. analyzed the study results; N.A.P., A.V.S. and N.I.D. wrote and edited the text of the article.
Corresponding author
Ethics declarations
All procedures performed in the course of research comply with the ethical standards of the national committee (permission to conduct the study was obtained from the ethical committee for research ethics (National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation, protocol no. 171 of 04/27/2023)). All studies were conducted in accordance with the principles of biomedical ethics as outlined in the 1964 Declaration of Helsinki and its later amendments.
Each participant in the study provided a voluntary written informed consent after receiving an explanation of the potential risks and benefits, as well as the nature of the upcoming study.
The authors of this work declare that they have no conflicts of interest.
Additional information
Publisher’s Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Petinati, N.A., Sadovskaya, A.V., Sats, N.V. et al. Molecular Changes in Immunological Characteristics of Bone Marrow Multipotent Mesenchymal Stromal Cells in Lymphoid Neoplasia. Biochemistry Moscow 89, 883–903 (2024). https://doi.org/10.1134/S0006297924050092
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297924050092