
Abstract
Takifugu species serve as a model system for evolutionary studies due to their compact genomes and diverse phenotypes. The ocellated puffer (Takifugu ocellatus), characterized by special colouration, is a scarce anadromous species in the genus Takifugu. As an ornamental and tasty fish species, T. ocellatus has moderate economic value. However, the available genomic resources for this pufferfish are still limited. Here, a chromosome-level reference genome, as well as two haploid genomes, was constructed by PacBio HiFi long sequencing and Hi-C technologies. The total length of the reference genome was 375.62 Mb with a contig N50 of 11.55 Mb. The assembled sequences were anchored to 22 chromosomes with an integration efficiency of 93.78%. Furthermore, 28,808 protein-coding genes were predicted. The haplotype-resolved reference genome of T. ocellatus provides a crucial resource for investigating the explosive speciation of the Takifugu genus, such as elucidating evolutionary histories, determining the genetic basis of trait evolution, and supporting future conservation efforts.
Measurement(s) | Whole Genome Sequencing • Chromosome assembly by Hi-C data • Whole Transcriptome Sequencing |
Technology Type(s) | PacBio Sequel System • Hi-C • Illumina HiSeq. 2500 • Illumina NovaSeq. 6000 |
Sample Characteristic - Organism | Takifugu ocellatus |
Similar content being viewed by others


Background & Summary
The genus Takifugu belongs to the family Tetraodontidae, which inhabits the northwest Pacific Ocean around the coastal area of east Asia1. Takifugu is composed of approximately 25 species2, which are well known for their inflation behaviour and potent neurotoxins. Meanwhile, the group exhibits diverse morphological characteristics and different ecological habits, as well as a compact genome, providing a great model for investigating species radiation. Four high-quality chromosome-level genomes of T. rubripes3, T. bimaculatus4, T. flavidus5, and T. obscurus6 have been completed in the genus Takifugu since the first teleost genome of T. rubripes was published in 20021, among which T. obscurus and T. ocellatus have the capacity for hypotonic adaptation7. The ocellated puffer Takifugu ocellatus in this study is harboured in China and Vietnam, and is commonly utilized as an ornamental fish species for culture. T. ocellatus exhibits saddle-shaped black dots profiled with orange in the dorsal region, in addition to featuring the capacity for euryhaline acclimation, both making it favour aquarium fish. Despite its deadly toxicity, it is also considered a delicacy in East Asia. Therefore, the species has considerable commercial value due to its ornamental value and edibleness. As an anadromous fish, T. ocellatus shares the same spawning sites and a similar diet to T. obscurus. They acclimate to a broad spectrum of saline water and migrate into freshwater to spawn, while the larvae remain there before emigrating to the seawater. Despite the abovementioned similarities, these two pufferfishes employ different reproductive strategies7. In addition, recent phylogenetic analyses in the Takifugu genus have shown that they belong to different sister groups, implying that they may be independent of each other in the evolutionary process of adapting to freshwater8. Therefore, a high-quality reference genome of T. ocellatus is essential to elucidate the speciation process during adaptive radiation, including clarifying the evolutionary histories and adaptation strategies.
In this work, we constructed a chromosome-level genome of T. ocellatus by combining PacBio HiFi (high fidelity) reads and Hi-C sequencing data. The genome assembly spanned 375.62 Mb consisting of 163 contigs with a contig N50 length of 11.55 Mb. After chromosome-level anchoring, 22 chromosomes with a total length of 352.28 Mb (93.78% of the draft assembly) were constructed corresponding to the karyotype. Moreover, 66.65 Mb (17.74% of the assembly) of repeat elements, and 28,808 protein-coding genes were annotated. Additionally, two chromosome-level haplotype genome assemblies were also constructed, which would serve as a baseline for future studies on allele-specific expression or conservation genomics. A contiguous and accurate reference genome is essential for basic genetic research and will facilitate evolutionary studies on this euryhaline and anadromous species. In addition, phylogenetic analysis indicated that T. ocellatus speciated from the common ancestor of Takifugu at approximately 21.4 (mya; 15.3–27.6 mya). We identified 789 gene families with expansion, 1,970 families with contraction, 1,034 rapidly evolving genes, and 767 positively selected genes in T. ocellatus. These results will help us to further explore the genetic basis of the freshwater adaptability of T. ocellatus and the explosive speciation mechanism of Takifugu species.
Methods
Sample collection and nucleic acid extraction
Healthy female T. ocellatus were collected from Fujian Takifugu Breeding Station in Zhangzhou, Fujian Province, China. Muscle, eye, skin, gill, kidney, liver, intestine, spleen, gonad, heart and stomach were sampled and frozen in liquid nitrogen immediately and then transferred to −80 °C for storage. Genomic DNA (gDNA) of T. ocellatus was extracted from enough muscle tissues following the manufacturer’s protocol by an AMPure bead cleanup kit (Beckman Coulter, High Wycombe, UK), while total RNA was extracted from all tissues by a TRIzoL kit and mixed equally for transcriptome sequencing. The quality of gDNA and RNA was detected by 1.5% agarose gel electrophoresis and DNA was quantified by a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA).
Library construction and sequencing
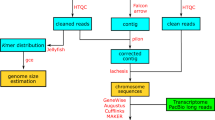
According to the manufacturer’s instructions, paired-end libraries for genome surveys with a 350 bp insert size were constructed using gDNA. Then, we sequenced this library with a strategy of 2 × 150 bp on the Illumina HiSeq. 2500 platform and obtained the raw data. For HiFi read generation, high-molecular-weight (HMW) gDNA was sheared to 15 Kbp before preparing a PacBio HiFi library. The genomic library was sequenced in CCS mode on the PacBio Sequel II system at Novogene (Tianjin). Subsequently, HiFi reads were generated from raw subreads using CCS workflow9 (v4.2.0) with a recommended setting. Finally, 31.20 Gb of CCS reads were yielded with a mean read length of 16.1 Kb resulted in 84-fold coverage of the T. ocellatus genome. The coverage was sufficient for haplotype-resolved assembly according to recommendations10. For Hi-C library construction, the MboI restriction enzyme was used to digest the cross-linked high molecular weight (HMW) gDNA. After 5′ overhang biotinylated and blunt-end ligation, the DNA was physically sheared into 300–500 bp fragments. Finally, the Hi-C library was sequenced with a strategy of 2 × 150 bp on the Illumina HiSeq. 2500 platform. In addition, 29.16 Gb of paired-end clean reads were generated from the Hi-C library. The RNA-seq library was constructed using the Illumina standard protocol (San Diego, CA, United States) and sequenced on the Illumina HiSeq. 6000 platform. In total, 32.47 Gb of paired-end short clean reads were generated from the RNA-seq library (Table 1).
Genome survey and assembly
Before assembly, the adapter sequences and low-quality reads generated from the Illumina platform were filtered using fastp (v. 0.23.1) software11., and the remaining reads were used for subsequent genome survey and assembly. To estimate the major characteristics of the genome, such as genome size, heterozygosity, and repeatability, genome surveys were performed using SOAPec (v. 2.01) and GenomeScope (v. 2.0) software with 17 K-mer frequencies. With a dominant peak depth of 124.92, the estimated genome size of T. ocellatus was 369.48 Mb, and the heterozygosity and repetitive sequence content were approximately 0.47 and 27.29%, respectively (Supplementary Table 1 and Supplementary Fig. 1). The estimated genome size is slightly smaller than that of other Takifugu species that were assembled by PacBio (previously reported; 373∼404 Mb)4,6. Then, the HiFi long reads along with paired-end Hi-C short reads were provided to HiFiasm12 (v0.16.1) to generate the monoploid and a pair of haplotype-resolved assembly contig graphs with default parameters. Using the Hi-C integrated algorithm, HiFiasm takes full advantage of phased graphs and long-range information to generate a haplotype-resolved assembly. Finally, three preliminary assemblies, including one monoploid assembly and two haploid assemblies, were yielded, which spanned 375.62 Mb (monoploid), 373.25 Mb (Haploid-1) and 372.15 Mb (Haploid-2), with a contig N50 length of 11.55 Mb, 4.86 Mb and 4.87 Mb, respectively (Table 2). The genome assembly was slightly larger than the estimated genome size of 369.48 Mb (Table 1) because some repeat fragments could be assembled by high-precision CCS reads13. Juicer14 and 3D-DNA15 were implemented to obtain the chromosome-level whole genome assembly for T. ocellatus. Afterwards, scaffolds were fine-tuned, and discordant contigs were removed from scaffolds by Juicebox16 assembly tools (Table 3; Fig. 1 and Supplementary Fig. 2).

Circos plots of the reference genome of T. ocellatus and syntenic relationship with other Takifugu species. (a): Circos plot of 22 chromosome-level scaffolds, representing annotation results of genes and transposable elements on these scaffolds. The tracks are: (1) 22 chromosome-level scaffold, (2) contigs which comprised the scaffolds (adjacent contigs on a scaffold are painted in different colors, (3) TE abundance of negative strand (grey), (4) TE abundance of positive-strand (purple), (5) gene length of negative strand (blue), (6) gene length of positive strand (green), (7) gene number of negative strand (yellow), (8) gene number of positive-strand (orange); (b): Circos diagrams showing T. ocellatus chromosome synteny relations with T. rubripes; (c): Circos diagrams showing T. ocellatus chromosome synteny relations with T. flavidus; (d): Circos diagrams showing T. ocellatus chromosome synteny relations with T. bimaculatus. Each coloured line in (b), (c) and (d) represents a 1 Kbp fragment match between two species. Chromosome numbers of T. ocellatus were re-ordered for better illustration.
Genomic repeat annotation
Repeat sequences of the T. ocellatus genome were identified with both homology-based and de novo strategies, following a previously established protocol17. First, RepeatModeler was utilized to detect repeats and generate a de novo repeat library. Combined with Repbase18, an ultimate repeat sequence library was constructed. Finally, RepeatMasker19 was employed to forecast repeat elements based on the library. TEclass (v2.1.3) was utilized to further identify unclassified repeats. To summarize the transposable element (TE) annotation results, buildSummary.pl of RepeatMasker was adopted. In addition, the Kimura divergence value of TE was calculated by calcDivergenceFromalign.pl, and TE landscapes were drawn by createRepeatLandscape.pl. Eventually, all repetitive regions were masked in the process of protein-coding gene annotation. By combining Repbase and de novo datasets, we obtained a total of 66.65 Mb of consensus and nonredundant repetitive sequences, which occupied more than 17.74% of the monoploid genome (Table 4 & Fig. 2a).

Gene and repeat annotations of the T. ocellatus genome. (a): Distribution of divergence rate for TEs in the T. ocellatus genome. (b): Veen diagram of fuctional annotation based on different databases.
Protein-coding gene finding and function annotation
For noncoding RNA (ncRNA) annotation, RNAmmer (v1.2) and tRNAScan (v1.3) were executed for rRNA and tRNA prediction, respectively. Other noncoding RNAs were detected by alignment against the Rfam database. Four types of noncoding RNAs, including 1,000 miRNAs, 810 tRNAs, 1,573 rRNAs and 775 sRNAs, were identified from the T. ocellatus genome (Table 4).
Structural annotation of the protein-coding genes was conducted using ab initio, homology-based and RNA-seq-based approaches, after all repeat sequences in the T. ocellatus genome were soft-masked. For homology-based gene prediction, the protein sequences of D. rerio20, O. latipes21, T. rubripes22, T. flavidus23 and T. bimaculatus24 were downloaded from the European Nucleotide Archive and provided to GenomeThreader (v.1.7.0)25. In addition, the RNAseq clean data were de novo assembled using Trinity software (v.2.10.0). Braker226 was employed to perform ab initio gene prediction using the transcripts assembled from RNAseq and known genes of D. rerio20, O. latipes21, T. rubripes22, T. flavidus23 and T. bimaculatus24. The optimal parameters were obtained after two rounds of model training. For another gene prediction approach, RNA-seq data were aligned to the T. ocellatus genome to assemble the transcriptome via hisat227 and stringtie28 (v2.1.4). Then, TransDecoder (v5.5.0) was adopted to predict the open reading frame (ORF) region. Last, a comprehensive gene set was produced by EvidenceModeler and annotated for protein-coding gene structure by PASA (v2.4.1)29.
For functional annotation of protein-coding genes, Diamond (v2.0.6) was applied to align protein-coding genes to the NR, TrembBL (http://www.uniprot.org/) and Swiss-Prot (http://www.uniprot.org/) protein databases with E-values < 1*10-5. The annotation of GO and KEGG pathways was performed using InterProScan (v4.8) and KEGG Automatic Annotation Server (KAAS).
Gene family identification and phylogenetic tree construction
To identify gene families among T. ocellatus and other representative species, the protein sequences of D. rerio (outgroup), G. aculeatus, H. comes, L. litulon, M. mola, O. latipes, T. bimaculatus, T. flavidus, T. nigroviridis, T. palembangensis, T. rubripes and T. septentrionalis were downloaded (Supplementary Table 2). Protein sequences shorter than 30 amino acids were filtered out in the above 13 proteome sets and provided to Orthofinder30 (v2.5.2) to construct orthologous groups. To reveal the phylogenetic relationships among T. ocellatus and 12 other species, single-copy orthologous genes were identified and used for the construction of the phylogenetic tree (Supplementary Table 3). The single-copy orthologues were further aligned using MUSCLE (v3.8.31). Then, RAxML31 (v8.2.12) with 1000 bootstrap replicates was executed to generate phylogenetic trees. The divergence time was estimated using MCMCTREE (PAML32 package) based on the molecular clock data in the TimeTree33 database (fossil time: zebrafish and medaka divided at 230 (million years ago (mya); 180.0–264.0 mya). The expansion and conversion gene families of T. ocellatus were identified by CAFÉ (v. 4.2).
Positive selection and rapidly evolving gene identification
To identify positively selected genes (PSGs) and rapidly evolving genes (REGs) in the T. ocellatus genome, the protein sequences of L. litulon, T. bimaculatus, T. palembangensis, T. rubripes, and T. septentrionalis were downloaded (Supplementary Table 2). We employed the software PRANK-MSA (v140110)34 with the parameters gaprate = 0.025 and gapext = 0.75 for coding sequence alignment of each homologous group. To examine the selective constraints on the genes, we estimated the dN/dS ratio (ω) using PAML (v4.4b)32. We tested three hypotheses: (1) H0, all branches have the same ω; (2) H1, the branch leading to T. ocellatus has a different ω, whereas the other branches have the same ω; and (3) H2, all branches have an independent ω. We used likelihood values and degrees of freedom of the three hypotheses to perform a likelihood-ratio test (LRT). We selected genes whose likelihood values for H1 were significantly larger (adjusted LRT p value of < 0.05) than those for H0 and genes whose likelihood values of H2 were not significantly larger than those of H1. In addition, we also ran branch-site models (model = 2; NSsite = 2) to detect the genes with positively selected sites in T. ocellatus. For the null hypothesis, we set ‘fix_omega = 1; omega = 1’, whereas for the alternative hypothesis, we set ‘fix_omega = 0; omega = 1.5’ with the tree ‘(((((T.bimaculatus,T. rubripes), T. ocellatus #1), T. palembangensis), T. septentrionalis), L. litulon)’.
In this study, a high-quality reference genome and two haplotype genomes of T. ocellatus were generated, which could contribute to further research on the genetic mechanism of freshwater adaptability and anadromous characteristics. The comparison between the genomes of freshwater-adapted T. obscurus and T. ocellatus will help us to understand whether there is convergent evolution for freshwater adaptation between these two species. In addition, as the first haplotype-level genome of Takifugu species, the T. ocellatus genome assembly constructed in this study will facilitate the wide use of T. obscurus as a valuable model species to investigate the evolutionary process of adaptive radiation and genetic mechanisms hidden within the compact genome. Combining such information with gene expression data and Hi-C data from different Takifugu species, we could deeply explore whether allele-specific gene expression and the 3D structure of the genome would accelerate speciation. Finally, the genome of T. ocellatus, as a potential freshwater aquaculture fish, will build a foundation for breeding projects, whose goal is excellent growth traits and freshwater breeding.
Data Records
The raw sequencing reads of all libraries are available from NCBI via the accession number of SRP40798435. The assembled genome is available in the NCBI with the accession number JAPVLW000000000 via the project PRJNA90163736. Besides, the assembled genome and sequence annotations are available in the figshare database with the DOI number: https://doi.org/10.6084/m9.figshare.20128412.v137.
Technical Validation
Evaluating the completeness of the genome assembly and annotation
To verify the integrity and accuracy of these assemblies, the completeness of the final genome assembly was assessed using Benchmarking Universal Single-Copy Orthologues (BUSCO)38 with the lineage database Actinopterygii_odb10. From 3,640 single-copy orthologues, ∼97.5% were fully discovered in the monoploid genome, ∼97.3%, and ∼97.1% were fully found in the Haploid-1 and Haploid-2 genomes (Supplementary Table 4). In addition, the Illumina short reads used for the genome survey were mapped to the genome using BWA39 and counted for mapping ratio determination using SAMtools40. As a result, the mapping ratios of the three assemblies were 96.19%, 95.65% and 96.03%, and the genome coverages of the three assemblies were 99.84%, 99.86% and 99.84%, respectively (Supplementary Table 5). The consensus quality value (QV) of genomes representing per-base consensus accuracy was estimated by Merqury41, and that of all three assemblies exceeded 45 (Supplementary Table 4). In addition, a total of 28,808 nonredundant protein-coding genes were successfully produced by combining de novo, homologous searching and transcriptome-assisted predictions. A total of 22,531 genes were successfully functionally annotated (Fig. 2b & Table 5). The number of genes of T. ocellatus (27,015) predicted through de novo prediction and homolog annotation was slightly greater than that of other species of Takifugu, such as T. bimaclatus (21,117)4 and T. obscurus (22,105)6, but slightly lower than that of T. flavidus (29,416)5. Hence, the high integration efficiency, mapping ratio, recognition rate of single-copy orthologues and gene number showed that three assemblies of T. ocellatus were of high quality.
To verify the accuracy of the contig anchoring, three chromosome-level assembled genomes (monoploid genome and two haploid genomes) were first aligned and named after the chromosome number of the published T. rubripes genome. Then, the monoploid assembly was aligned to 3 other species in the genus Takifugu, including T. bimaculatus, T. flavidus and T. rubripes. Two haploid assemblies were aligned mutually with a unit of 1 Kbp. The 22 chromosomes we identified in the T. ocellatus genome aligned exactly against the chromosomes of the other three Takifugu species, which suggested a high degree of concordance among them (Fig. 1). The haplotypes also showed strongly reciprocal collinearity (Supplementary Fig. 3).
Phylogenetic and evolutionary analysis
A total of 21,446 orthologous gene families were identified from the 13 related species (Supplementary Table 3). A total of 2,698 single-copy orthologous gene families in a 1:1:1 manner was identified and used for phylogenetic analysis (Supplementary Table 3). Phylogenetic analysis indicated that T. ocellatus speciated from the common ancestor of Takifugu at approximately 21.4 (mya; 15.3–27.6 mya) (Fig. 3), which was located at the base of the phylogenetic tree of the Takifugu genus, which was consistent with the previous phylogenetic relationship of the Takifugu genus based on mitochondrial and whole-genome resequencing8. In addition, our results showed that the divergence time between the Takifugu genus and the other freshwater Tetraodontidae species was 38.4 mya (27.9–51.0 mya). In addition, the phylogenetic relationship between Tetraodontiformes and other fish was also consistent with previous taxonomic studies4,5. Moreover, we uncovered 789 T. ocellatus gene families with expansion and 1,970 families with contraction (Fig. 3). GO enrichment analysis showed that the expanded gene families were mainly involved in the extracellular region (GO:0005576), lipid transport (GO:0006869), single-organism transport (GO:0044765) and growth factor activity (GO:0008083) (Supplementary Fig. 4 and Supplementary Table 6). On the other hand, the contracted gene families were mainly involved in xenobiotic transporter activity (GO:0042910), transmembrane transport (GO:0022857), secondary active transmembrane transporter activity (GO:0015291), and hydrolase activity (GO:0016787) (Supplementary Fig. 4 & Supplementary Table 7).

Phylogenetic analysis and divergence time tree of T. ocellatus and other representative species. (D. rerio, G. aculeatus, H. comes, L. litulon, M. mola, O. latipes, T. bimaculatus, T. flavidus, T. nigroviridis, T. palembangensis, T. rubripes, T. septentrionalis). The expansion (red) and contraction (blue) gene numbers were listed under the species name.
We identified 1,034 rapidly evolving genes with significant false discovery rate (FDR)-corrected p values (<0.05) in T. ocellatus (Supplementary Table 8). GO enrichment analysis showed that the REGs were mainly involved in RNA metabolic process (GO:0016070), cellular aromatic compound metabolic process (GO:0006725), nitrogen compound metabolic process (GO:0006807), and gene expression (GO:0010467) (Fig. 4 and Supplementary Table 9). In addition, using an FDR-corrected LRT p-value (adjusted LRT p value) cut-off of 0.05, we identified 767 PSGs in T. ocellatus (Supplementary Table 10). GO enrichment analysis showed that the REGs were mainly involved in intracellular (GO:0005622), small molecule metabolic process (GO:0044281), nitrogen compound metabolic process (GO:0006807), and oxoacid metabolic process (GO:0043436) (Fig. 4 & Supplementary Table 11).

The bubble diagram of GO enrichment of positive selection and rapidly evolving genes in T. ocellatus; (a–c): GO enrichment of the positive selection; (d–f): GO enrichment of the rapidly evolving genes.
For marine fishes, K+/Cl− cotransporters (KCCs) play important roles in epithelial ion transport and osmotic homeostasis6. In another freshwater-adapted pufferfish, T. obscurus, “potassium:chloride symporter activity (GO0015379)” was observed as an expanded gene family, which may be associated with the unique anadromous characteristics of T. obscurus6. For T. ocellatus, we identified several REGs that participated in the osmotic pressure regulation of K+/Cl−, such as potassium channel subfamily K member 3 (kcnk3), potassium channel subfamily K member 1 (kcnk1) and potassium channel subfamily K member 10 (kcnk10) (Supplementary Table 8). In addition, we identified several PSGs that were also involved in K+/Cl- transport, such as potassium channel subfamily K member 5 (kcnk5), solute carrier family 12 member 3 (slc12a3), and solute carrier family 26 member 6 (slc26a6) (Supplementary Table 10). These candidate genes may enhance the osmotic pressure regulation ability so that T. ocellatus can adapt to the freshwater environment.
Code availability
Genome annotation:
(1) RepeatMasker: parameters: -e ncbi -a -nolow -no_is -norna
(2) TE-class: parameters: all parameters were set as default
(3) Braker2: parameters: all parameters were set as default
(4) PASA:–ALIGNERS blat
(5) EvidenceModeler: parameters: all parameters were set as default
Genome assembly:
(1) CCS: parameters: all parameters were set as default
(2) HiFiasm: parameters: hifiasm -u -o genome.asm–h1 R1.fq.gz–h2 R2.fq.gz ccs.fa.gz
Gene family identification and phylogenetic analysis:
(1) RAxML: parameters: -f a -m PROTGAMMAAUTO
(2) MCMCTREE: parameters: all parameters were set as default
The parameters of other not mentioned analysis modules were used as default parameters. The other custom codes used in this analysis were mentioned in the methods sections.
References
Aparicio, S. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297, 1301–1310 (2002).
Santini, F. et al. Do habitat shifts drive diversification in teleost fishes? An example from the pufferfishes (Tetraodontidae). Journal of Evolutionary Biology 26, 1003–1018 (2013).
Kai, W. et al. Integration of the Genetic Map and Genome Assembly of Fugu Facilitates Insights into Distinct Features of Genome Evolution in Teleosts and Mammals. Genome Biology and Evolution 3, 424–442 (2011).
Zhou, Z. X. et al. The sequence and de novo assembly of Takifugu bimaculatus genome using PacBio and Hi-C technologies. Scientific Data 6 (2019).
Zhou, Y. T. et al. Chromosome genome assembly and annotation of the yellowbelly pufferfish with PacBio and Hi-C sequencing data. Scientific Data 6 (2019).
Kang, S. et al. Chromosomal-level assembly of Takifugu obscurus (Abe, 1949) genome using third-generation DNA sequencing and Hi-C analysis. Molecular Ecology Resources 20, 520–530 (2020).
Yang, Z. & Chen, Y. F. Differences in reproductive strategies between obscure puffer Takifugu obscurus and ocellated puffer Takifugu ocellatus during their spawning migration. Journal of Applied Ichthyology 24, 569–573 (2008).
Liu, B. et al. Genome-Scale Phylogenetic and Population Genetic Studies Provide Insight Into Introgression and Adaptive Evolution of Takifugu Species in East Asia. Frontiers in Genetics 12 (2021).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology 37, 1155-+ (2019).
Cheng, H. Y. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170-+ (2021).
Harder, A. M., Walden, K. K. O., Marra, N. J. & Willoughby, J. R. High-Quality Reference Genome for an Arid-Adapted Mammal, the Banner-Tailed Kangaroo Rat (Dipodomys spectabilis). Genome Biol Evol 14 (2022).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Systems 6, 256-+ (2018).
Bai, Y. L. et al. Chromosome-Level Assembly of the Southern Rock Bream (Oplegnathus fasciatus) Genome Using PacBio and Hi-C Technologies. Frontiers in Genetics 12 (2021).
Bao, W. D., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6 (2015).
Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 5, 4.10.1–4.10.14 (2004).
European Nucleotide Archive. http://identifiers.org/ena.embl:PRJNA11776 (2022).
European Nucleotide Archive. http://identifiers.org/ena.embl:PRJNA325079 (2022).
European Nucleotide Archive. http://identifiers.org/ena.embl:PRJEB31988 (2021).
European Nucleotide Archive. http://identifiers.org/ena.embl:PRJNA491395 (2022).
European Nucleotide Archive. http://identifiers.org/ena.embl:PRJNA508537 (2022).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Information and Software Technology 47, 965–978 (2005).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP plus and AUGUSTUS supported by a protein database. Nar Genomics and Bioinformatics 3 (2021).
Kim, D., Landmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–U121 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290-+ (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9 (2008).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20 (2019).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Yang, Z. H. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences 13, 555–556 (1997).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Loytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol Biol 1079, 155–70 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP407984 (2022).
Zhou, Z. Takifugu ocellatus breed Ocellated Puffer, whole genome shotgun sequencing project. GenBank https://www.ncbi.nlm.nih.gov/nuccore/JAPVLW000000000 (2022).
Zhou, Z. The genome of Takifugu ocellatus. figshare https://doi.org/10.6084/m9.figshare.20128412.v1 (2022).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
Acknowledgements
We acknowledge financial support from the Innovation and Industrialization Project of Takifugu breeding Industry (No.2017FJSCZY03).
Author information
Authors and Affiliations
Contributions
Q.Z. and P.X. conceived and supervised the study. Z.Z., M.Y. and L.L. collected the sample. Z.Z. and Q.H. performed bioinformatics analysis. Z.Z. and Q.H. drafted the manuscript. F.P. helped with manuscript preparation.
Corresponding author
Ethics declarations
Ethics statement
This work was approved by the Animal Care and Use Committee at the College of Ocean and Earth Sciences, Xiamen University. All the methods used in this study were carried out following approved guidelines.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, Q., Zhou, Z., He, Q. et al. Chromosome-level haplotype-resolved genome assembly for Takifugu ocellatus using PacBio and Hi-C technologies. Sci Data 10, 22 (2023). https://doi.org/10.1038/s41597-023-01937-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-01937-2
- Springer Nature Limited
This article is cited by
-
Decoding the fish genome opens a new era in important trait research and molecular breeding in China
Science China Life Sciences (2024)