
Abstract
We conducted a discovery genome-wide association study with expression quantitative trait loci (eQTL) annotation of new-onset diabetes (NOD) among European Americans, who were exposed to a calcium channel blocker-based strategy (CCB strategy) or a β-blocker-based strategy (β-blocker strategy) in the INternational VErapamil SR Trandolapril STudy. Replication of the top signal from the SNP*treatment interaction analysis was attempted in Hispanic and African Americans, and a joint meta-analysis was performed (total 334 NOD cases and 806 matched controls). PLEKHH2 rs11124945 at 2p21 interacted with antihypertensive exposure for NOD (meta-analysis P=5.3 × 10−8). rs11124945 G allele carriers had lower odds for NOD when exposed to the β-blocker strategy compared with the CCB strategy (Odds ratio OR=0.38(0.24−0.60), P=4.0 × 10−5), whereas A/A homozygotes exposed to the β-blocker strategy had increased odds for NOD compared with the CCB strategy (OR=2.02(1.39−2.92), P=2.0 × 10−4). eQTL annotation of the 2p21 locus provides functional support for regulating gene expression.
Similar content being viewed by others

Introduction
Hypertension affects ~80 million Americans and 22% of adults worldwide,1, 2 putting those affected at increased risk for cardiovascular morbidity, mortality and diabetes mellitus (DM).1, 3 Blood pressure reduction with antihypertensive medications is crucial for reducing risk for adverse cardiovascular outcomes. Thiazide diuretics and β-blockers are commonly prescribed and effective antihypertensive drug classes, but consistent evidence suggests increased risk for new-onset diabetes (NOD) with these classes compared with placebo and other antihypertensives.4, 5, 6, 7, 8, 9, 10 The direct association between NOD and adverse cardiovascular outcomes remains controversial.10, 11, 12, 13, 14, 15, 16 However, individuals with concomitant prevalent DM and hypertension face an increased risk for adverse cardiovascular outcomes compared with those with hypertension alone.12, 13, 15
Among individuals treated with antihypertensive agents, there is substantial inter-individual variability with regard to NOD development. In addition to clinical risk factors, such as race, baseline glucose, dyslipidemia and body mass index (BMI),5, 6, 17 genetic variation may contribute to the inter-individual variability for risk of developing NOD among those treated with antihypertensive agents. Genetic risk for type 2 DM has been well studied, and an enrichment of type 2 DM associations among expression quantitative trait loci (eQTL) variants has been observed.18, 19 In addition, treatment with some classes of antihypertensives has been shown to influence the susceptibility of NOD and several susceptibility genes have been suggested to play a role. Candidate gene studies have associated previously identified type 2 DM or fasting glucose susceptibility genes, such as KCNJ1 and TCF7L2, and NOD in individuals exposed to the thiazide diuretic hydrochlorothiazide.20, 21 However, the genetic contribution to development of NOD, its interaction with antihypertensive drugs and, most importantly, the underlying mechanisms remain poorly understood.
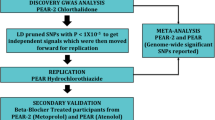
Therefore, we performed the first genome-wide association study (GWAS) to investigate single-nucleotide polymorphisms (SNPs) associated with differential risk for NOD in response to exposure to two commonly prescribed antihypertensive treatment strategies. In addition, eQTL enrichment analyses were performed. The research was conducted in a case–control sample created from the INternational VErapamil SR Trandolapril STudy GENEtic Substudy (INVEST-GENES), which consists of elderly hypertensive individuals with documented coronary artery disease.22
Materials and methods
Study design and participants
INVEST was a multicenter, randomized clinical trial (clinicaltrials.gov: NCT00133692) that has been described previously.23 Briefly, hypertensive individuals ⩾50 years old and with documented coronary artery disease were randomized to either a calcium channel blocker (Verapamil SR)-based treatment strategy (CCB strategy) or a β-blocker (atenolol)-based treatment strategy (β-blocker strategy), with hydrochlorothiazide and trandolapril available as add-on agents in both strategies. Participants were followed for an average of 2.7 years for NOD, which was a pre-specified outcome. Among those who were DM-free at baseline, development of NOD was defined as any new occurrence of self/physician reported-DM, or new use of a DM medication during the follow-up period.6 INVEST-GENES includes deoxyribonucleic acid (DNA) samples from 5979 INVEST participants from the United States, including Puerto Rico.22 All study protocols were approved by local or central institutional review boards and all participants provided separate, voluntary, written informed consent for participation in INVEST and INVEST-GENES.
Within INVEST-GENES, a nested NOD case–control study was conducted. Cases were participants who developed NOD as defined above, and controls were those who remained DM-free (were not diagnosed with DM and were not taking DM medications) during the entire follow-up period. Cases and controls were frequency-matched based on gender, race/ethnicity and age (by decade). Race/ethnicity information was self-identified and confirmed by principal component analysis defined genetic ancestry (described below). From now onwards, race/ethnicity will be referred to as race throughout the rest of the document. Among those who were genetically confirmed to be of European ancestry, each case was frequency matched to three controls. For those who were genetically confirmed as Hispanic or African ancestry, each case was matched to two controls.
Genotyping, quality control and imputation
DNA samples were genotyped at the RIKEN Center for Integrative Medical Sciences (Yokohama, Japan) using the Illumina OmniExpressExome Beadchip. Subsequently, individual and SNP-level quality control procedures were performed using PLINK (v1.07).24 Minor allele frequency (MAF) and genotyping call rates were assessed. Concordance of genetic sex to pedigree sex was evaluated via X-chromosome heterozygosity. Cryptic relativeness or sample duplication was assessed through genome-wide identity-by-descent analysis. Potential sample contamination was tested using the inbreeding coefficient. SNPs or individuals were removed if any of the following criteria were met: genotyping call rate <95%, mismatch of genetic sex with pedigree sex, sample duplication or potential sample contamination. A principal component (PC) analysis was performed using EIGENSTRAT on a linkage-disequilibrium pruned set of high-quality SNPs that passed quality control, and genetic continental ancestry was determined based on PC analysis clustering.25 PC analysis was then performed within each defined genetic ancestry group to identify PCs that best summarized genetic structure and ancestry clusters for each race group. Hardy–Weinberg equilibrium was assessed for each SNP, and deviations were flagged.
Genome-wide imputation was conducted at the RIKEN Center for Integrative Medical Sciences using the 1000 Genomes phase I, release 3, multiethnic haplotype data set as a reference (released 30 April 2012) for all study participants. SNPs that passed quality control with MAF>0.01 in any of the INVEST race groups or 1000 Genome reference populations (EUR/AFR/AMR) were included for imputation. Called SNPs were aligned to the forward strand on the human genome reference Build 37 and oriented to the 1000 Genomes reference and alternate alleles. SNPs with alleles that did not match those in 1000 Genomes were removed. The oriented genotypes were then phased using SHAPEIT2 (v.778)26 and genotypes were imputed using IMPUTE2 (v.2.3.0).27 After genome-wide imputation, SNPs with a quality information metric <0.4, which demonstrated lower imputation certainty, were excluded. In addition, imputed SNPs with MAF<0.03 in each race group were excluded.
Statistical analysis
Continuous characteristics are presented as mean and standard deviation, and categorical characteristics are presented as frequency and percentages. Unpaired two-sample Student t-test and chi-square tests were conducted to compare the baseline characteristics between cases and controls by race groups.
Genome-wide SNP*treatment interaction analyses with NOD development were conducted under an additive genetic model by race groups using logistic regression modeling. Analyses were performed using PLINK [ref. 24] adjusted for age, gender, principal components for ancestry, INVEST treatment strategy, genotyping call rate between 95 and 98%, and use of hydrochlorothiazide and/or trandolapril add-on therapy. Because both hydrochlorothiazide and trandolapril have been observed to affect risk for NOD development,8 exposure to these agents was included in the model. Genotype call rate between 95 and 98% was also included as a covariate in the model to prevent confounding from individual DNA quality. The discovery sample consisted of European Americans as they made up the majority of the population. Genome-wide significance was specified at an alpha level of 5 × 10−8. SNPs with an interaction P<1 × 10−6 in European Americans were considered to be of suggestive significance and were tested for evidence of association in Hispanics and African Americans. A replication was defined as SNPs having consistent direction of effect in Hispanics and African Americans with an interaction P-value that achieved the Bonferroni corrected significance level under a one-sided hypothesis.
A meta-analysis of all three race groups was performed on SNPs that replicated in Hispanics and African Americans using METAL.28 Heterogeneity across race groups was assessed using Cochran’s Q test. Follow-up analyses for adjusted odds of NOD development by genotype were conducted using SAS 9.3 (Cary, NC, USA) for top-association SNPs under additive or dominant models as appropriate. We also performed analyses adjusting for BMI in addition to the prespecified covariates listed above for the top SNPs to determine whether the identified associations were driven by BMI.
Cis-eQTL annotation and enrichment analysis
Since eQTL studies are generally better powered than GWAS to detect functional SNPs,29, 30, 31 we annotated our findings in INVEST European Americans with eQTL results from the Genotype-tissue expression (GTEx) pilot analysis. The GTEx pilot results were acquired from postmortem donors.31, 32 In accordance with the GTEx project, only eQTLs that act cis to the target gene (within ±1 MB of the transcriptional start site) were evaluated due to power considerations.31, 32 Three adequately powered GTEx eQTL tissues with sample size >80 were analyzed, including subcutaneous adipose and skeletal muscle, which are peripheral tissues sensitive to insulin, as well as whole blood, which is the most accessible tissue. SNPs with a nominal SNP–gene expression association (P<0.05) were defined as eSNPs in each selected tissue, and were included for evaluation of enrichment. This lenient eSNP definition allowed inclusion of more eSNPs and improved power to detect enrichment. On the basis of results from the genome-wide interaction analysis, cis-eQTL enrichment was assessed in each tissue separately. In addition, the proportion of SNPs with an interaction P<0.05 were compared among the sets of eSNPs and non-eSNPs, and enrichment P-value was estimated using the Z statistic.
Results
Baseline demographics
Baseline characteristics of individuals included in this study (334 NOD cases and 806 controls) are summarized in Table 1. In general, baseline characteristics were similar between cases and controls in each race group, with the exception of age (which was matched by decade) and BMI.
Genome-wide interaction analysis and between race confirmations
Genome-wide interaction analysis in European Americans did not identify SNPs that achieved genome-wide significance. However, two loci (containing 17 SNPs) demonstrated evidence of a suggestive association (interaction P<1 × 10−6) with risk for NOD (Supplementary Figure 1, Supplementary Table 1). Within each locus the SNPs are in high linkage disequilibrium (r2>0.8) with each other, and thus represent two independent signals. The chromosome 2p21 locus contains THADA (thyroid adenoma associated) and PLEKHH2 (Pleckstrin Homology Domain-Containing Family H Member 2) (Figure 1). The 4p14 locus consists of a cluster of toll-like receptor (TLR) genes (TLR1, TLR6 and TLR10) (Supplementary Figure 2), and contains the strongest signal (rs4833103, interaction P=1.6 × 10−7), which is located 8 kb 5′ of TLR1 and 10 kb 3′ of TLR6.

Regional plot at chromosome 2p21 locus for new-onset diabetes (NOD) association in INVEST European Americans. Regional single-nucleotide polymorphism (SNP)*treatment interaction analysis results (−log P-value) are plotted for INVEST European Americans. Figure is generated using LocusZoom.60 The diamond-shaped dot represent the SNP with lowest interaction P-value in the loci. The lower panel demonstrated the RefSeq genes.
When evaluating these two independent signals in Hispanics and African Americans, rs11124945 at 2p21 was directionally consistent across race groups and achieved the Bonferroni corrected one-sided alpha of 0.05 in Hispanics (one-sided interaction P=1.56 × 10−2) and African Americans (one-sided interaction P=2.47 × 10−2). Meta-analysis of 2p21 rs11124945 of all three race groups resulted in a P-value approaching genome-wide significance (meta-analysis P=5.33 × 10−8, Table 2). Overall, rs11124945 G allele carriers had lower odds for NOD when exposed to the β-blocker strategy compared with exposure to the CCB strategy (Odds ratio (OR, 95% CI)=0.38(0.24−0.60), P=4.02 × 10−5), whereas A/A homozygotes exposed to the β-blocker strategy had increased odds for NOD compared with exposure to CCB strategy (OR=2.02(1.39−2.92), P=2.0 × 10−4) (Figure 2). Adjusting for BMI in the logistic regression model did not affect the level of significance (Supplementary Table 2). Supplementary Table 3 describes allele counts and Hardy–Weinberg equilibrium result of rs11124945. The 4p14 signal observed in European Americans, although directionally consistent, did not achieve the corrected significance level in the Hispanics and African Americans.

Forest plot of the PLEKHH2 rs11124945 pharmacogenomics association with new-onset diabetes (NOD). This figure shows PLEKHH2 rs11124945 genotypes specific odds ratio (OR) and 95% confidence interval for NOD in each race group and combined meta-analysis from the INVEST NOD case–control study (n=1140). Point estimate was reported under a dominant genetic model for each genotype group, which represents higher risk for NOD in the CCB strategy or the β-blocker strategy. Interaction P-values for Hispanics and African Americans are one-sided.
Cis-eQTL enrichment of NOD GWAS
Enrichment of cis-eQTL for pharmacogenomic associations was observed in subcutaneous adipose tissue, skeletal muscle tissue and whole blood for NOD, as evidenced by the deviation from the null distribution in the Q–Q plots for all three tissues (Supplementary Figure 3). In addition, a significantly higher proportion of SNPs with interaction P<0.05 was observed among eSNPs compared with non-eSNPs in all three tissues (subcutaneous adipose: 5.0 vs 4.3%, Z=12.25, P<1.0 × 10−5; muscle skeletal: 5.0 vs 4.3%, Z=12.69, P<1.0 × 10−5; and whole blood: 5.0 vs 4.3%, Z=11.67, P<1.0 × 10−5, Supplementary Table 4). These results represent a 1.16-fold enrichment among eSNPs compared with non-eSNPs in each tissue. Moreover, the two strongest loci in European Americans (2p21 and 4p14) displayed evidence of a tissue-dependent eQTL–gene relationship among the tested tissues (Table 3).
Discussion
Among European Americans in INVEST-GENES exposed to CCB-based and β-blocker-based antihypertensive strategies, we performed the first genome-wide investigation of SNPs associated with NOD development. SNP rs11124945, located in the second intron of PLEKHH2, was discovered in European Americans, was replicated in Hispanics and African Americans, and had the strongest evidence of an interaction with treatment strategy exposure for NOD development. Furthermore, genome-wide annotation with eQTLs information from the GTEx project provided functional support for the top GWAS signals and evidence for the notion that the identified signals are more likely to be true-positive associations. The GWAS was performed using well-characterized phenotypes and medication exposure information from the randomized INVEST clinical trial, which greatly reduces potential confounding. Moreover, our study contains a racially diverse population, providing a unique opportunity to assess genetic associations in Hispanics and African Americans, who are at higher risk for DM [ref. 33] and NOD compared with individuals of European ancestry.6, 17, 34
PLEKHH2 may play roles in the linkage of kidney podocytes to the basement membrane, and actin stabilization,35 and SNPs within PLEKHH2 have been associated with diabetic nephropathy.36 Although the functional consequences of rs11124945 are incompletely understood, we observed differential risk for NOD by rs11124945 genotype and antihypertensive exposure across multiple race groups. When exposed to the β-blocker strategy vs the CCB strategy, G allele carriers had decreased risk for NOD, whereas A/A homozygotes had increased risk. This observation may suggest that for rs11124945 G allele carriers, treatment with a β-blocker-based strategy would be preferred, whereas for A/A homozygotes treatment with a CCB-based strategy would be preferred. Within this INVEST-GENES GWAS, our observation that major allele carriers had increased risk for NOD when exposed to a β-blocker strategy is in line with data from other clinical trials, including the overall INVEST trial (n=22 576), which demonstrated that risk for NOD was higher in individuals treated with regimens containing a β-blocker.5, 6, 8
PLEKHH2 rs11124945 is ~50 kb 5’ upstream of THADA, a gene with well-established associations with type 2 DM,37 polycystic ovary syndrome (a phenotype characterized by insulin resistance and risk for type 2 DM),38, 39 and prostate cancer40 in previous GWAS. THADA may play a role in apoptosis and death receptor signaling, but its function requires further characterization.41 The THADA rs7578597 T allele has been associated with risk for type 2 DM and lower β-cell function, potentially via reduced β-cell mass.42 Although our observed association between rs11124945 and NOD risk is not driven by rs7578597 (r2=0.007, D’=1), rs11124945 is in linkage disequilibrium with rs6544683 (r2=0.9, D’=1) that has tissue-dependent eQTL associations, and could be in linkage disequilibrium with other unknown functional variants. Given the association between THADA and type 2 DM, and its proximity to rs11124945, THADA represents a plausible candidate gene for NOD. Previous type 2 DM and polycystic ovary syndrome GWAS reported disease associations with THADA independent of BMI.37, 38 Similarly, we did not observe an effect of BMI on NOD association with rs11124945.
The 4p14 locus consists of TLR1, TLR6 and TLR10. TLRs are a family of transmembrane receptors that recognize pathogen- or damage-associated molecular patterns and are crucial in mediating the innate immune response.43 TLR2 and TLR4 play important roles in type 2 DM pathogenesis,44 and TLR1 and TLR6 form heterodimers with TLR2 essential for TLR2 signaling.45 Activation of the TLR/NFĸB pathway in conjunction with the NLRP3 inflammasome triggers IL-18- and IL-1β-mediated inflammatory cascade, which results in monocyte recruitment and production of additional cytokines with deleterious effects on pancreatic β-cell insulin secretion and systemic insulin resistance.43, 44, 45, 46 Although the observed pharmacogenomic association at 4p14 needs further replication, the biology is compelling.
Our eQTL-based enrichment results demonstrate a deviation from the null hypothesis, which suggests higher probability that signals at 2p21 and 4p14 are true positive associations. Moreover, SNPs within 2p21 and 4p14 had functional support for regulating expression in a tissue-dependent manner, suggesting that they might regulate gene expression in other relevant tissues. It has been demonstrated that drug response-associated SNPs are likely to be eQTLs and to regulate expression of multiple genes.30 We observed enrichment of NOD association among eSNPs compared with non-eSNPs. Previous studies demonstrated that eQTL SNPs are more likely to be associated with type 2 DM in relevant tissues,19, 47 and a trans-regulatory characteristic has been reported.48, 49, 50 Currently, GTEx is not sufficiently powered to provide a trans-eQTL reference panel. We examined cis-eQTL enrichment in available tissues relevant to DM pathophysiology with appropriate power. Exploration of eQTL enrichment using other key DM-related tissues, such as pancreatic islet, liver, and brain, can be done when the information becomes available from GTEx.
We observed evidence of interaction with treatment strategies at the 2p21 locus, which has been previously associated with type 2 DM and related phenotypes, suggesting that there are potential overlapping pathways between NOD and type 2 DM. These conditions share clinical risk factors, such as race and BMI.5, 6, 17 Although the association between NOD and adverse cardiovascular outcome is in debate,10, 11, 12, 13, 14, 15, 16 DM of other etiologies independently contributes a two-fold excess risk of cardiovascular disease,51 and an increased cardiovascular risk has been observed in individuals with NOD.12, 15 The majority of genetic markers for type 2 DM risk have been associated with primary defects in pancreatic β-cells,18, 52, 53 which could lead to a reduced ability to produce insulin and maintain glucose homeostasis in the presence of environmental risk factors. Reduced pancreatic β-cell mass as a result of increased apoptosis has been hypothesized as a potential mechanism by which THADA affects type 2 DM risk.42 Taken together, our findings may suggest that when an individual has existing genetic risk for type 2 DM, introducing diabetogenetic antihypertensives like β-blockers and thiazide diuretics could serve as environmental risk factors, and promote deterioration of glucose homeostasis. Currently, the exact mechanism for the observed interaction between the 2p21 locus and treatment strategies remains unclear. Future studies, including functional characterization of these genetic signals, are needed to elucidate the mechanism. A better understanding of the genetic etiology of NOD in addition to other known risk factors could provide guidance for personalization of pharmacotherapy tailored to optimize benefit and minimize risk for adverse outcomes like NOD.
Study limitations include a relatively small number of NOD cases with no external replication since INVEST-GENES is one of the only genetic cohorts that consist of elderly adults with documented hypertension and coronary artery disease. Although sample size was limited, we conducted this study because pharmacogenomic SNPs generally have larger effect size than disease genetics SNPs.54 Although our study had limited power, we included additional evidence provided from the cross race confirmation and the eQTL analysis to minimize the chance of reporting a spurious result. Even though the magnitude of effect of PLEKHH2 rs11124945 is smaller in Hispanics and African Americans, the direction of effect is consistent with what we observed in European Americans. In published pharmacogenomic studies, reported pharmacogenomic associations are often observed across race groups,55, 56, 57 although the magnitude of effect could be different due to population substructure. A cross race replication approach provides the advantage of increased power and the possibility to identify functional SNPs, as truly functional SNP associations should replicate across race groups.58, 59 Although we detected no genome-wide significant associations in European Americans, our eQTL-based enrichment results demonstrate an improvement of the rate of false discovery when using eQTL information in disease-relevant tissues. Our results also show that our most highly ranked SNPs can be distinguished from ‘noise’, and that these SNPs have prior functional support for regulating expression in a tissue-dependent manner. Despite lacking an appropriate external replication, we observed associations in all race groups, and together with the eQTL results, we believe it is less likely to be a chance finding. Despite the limited power, this study represents the first investigation of SNP–treatment interactions for NOD, and provides valuable initial data to the field. Finally, there are differences in age and BMI comparing cases and controls in some race groups. However, age was included as a covariate in the analyses and additional analyses were conducted adjusting for BMI for the top association signal.
In conclusion, we conducted the first GWAS for NOD in a cohort with hypertension and coronary artery disease, and identified SNP rs11124945 at the 2p21 locus as having the strongest evidence of interaction with antihypertensive exposure and NOD development. rs11124945 G allele carriers had lower odds for NOD when exposed to the β-blocker strategy compared with the CCB strategy, whereas A/A homozygotes exposed to the β-blocker strategy had increased odds for NOD compared with the CCB strategy. The 2p21 locus has functional support for regulating gene expression and contains both PLEKHH2 and THADA that are relevant to DM and related phenotypes. Genes and pathways influencing DM susceptibility could be important in determining an individual’s genetic predisposition for NOD. These results along with enrichment of pharmacogenomic association among eQTLs, shed light on the underlying mechanism of NOD, and provide useful information for future studies. Further replication and functional characterization is warranted.
References
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 2015; 131: e29–322.
WHO Global Status Report on Noncommunicable Diseases 2014. World Health Organization: Switzerland, 2014.
Weycker D, Nichols GA, O'Keeffe-Rosetti M, Edelsberg J, Vincze G, Khan ZM et al. Excess risk of diabetes in persons with hypertension. J Diabetes Complicat 2009; 23: 330–336.
Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA 2002; 288: 2981–2997.
Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NR . Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—blood pressure lowering arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31: 982–988.
Cooper-Dehoff R, Cohen JD, Bakris GL, Messerli FH, Erdine S, Hewkin AC et al. Predictors of development of diabetes mellitus in patients with coronary artery disease taking antihypertensive medications (findings from the INternational VErapamil SR-Trandolapril STudy [INVEST]). Am J Cardiol 2006; 98: 890–894.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U et al. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359: 995–1003.
Elliott WJ, Meyer PM . Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369: 201–207.
Shen L, Shah BR, Reyes EM, Thomas L, Wojdyla D, Diem P et al. Role of diuretics, beta blockers, and statins in increasing the risk of diabetes in patients with impaired glucose tolerance: reanalysis of data from the NAVIGATOR study. BMJ 2013; 347: f6745.
Rizos CV, Elisaf MS . Antihypertensive drugs and glucose metabolism. World J Cardiol 2014; 6: 517–530.
Barzilay JI, Davis BR, Cutler JA, Pressel SL, Whelton PK, Basile J et al. Fasting glucose levels and incident diabetes mellitus in older nondiabetic adults randomized to receive 3 different classes of antihypertensive treatment: a report from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). Arch Intern Med 2006; 166: 2191–2201.
Verdecchia P, Reboldi G, Angeli F, Borgioni C, Gattobigio R, Filippucci L et al. Adverse prognostic significance of new diabetes in treated hypertensive subjects. Hypertension 2004; 43: 963–969.
Almgren T, Wilhelmsen L, Samuelsson O, Himmelmann A, Rosengren A, Andersson OK . Diabetes in treated hypertension is common and carries a high cardiovascular risk: results from a 28-year follow-up. J Hypertens 2007; 25: 1311–1317.
Kostis JB, Wilson AC, Freudenberger RS, Cosgrove NM, Pressel SL, Davis BR . Long-term effect of diuretic-based therapy on fatal outcomes in subjects with isolated systolic hypertension with and without diabetes. Am J Cardiol 2005; 95: 29–35.
Aksnes TA, Kjeldsen SE, Rostrup M, Omvik P, Hua TA, Julius S . Impact of new-onset diabetes mellitus on cardiac outcomes in the Valsartan Antihypertensive Long-term Use Evaluation (VALUE) trial population. Hypertension 2007; 50: 467–473.
Karagiannis A, Tziomalos K, Pagourelias ED, Gossios TD, Athyros VG . Effect of antihypertensive drug-associated diabetes on cardiovascular risk. Hellenic J Cardiol 2010; 51: 195–199.
Aksnes TA, Kjeldsen SE, Rostrup M, Storset O, Hua TA, Julius S . Predictors of new-onset diabetes mellitus in hypertensive patients: the VALUE trial. J Hum Hypertens 2008; 22: 520–527.
Billings LK, Florez JC . The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci 2010; 1212: 59–77.
Zhong H, Beaulaurier J, Lum PY, Molony C, Yang X, Macneil DJ et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet 2010; 6: e1000932.
Karnes JH, Gong Y, Pacanowski MA, McDonough CW, Arwood MJ, Langaee TY et al. Impact of TCF7L2 single nucleotide polymorphisms on hydrochlorothiazide-induced diabetes. Pharmacogenet Genomics 2013; 23: 697–705.
Karnes JH, McDonough CW, Gong Y, Vo TT, Langaee TY, Chapman AB et al. Association of KCNJ1 variation with change in fasting glucose and new onset diabetes during HCTZ treatment. Pharmacogenomics J 2013; 13: 430–436.
Beitelshees AL, Gong Y, Wang D, Schork NJ, Cooper-Dehoff RM, Langaee TY et al. KCNMB1 genotype influences response to verapamil SR and adverse outcomes in the INternational VErapamil SR/Trandolapril STudy (INVEST). Pharmacogenet Genomics 2007; 17: 719–729.
Pepine CJ, Handberg-Thurmond E, Marks RG, Conlon M, Cooper-DeHoff R, Volkers P et al. Rationale and design of the International Verapamil SR/Trandolapril Study (INVEST): an Internet-based randomized trial in coronary artery disease patients with hypertension. J Am Coll Cardiol 1998; 32: 1228–1237.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Alkes L, Price NJP, Plenge Robert M, Weinblatt Michael E, Shadick Nancy A, Reich David . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Delaneau O, Marchini J, Zagury JF . A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181.
Howie BN, Donnelly P, Marchini J . A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
Willer CJ, Li Y, Abecasis GR . METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–2191.
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ . Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010; 6: e1000888.
Gamazon ER, Huang RS, Cox NJ, Dolan ME . Chemotherapeutic drug susceptibility associated SNPs are enriched in expression quantitative trait loci. Proc Natl Acad Sci USA 2010; 107: 9287–9292.
Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–660.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S et al. The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013; 45: 580–585.
Narayan KM, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF . Lifetime risk for diabetes mellitus in the United States. JAMA 2003; 290: 1884–1890.
Barner JC, Worchel J, Yang M . Frequency of new-onset diabetes mellitus and use of antipsychotic drugs among Central Texas veterans. Pharmacotherapy 2004; 24: 1529–1538.
Perisic L, Lal M, Hulkko J, Hultenby K, Onfelt B, Sun Y et al. Plekhh2, a novel podocyte protein downregulated in human focal segmental glomerulosclerosis, is involved in matrix adhesion and actin dynamics. Kidney Int 2012; 82: 1071–1083.
Greene CN, Keong LM, Cordovado SK, Mueller PW . Sequence variants in the PLEKHH2 region are associated with diabetic nephropathy in the GoKinD study population. Hum Genet 2008; 124: 255–262.
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008; 40: 638–645.
Chen ZJ, Zhao H, He L, Shi Y, Qin Y, Li Z et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet 2011; 43: 55–59.
Goodarzi MO, Jones MR, Li X, Chua AK, Garcia OA, Chen YD et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet 2012; 49: 90–95.
Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, Severi G et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet 2009; 41: 1116–1121.
Rippe V, Drieschner N, Meiboom M, Murua Escobar H, Bonk U, Belge G et al. Identification of a gene rearranged by 2p21 aberrations in thyroid adenomas. Oncogene 2003; 22: 6111–6114.
Simonis-Bik AM, Nijpels G, van Haeften TW, Houwing-Duistermaat JJ, Boomsma DI, Reiling E et al. Gene variants in the novel type 2 diabetes loci CDC123/CAMK1D, THADA, ADAMTS9, BCL11A, and MTNR1B affect different aspects of pancreatic beta-cell function. Diabetes 2010; 59: 293–301.
Santoni M, Andrikou K, Sotte V, Bittoni A, Lanese A, Pellei C et al. Toll like receptors and pancreatic diseases: From a pathogenetic mechanism to a therapeutic target. Cancer Treat Rev 2015; 41: 569–576.
Donath MY, Shoelson SE . Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 2011; 11: 98–107.
Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA . Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol 2014; 92: 314–323.
Grant RW, Dixit VD . Mechanisms of disease: inflammasome activation and the development of type 2 diabetes. Front Immunol 2013; 4: 50.
Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci USA 2014; 111: 13924–13929.
Below JE, Gamazon ER, Morrison JV, Konkashbaev A, Pluzhnikov A, McKeigue PM et al. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia 2011; 54: 2047–2055.
Elbein SC, Gamazon ER, Das SK, Rasouli N, Kern PA, Cox NJ . Genetic risk factors for type 2 diabetes: a trans-regulatory genetic architecture? Am J Hum Genet 2012; 91: 466–477.
Das SK, Sharma NK . Expression quantitative trait analyses to identify causal genetic variants for type 2 diabetes susceptibility. World J Diabetes 2014; 5: 97–114.
Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010; 375: 2215–2222.
Torres JM, Cox NJ, Philipson LH . Genome wide association studies for diabetes: perspective on results and challenges. Pediatr Diabetes 2013; 14: 90–96.
Prasad RB, Groop L . Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel) 2015; 6: 87–123.
Maranville JC, Cox NJ . Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharmacogenomics J 2015; 16: 388–392.
Shuldiner AR, O'Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009; 302: 849–857.
Niu Y, Gong Y, Langaee TY, Davis HM, Elewa H, Beitelshees AL et al. Genetic variation in the beta2 subunit of the voltage-gated calcium channel and pharmacogenetic association with adverse cardiovascular outcomes in the INternational VErapamil SR-Trandolapril STudy GENEtic Substudy (INVEST-GENES). Circ Cardiovasc Genet 2010; 3: 548–555.
McDonough CW, Gong Y, Padmanabhan S, Burkley B, Langaee TY, Melander O et al. Pharmacogenomic association of nonsynonymous SNPs in SIGLEC12, A1BG, and the selectin region and cardiovascular outcomes. Hypertension 2013; 62: 48–54.
Limdi NA, Wadelius M, Cavallari L, Eriksson N, Crawford DC, Lee MT et al. Warfarin pharmacogenetics: a single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 2010; 115: 3827–3834.
Yin T, Miyata T . Pharmacogenomics of clopidogrel: evidence and perspectives. Thromb Res 2011; 128: 307–316.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–2337.
Acknowledgements
This project was supported by the National Institute of Health Pharmacogenetics Research Network Grant U01-GM074492, NIH R01 HL074730, UF Opportunity Fund and Abbott Laboratories. The genotyping and imputation were performed by the RIKEN Center for Integrative Medical Sciences. INVEST was supported by the University of Florida and Grants from BASF Pharma and Abbott Laboratories.
Author contributions
S-WC wrote the manuscript and all co-authors critically evaluated and reviewed the manuscript. CJP, JAJ and RCD designed and conducted the INVEST-GENES and secured funding. S-WC, YG, CWM, JAJ and RCD designed the research. TAJ, TT, AT, TT and MK facilitated genotyping and imputation. ERG and MAP provided guidance on the eQTL analysis. S-WC, YG, CWM, RMC-D, CJP and JAJ performed the research, and S-WC analyzed the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CWM, YG, MAP, JAJ, CJP and RMC-D have received support from NIH. CJP, JAJ and RMC-D also received support for this project from Abbott Laboratories. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website
PowerPoint slides
Rights and permissions
About this article

Cite this article
Chang, SW., McDonough, C., Gong, Y. et al. Genome-wide association study identifies pharmacogenomic loci linked with specific antihypertensive drug treatment and new-onset diabetes. Pharmacogenomics J 18, 106–112 (2018). https://doi.org/10.1038/tpj.2016.67
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2016.67
- Springer Nature Limited
This article is cited by
-
AC010883.5 promotes cell proliferation, invasion, migration, and epithelial-to-mesenchymal transition in cervical cancer by modulating the MAPK signaling pathway
BMC Cancer (2023)
-
Intergenerational effects of preconception opioids on glucose homeostasis and hepatic transcription in adult male rats
Scientific Reports (2022)
-
Drug-induced hyperglycaemia and diabetes: pharmacogenomics perspectives
Archives of Pharmacal Research (2018)