

Abstract
Methanogenic archaea are main contributors to methane emissions, and have a crucial role in carbon cycling and global warming. Until recently, methanogens were confined to Euryarchaeota, but metagenomic studies revealed the presence of genes encoding the methyl coenzyme M reductase complex in other archaeal clades1,2,3,4, thereby opening up the premise that methanogenesis is taxonomically more widespread. Nevertheless, laboratory cultivation of these non-euryarchaeal methanogens was lacking to corroborate their potential methanogenic ability and physiology. Here we report the isolation of a thermophilic archaeon LWZ-6 from an oil field. This archaeon belongs to the class Methanosuratincolia (originally affiliated with ‘Candidatus Verstraetearchaeota’) in the phylum Thermoproteota. Methanosuratincola petrocarbonis LWZ-6 is a strict hydrogen-dependent methylotrophic methanogen. Although previous metagenomic studies speculated on the fermentative potential of Methanosuratincolia members, strain LWZ-6 does not ferment sugars, peptides or amino acids. Its energy metabolism is linked only to methanogenesis, with methanol and monomethylamine as electron acceptors and hydrogen as an electron donor. Comparative (meta)genome analysis confirmed that hydrogen-dependent methylotrophic methanogenesis is a widespread trait among Methanosuratincolia. Our findings confirm that the diversity of methanogens expands beyond the classical Euryarchaeota and imply the importance of hydrogen-dependent methylotrophic methanogenesis in global methane emissions and carbon cycle.
Similar content being viewed by others


Main
Methanogenesis, exclusively performed by methanogenic archaea, represents one of the oldest metabolisms on Earth5. Methanogens are major contributors to global methane emissions, and have a substantial role in climate change6,7. For many decades, it was widely believed that methanogens were restricted to the Euryarchaeota—an archaeal phylum that was originally proposed by C. Woese8. Yet, the emergence of technologies enabling deep sequencing of metagenomes from uncultured microbial communities challenged this view. In 2015, the key genes encoding the methyl-coenzyme M reductase (MCR) complex—a hallmark enzyme in methanogens9—as well as other genes of methanogenic pathways were identified in two metagenome-assembled genomes (MAGs) of ‘Candidatus Bathyarchaeota’, suggesting methanogenic potential in this non-euryarchaeal lineage1. Subsequently, the MCR complex genes and methanogenic pathway genes were identified in several uncultured non-euryarchaeal taxa, including ‘Candidatus Korarchaeota’3, ‘Candidatus Nezhaarchaeota’4,10 and ‘Ca. Verstraetearchaeota’2,11,12 and Thaumarchaeota13. Although introduced as phyla, ‘Ca. Bathyarchaeota’, ‘Ca. Korarchaeota’, ‘Ca. Verstraetearchaeota’ and Thaumarchaeota were later proposed to be classes within the phylum Thermoproteota as ‘Candidatus Bathyarchaeia’, ‘Candidatus Korarchaeia’, ‘Candidatus Methanomethylicia’ and Nitrososphaeria, respectively. ‘Ca. Nezhaarchaeota’ was reclassified as an order (‘Candidatus Nezhaarchaeales’) within ‘Ca. Methanomethylicia’14. However, whether these archaea actually perform methanogenesis is untestable for lacking cultivation.
It is known that methanogens use methanogenesis as the sole energy conservation mechanism15. Until now, five methanogenesis pathways have been characterized on the basis of the type of substrate metabolized, that is, aceticlastic, alkylotrophic, hydrogenotrophic, methoxydotrophic and methylotrophic16,17,18. Fermentative metabolism has been speculated for potential methanogens from the ‘Ca. Bathyarchaeia’1 and ‘Ca. Methanomethylicia’2 based on genomic analyses. Furthermore, MCR-containing MAGs from ‘Ca. Korarchaeia’ encode the genes required for dissimilatory sulfur metabolism3. However, no cultivated representatives of methanogens from these new lineages have been obtained and their physiology remains unclear.
‘Ca. Methanomethylicia’ has been predicted to possess the potential not only for methanogenesis, but also for the fermentation metabolism of sugars, peptides and amino acids2,19. This taxon is widely distributed, primarily in sediments of various anoxic environments, including hot springs, hydrothermal systems, marine and freshwater bodies, wetlands, and oil and gas fields2,20,21. Using a hybrid cultivation comprising both classical and high-throughput approaches over a 7 year period, we successfully obtained a stable co-culture containing one archaeal species from ‘Ca. Methanomethylicia’ (which we renamed ‘Candidatus Methanosuratincolia’), and also the pure culture. These cultures were used to investigate the physiology of methanogens beyond those affiliated with Euryarchaeota.
Isolation of Methanosuratincolia
Microorganisms affiliated with ‘Ca. Methanosuratincolia’ were rare in the original samples of oily sludge and oil-produced water in the Shengli oilfield (China), with a relative abundance of less than 1% of total archaea, as identified by 16S rRNA gene amplicon sequencing (Fig. 1a). After anaerobic incubation at temperatures ranging from 25 to 75 °C in basal medium with substrates such as acetate, propionate, butyrate, alkanes or crude oil, Methanosuratincolia members reached relative abundances of 8.1–82.5% at 45–65 °C (Extended Data Fig. 1). Subsequently, cultures showing a high abundance of Methanosuratincolia were tenfold serially diluted in 96-well microplates with mixed substrates and incubated at 55 °C. After several transfers over consecutive years, the archaeal diversity was reduced to one Methanosuratincolia-related species and a hydrogenotrophic methanogen closely related to Methanoculleus receptaculi22 (Fig. 1a (stage 2)). The simplified culture was then transferred into Hungate tubes supplemented with methanol without hydrogen and carbon dioxide, and M. receptaculi was eventually eliminated after subculturing 3–4 times (Extended Data Fig. 2). At this point, the retained single-archaeon culture was still accompanied by several bacterial taxa, which were again serially diluted and supplemented with antibiotics and lysozyme (Methods). A co-culture containing one Methanosuratincolia archaeon (strain LWZ-6) and Acetomicrobium sp. CY-2 was obtained (Fig. 1a,b (stage 4)) and maintained after consecutive transfers (Extended Data Fig. 3). Ultimately, the bacterium was removed by several subcultures with antibiotics (Fig. 1a,b and Extended Data Fig. 4d–f). The physiological characteristics of LWZ-6 were mainly investigated by using the co-culture before the isolation of the pure culture. On the basis of the pairwise 16S rRNA gene and genomic average nucleotide identity (ANI) analysis, strain LWZ-6 displays 100% and 97.2% identity to ‘Candidatus Methanosuratincola petrocarbonis’ V4 (MAGV00000000)2,23 (Supplementary Fig. 2), respectively. We here propose the Methanosuratincolia archaeon as Methanosuratincola petrocarbonis LWZ-6 (CCAM 1872; a taxonomic description is provided in the Methods).

a,b, Comparison of archaeal (a) and bacterial (b) communities at the genus level in original samples (OS) and the four isolation stages determined by 16S rRNA gene amplicon sequencing. The pure culture was obtained in stage 5. ND, not detected. c, CARD-FISH images of cells from the co-culture hybridized with nucleotide probes that target archaea ARCH-915 (green) and EUB-338 (red). Representative images are from 18 recorded images of n = 3 replicates. d,e, SEM images of the co-culture; the white arrows indicate LWZ-6 cells. f, TEM showing an ultrathin section of the coculture. The black arrows indicate LWZ-6 cells. SEM and TEM representative images are from 8 recorded images of n = 2 replicates. Scale bars, 10 μm (c), 5 μm (d), 500 nm (e) and 200 nm (f).
Analyses using catalysed reporter deposition fluorescence in situ hybridization (CARD-FISH) and scanning electron microscopy (SEM) of the co-culture revealed that LWZ-6 cells were small (approximately 0.5 μm in diameter) cocci, and that Acetomicrobium sp. CY-2 cells were rod shaped (Fig. 1c–e and Extended Data Fig. 5a–f). These two microorganisms did not seem to aggregate during growth observed under the light microscope (data not shown). Archaella were not observed under transmission electron microscopy (TEM) (Fig. 1f and Extended Data Fig. 5g–h). LWZ-6 lacked cofactor F420 autofluorescence with the blue-green light under the ultraviolet microscope, which is consistent with the absence of F420 biosynthetic genes in its genome (see below; Supplementary Table 4).
Methyl-reducing methanogenesis of LWZ-6
To study the methanogenic ability of strain LWZ-6, the co-culture of strain LWZ-6 and CY-2 was incubated in fresh medium supplemented with methanol and hydrogen in the headspace. After 18 days of incubation at 55 °C, 0.36 ± 0.05 mmol methanol was used by the culture and methane accumulated up to 0.41 ± 0.02 mmol (Fig. 2a and Extended Data Fig. 6a,b). At this point, strain LWZ-6 reached 109 cells per ml as determined by quantitative PCR (qPCR) analysis of the mcrA gene (Fig. 2a). The methanogenesis rate was calculated at about 150 fmol per cell per day. Both methane production and growth of strain LWZ-6 were inhibited by the addition of the common methanogenesis inhibitor of 2‐bromoethanesulfonate (BES) (Fig. 2b). Accumulation of hydrogen in the presence of BES was most probably generated from the fermentation of yeast extract by the coexisting bacterium Acetomicrobium sp. CY-2 (Extended Data Fig. 6c,e and Supplementary 1d). An hydrogen consumption of 0.41 ± 0.07 mmol by the archaeon was estimated by subtracting the amount of hydrogen present in the incubation without added BES from the total amount in the incubation with BES (Fig. 2b). Together, 1 mol methanol and 1 mol hydrogen converted to 1 mol methane (CH4) could be calculated, in accordance with the stoichiometry of the reaction: CH3OH + H2 → CH4 + H2O. Methane was still produced when Acetomicrobium sp. CY-2 was removed (Extended Data Fig. 4), and identical maximum specific methanogenesis rates were observed in the co-culture or in pure culture of LWZ-6 alone (0.29 per day and 0.24 per day, respectively).

a, Methanogenesis activity when the M. petrocarbonis LWZ-6–Acetomicrobium sp. CY-2 co-culture was incubated with methanol and hydrogen. b, Methane (CH4) production in the presence of BES. Data are mean ± s.d. of triplicates. Methane and hydrogen were measured in the stationary phase. The co-culture was incubated in 50 ml fresh medium at 55 °C. Methanol, 10 mM; BES, 10 mM; H2, 10 kPa. µ represents the specific growth rate of strain LWZ-6. c, Stable isotope tracer analysis of the 13C-methanol produced when supplemented with different ratios (1, 2, 4, 8%) of 13C-methanol (100% purity) addition. d, SEM images before NanoSIMS analysis targeting strain LWZ-6. The white arrow indicates strain LWZ-6. SEM images are representative of n = 26 recorded images from 3 samples. e,f, NanoSIMS ion images of 12C and 13C in strain LWZ-6 when incubated with labelled methanol. 13C/12C = 1.1% represents the relative abundance in cells. Cts, counts. The white arrow indicates strain LWZ-6. The NanoSIMS representative images are from n = 7 recorded images from 3 samples. Scale bars, 2 μm (d), 2 μm (e) and 2 μm (f). g, Lipid-SIP analysis showing the 13C-labelled archaeol (dark green) and Me-GMGT (brown) determined in strain LWZ-6 when the co-culture was grown with 13C-labelled acetate, methanol, NaHCO3 or yeast extract (YE). DoL, the total degree of 13C-labelled lipids. Data are mean ± s.d. of triplicates. All symbols represent the mean of three individual incubations, and the error bars represent the s.d. of triplicates; invisible error bars are smaller than the symbols.
To further verify methyl-reducing methanogenesis by strain LWZ-6, stable-isotope-tracing experiments supplemented with different amounts of 13C-labelled methanol were performed in the co-culture. The heavy-carbon fraction in the produced methane shows a linear increase with the concentration of initial 13C-methanol, with a slope of 0.99 (Fig. 2c). Meanwhile, the 13C/12C ratio of the generated CO2 had a slight change from 1.08% to 1.44% (Extended Data Fig. 7), indicating that methanol is almost completely converted into methane. No labelled methane was produced when the co-culture was incubated with 13C-labelled acetate or 13C-labelled carbon dioxide (data not shown). Moreover, nanoscale secondary ion mass spectrometry (NanoSIMS) scanning and lipid stable isotope probing (lipid-SIP) analysis revealed that the biomass and lipid of strain LWZ-6 was not labelled during 13C-methanol incubations (Fig. 2d–g). Lipid-SIP detected a 13C-labelled archaeol and methylated glycerol monoalkyl glycerol tetraether (Me-GMGT) fraction when strain LWZ-6 was incubated with 13C-labelled acetate, CO2 or yeast extract, indicating that they were required as carbon sources (Fig. 2g). Strain LWZ-6 also produced methane when grown on monomethylamine, but not on other C1-methylated compounds of dimethylamine, trimethylamine and methanethiol (Extended Data Fig. 8a). Thus, these results confirmed that strain LWZ-6 uses hydrogen-dependent methylotrophic methanogenesis as its energy-conservation pathway.
In the co-culture, yeast extract or casamino acids (minimum at 0.05 g l−1) were essential for the growth of strain LWZ-6 (Extended Data Fig. 8b). The methanogenesis conditions of strain LWZ-6 in the coculture were observed, with an optimal temperature at 55 °C, a pH at 6.0–7.0 and NaCl of 9 g l−1 (Extended Data Fig. 8c–e).
Metabolic pathways of LWZ-6
The complete 1.54 Mb circular genome of strain LWZ-6 (DNA G+C content, 54.42 mol%; 1,606 open reading frames) was obtained by reassembly of Nanopore and Illumina sequencing data (Supplementary Table 3). The genome contains the whole suite of genes encoding methanol methyltransferase (MtaABC) for methanol use and McrABG for methane production (Fig. 3 and Supplementary Table 1). The genome contains two copies of the monomethylamine transferase subunit (mtmB) but not the methylcobamide: CoM methyltransferase gene (mtbA), which is most likely replaced by mtaA for transferring monomethylamine to CH3-S-CoM24 (Fig. 3 and Supplementary Table 1). The genome lacks the archaeal methyl branch of Wood–Ljungdahl pathway genes, which are involved in carbon dioxide-reductive methanogenesis or methanol oxidation in canonical methylotrophic methanogenesis, and N5-methyltetrahydromethanopterin: coenzyme M methyltransferase (Mtr) complex (Fig. 3 and Supplementary Tables 1 and 2). This provides the genetic basis for strain LWZ-6 to exclusively perform obligately hydrogen-dependent methylotrophic methanogenesis.

The bold arrows indicate the identified genes in the complete circular genome, and the dotted line represents the genes that are not found in the genome. Proteins labelled in red, blue, green, orange and brown are involved in methanogenesis, gluconeogenesis, the nucleotide salvage pathway, amino acid biosynthesis and the TCA cycle, respectively. The transcriptome data of M. petrocarbonis LWZ-6 was collected at the log phase of methane production with the addition of methanol and hydrogen. The transcript abundance rank was based on the log2[fragments per kilobase million (FPKM)] values as indicated by coloured squares referenced to that at the bottom right corner. The genes described in the figure and their abbreviations are listed in Supplementary Table 2. Pi, inorganic phosphate, Fdred, reduced ferredoxin; Fdox, oxidized ferredoxin.
Strain LWZ-6 may use two mechanisms for heterodisulfide reduction. One possible mechanism involves a heterodisulfide reductase (HdrD) and cytoplasmic flavin adenine dinucleotide-containing dehydrogenase (GlcD) coupling an F420-methanophenazine-oxidoreductase-like complex for heterodisulfide reduction and ferredoxin reduction. The other is through an energy-converting complex (Ehd) formed by a NiFe hydrogenase and HdrBC, which may couple the exergonic heterodisulfide reduction with hydrogen oxidation to ion translocation over the membrane, thereby building an ion motive force12. An energy-converting hydrogenase B complex (Ehb) might be used for the oxidation of hydrogen and the reduction of ferredoxin25. A proton gradient could be generated by Ehd, F420-methanophenazine-oxidoreductase-like complex and Mnh (Na+/H+ antiporter) to drive V/A-type ATP synthase to produce ATP (Fig. 3 and Supplementary Tables 1 and 2). The absence of the methyl branch of Wood–Ljungdahl pathways in the LWZ-6 genome leaves all of the intermediate reactions in the methanogenic pathway unconnected with gluconeogenesis. We speculate that acetate conversion to acetyl-CoA, which is then combined with carbon dioxide to form pyruvate, could be the pathway channelled to gluconeogenesis for biomass generation in strain LWZ-6 (Fig. 3).
The exponential growth phase of strain LWZ-6 grown on methanol and hydrogen was analysed using transcriptomics. Genes of mcrABG, mtaABC, hdrD and glcD ranked in the top 1%, and the hydrogenases Ehb genes ranked at 13% to 47% and Ehd genes ranked at 35% to 62% among the transcribed ones (Fig. 3 and Supplementary Table 2); thus, we verified the hydrogen-dependent methylotrophic methanogenesis activity of strain LWZ-6.
Diversity of Methanosuratincolia
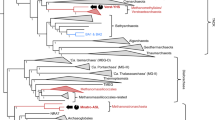
We obtained 23 Methanosuratincolia MAGs from our various enrichment cultures (Extended Data Fig. 1). One MAG could be assigned to strain LWZ-6 and was replaced by the latter’s complete genome sequence for the analyses. Genome completeness of the obtained MAGs ranged from 75% to 100% (Supplementary Table 3). On the basis of phylogenomic analysis and gene- and genome-level identities including publicly available MAGs from Methanosuratincolia, the 23 genomes could be assigned to three genera and five species in the order Methanosuratincolales. Of these, one MAG (cluster 4) represented the first representative of a new genus for which we propose the name ‘Candidatus Methanoluiimicrobium’. Two other MAGs (cluster 2) represented a new species within an existing genus (Fig. 4, Supplementary Fig. 2 and Supplementary Table 3; taxonomic descriptions are provided in the Methods). Analyses of five representative genomes—one from each of the five species-level clusters—indicated that they all have the MCR complex and key genes for hydrogen-dependent methylotrophic methanogenesis (Fig. 4c and Supplementary Table 4). Phylogenomic tree reconstruction supplemented with 16S rRNA gene sequence similarity26 and average amino acid identity (AAI)27 using high-quality reference genomes covering the entire Methanosuratincolia confirmed the existence of three order-level clades: Methanosuratincolales, ‘Ca. Nezhaarchaeales’ and ‘Candidatus Culexarchaeales’ (Fig. 4a). Moreover, two MAGs could be assigned to a newly proposed order that we tentatively name ‘Candidatus Aukarchaeales’, named after the Auka hydrothermal vent from which one of the MAGs was recovered28. Genome mining revealed that all Methanosuratincolales and ‘Ca. Aukarchaeales’ MAGs contained hydrogen-dependent methylotrophic methanogenesis pathways (Fig. 4c and Supplementary Table 4). The MAGs of ‘Ca. Nezhaarchaeales’ and ‘Ca. Culexarchaeales’ often lacked several methanogenesis key genes (Fig. 4c), although this could be due to genome incompleteness. Recovery of complete genomes from these lineages will reveal whether this pathway is also universally present in these orders. Notably, none of the ‘Ca. Culexarchaeales’ MAGs had the potential for hydrogen-dependent methylotrophic methanogenesis, indicating that this ability was lost when the ‘Ca. Culexarchaeales’ lineages split from the common ancestor of ‘Ca. Culexarchaeales’ and ‘Ca. Nezhaarchaeales’. Phylogenetic analysis of methyl-coenzyme M reductase subunit A (McrA) and McrA-like homologues showed a highly supported Methanosuratincolia clade in which distinct subclades of Methanosuratincolales and ‘Ca. Aukarchaeales’ McrA could be observed (Fig. 4b). These findings indicate that hydrogen-dependent methylotrophic methanogenesis is probably a widespread physiological property in Methanosuratincolia. We also identified the McrA-like homologue involved in the alkane metabolism in clusters 2 and 4. These clusters, along with the ethanogenesis29 and short-alkanes oxidization speculated previously30, expanded the metabolic repertoire in Methanosuratincolia. However, this needs further verification after cultivation.

a, The phylogenomic tree was constructed using IQ-TREE (LG+C60+F+G model) with 100 nonparametric bootstrap replicates. Five representative Methanosuratincolia MAGs were chosen for analysis (detailed information is provided in Supplementary Table 3). b, mcrA phylogeny of five clusters (coloured text). The tree was generated using IQ-TREE (Q.pfam+C40+F+G8 model) with 100 nonparametric bootstrap replicates. c, The methanogenetic features of five Methanosuratincolia clusters recovered in this study in comparison to other hydrogen-dependent methylotrophic methanogens. Circles that are one-quarter, one-half, three-quarters and completely filled correspond to genes/gene sets identified in at least 25, 50, 75 and 100% of MAGs, respectively. Circles with a star symbol correspond to genes/gene sets identified in less than 25% of MAGs. Numbers in brackets represent the numbers of MAGs analysed in the lineage. The genes described in the figure and their abbreviations are listed in Supplementary Table 4.
Screening the global distribution of Methanosuratincolia-specific 16S rRNA genes and genomes indicated that they are present in a variety of environments, particularly in subsurface sediments2,19,20 (Extended Data Fig. 9 and Supplementary Table 5). The environmental advantages of hydrogen-dependent methylotrophic methanogenesis over that of hydrogenotrophic methanogenesis can lie in the requirement for lower hydrogen partial pressures31. Moreover, methylated compounds such as plant-derived methanol are abundant in nature32,33. Along with the confirmation of hydrogen-dependent methylotrophic methanogenesis in Methanosuratincolia and their global distribution in subsurface environments, several other methanogenic lineages within the Euryarchaeota also implement hydrogen-dependent methylotrophic methanogenesis34,35,36,37, highlighting the significance of this methanogenic pathway in global methane emissions.
LWZ-6 is incapable of fermentative growth
The fermentative metabolism in methanogens from Methanosuratincolia was proposed based on metagenome analyses suggesting that they may grow and gain energy independent of methanogenesis2, which implies their alternative role in the global carbon cycle. However, strain LWZ-6 exhibited no active growth when incubated with glucose, pyruvate or acetate without BES addition. Similarly, no growth was observed with amino acids, casamino acids, keratin hydrolysates or yeast extract with BES in the absence of methane production (Extended Data Table 1). Strain LWZ-6 lacks sugar transporter genes and genes encoding pyruvate kinase and phosphofructokinase specific for the glycolysis pathway. By contrast, genes encoding phosphoenolpyruvate synthase and fructose-1,6-bisphosphatase, both specific in the gluconeogenesis pathway38, are present in the genome of strain LWZ-6. Genes encoding peptide and amino acids transporters, aminotransferases and 2-oxoacid:ferredoxin oxidoreductases (Kor and Ior) and aldehyde ferredoxin oxidoreductases (Aor) were found, but they may also be associated with the biosynthesis of amino acids12,39 (Fig. 3). Energy generation through fermentation was previously proposed through substrate-level phosphorylation by archaeal adenosine (ADP)-forming acetyl-CoA synthetase (Acd) converting acetyl-CoA to acetate2. AcdA and AcdB (the alpha and beta subunits of Acd, respectively) display 52.95% and 50.88% amino acid sequence similarity to the AcdA and AcdB of the hyperthermophilic euryarchaeaon Pyrococcus furiosus DSM 3638T, respectively. The P. furiosus Acd has been shown to exhibit an acetyl-CoA to acetate converting activity at 90 °C (ref. 40), whereas strain LWZ-6 grew optimally at 55 °C, a temperature at which Acd catalyses the reverse reaction, converting acetate to acetyl-CoA40. Thus, this might explain why strain LWZ-6 was unable to fermentatively grow.
Conclusion
The field of archaeal methanogenesis has expanded considerably over the past decade, many of these organisms are unrelated to the classical euryarchaeal methanogens. Comparative genomic and phylogenetic analyses of several as-yet-uncultured archaeal lineages indicate that methanogenesis and methane-associated pathways are ancient, potentially dating back to the last archaeal common ancestor29,41. However, relying solely on DNA analysis limits studies on the physiology of these microorganisms.
Here we isolated and characterized a methanogen outside Euryarchaeota—M. petrocarbonis LWZ-6 in the phylum Thermoproteota. We further demonstrated that it uses hydrogen-dependent methylotrophic methanogenesis as the exclusive energy-conservation pathway, rather than the alternative metabolism previously predicted from metagenomics analysis2. Strain LWZ-6 produces methane at a parallel rate and yield as the euryarchaeal methanogens42, further indicating that it represents an authentic methanogen. Other cultivation studies also confirmed that Methanosuratincolia43 and ‘Ca. Korarchaeia’ (Krukenberg, V. and Kohtz, A. J., unpublished data), outside the Euryarchaeota, are strict methanogens.
Analysis of metagenomic data revealed that potential hydrogen-dependent methylotrophic methanogens, including those that are part of the Methanosuratincolia, are globally distributed across various anoxic subsurface environments21. This indicates that the role of hydrogen-dependent methylotrophic methanogenesis in methane-emitting environments deserves re-evaluation as it is likely to impact efforts to model the global methane emissions.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Enrichment and cultivation conditions
The oily sludge and oil-produced water used as inoculum were collected from the Shengli oilfield in China (37° 54′ N, 118° 33′ E). After collection, the samples were stored anaerobically at 4 °C. Additional details and parameters of the oily sludge were described previously16. The basal medium for incubation was made of 9 g l−1 NaCl, 3 g l−1 MgCl2·6H2O, 0.15 g l−1 CaCl2·2H2O, 0.3 g l−1 NH4Cl, 0.2 g l−1 KH2PO4, 0.5 g l−1 KCl, 0.5 g l−1 cysteine-HCl, 1 ml l−1 resazurin solution and 2 ml l−1 trace element solution44. The medium was boiled for 30 min and then dispensed in serum bottles (Shuniu) with butyl rubber stoppers (Bellco Glass). The headspace was replaced by 99.999% oxygen-free N2. After sterilization at 121 °C for 30 min, the basal medium was supplied with filter-sterilized vitamin mixture (2 ml l−1)45, vitamin B12 (2 ml l−1) and vitamin B1 (2 ml l−1) solutions. Fresh medium was made of the basal medium supplied with 10 mg l−1 filter-sterilized 2-mercaptoethanesulfonic acid (CoM), 1% selenite-tungstate solution46, 0.5 g l−1 yeast extract and 20 mM 2-morpholinoethanesulfonic acid monohydrate (MES) buffer. Then, 1 M HCl or NaOH solutions were used to adjust the pH of the medium to 6.5–7.0 before use. The oil-produced water incubated with autoclaved 20 mM acetate, 20 mM propionate, 20 mM butyrate, and oily sludge incubated with n-hexadecane (1 ml l−1, Sigma-Aldrich), eicosane (1 ml l−1, Sigma-Aldrich), and alkane mix (including n-docosane (1 g l−1, Sigma-Aldrich), n-hexadecylcyclohexane (1 ml l−1, TCl) and n-hexadecylbenzene (1 ml l−1, TCl) or crude oil (1 g l−1) as substrates in basal medium were performed at temperatures ranging from 25 °C to 75 °C. The enrichments with the relative abundance of Methanosuratincolia above 20% were chosen for further isolation (Extended Data Fig. 1). These cultures were tenfold serially diluted in 96-well microplates with fresh medium of different substrates (10 mM acetate, 10 mM glucose, 10 mM methanol, 10 mM lactate, 10 mM pyruvate, 10 mM succinate, 10 mM formate, 0.5 g l−1 casamino acids, 0.5 g l−1 rumen fluid) at 55 °C. Detection of Methanosuratincolia in 96-well microplates was determined by PCR with specific- Methanosuratincolia primer (MSR4F/MSR4R; Supplementary Table 6). After transfer three times with a dilution of 1:100,000 in mixed substrates of acetate, methanol and lactate, Methanosuratincolia was detected. These positive Methanosuratincolia cultures in 96-well microplates were transferred into 10 ml Hungate tubes. Consecutive subcultures with dilutions varying from 0.1% to 10% in fresh medium supplemented with 10 mM methanol were then conducted in the Hungate tubes. To remove bacteria, tenfold serial dilutions supplemented with 500 mg l−1 lysozyme and several antibiotics including 100 mg l−1 ampicillin, 10 mg l−1 chloramphenicol, 100 mg l−1 gentamicin, 100 mg l−1 kanamycin, 100 mg l−1 streptomycin and 50 mg l−1 vancomycin separately or mixed together were used. To eliminate Acetomicrobium sp. CY-2, 10- to 100-fold serial dilutions supplemented with mixed 500 mg l−1 erythromycin, 200 mg l−1 streptomycin and 200 mg l−1 vancomycin were performed in Hungate tube with 5 ml fresh medium, supplemented with 0.5 g l−1 casamino acids, 50 mM methanol, 10 kPa H2, 2 mM sodium acetate and 1 mM NaHCO3 at 55 °C.
If not mentioned otherwise, the Methanosuratincolia cultures were incubated in 120 ml serum bottles with 50 ml fresh medium, 10 mM methanol, and 10 kPa H2 at 55 °C. BES was added to a concentration of 10 mM. Methylated compound use was examined by adding 5 mM dimethylamine, 5 mM methanol, 5 mM methanethiol, 5 mM monomethylamine or 5 mM trimethylamine into fresh medium at 55 °C. Temperature, pH, salt tolerance and essential growth factor tests were performed as previously described47. All incubations were performed triplicate in the dark without shaking.
In the stable isotope tracer experiment, the Methanosuratincolia co-culture (strain LWZ-6/CY-2) was incubated with labelled substrates. To determine the methanogenesis pathway, 13C-labelled methanol (100% purity) (Sigma-Aldrich) was added to the medium with 13C-labelling ratios of 1, 2, 4 and 8%, unlabelled methanol was used as the control group. Methanol was added to a final concentration of 10 mM. The actual content of the initial 13C-labelled methanol was measured by stable isotope mass spectrometer (Delta V advantage, Thermo Fisher Scientific). To determine the use of carbon sources, 5 mM 1,2-13C-acetate (Sigma-Aldrich), 1% (w/w) 13C-NaHCO3 (Sigma-Aldrich) or 0.5 g l−1 13C-yeast extract were used in fresh medium with 10 mM unlabelled methanol. 10 mM 13C-methanol was used in fresh medium. The 13C-yeast extract was made from the labelled 13C cell extract of Pichia pastoris48. In brief, P. pastoris was incubated in a minimal medium48 with 2 g l−1 13C6-d-glucose (Sigma-Aldrich) as the sole carbon source. After incubation for 4 days at 28 °C, the cells were centrifuged at 8,000g for 10 min and washed three times with PBS (1 M, pH 6.8). The washed cells were then ultrasonicated on ice for 50 min. The supernatant from the cells was collected, and the precipitate was ultrasonically lysed in 50% methanol for 10 min twice and centrifuged. All of the supernatants were combined and freeze-dried for further use. All of the incubations were performed in triplicate at 55 °C in the dark without shaking.
To determine fermentative growth, the Methanosuratincolia co-culture (strain LWZ-6/CY-2) was incubated with different substrates including 20 mM acetate, 20 mM glucose, 20 mM pyruvate or 5 g l−1 yeast extract in fresh medium. The Methanosuratincolia co-culture was also incubated with amino acid mixtures (concentration for each amino acid of 4 mM), 5 g l−1 casamino acids, 5 g l−1 keratin hydrolysates or 5 g l−1 yeast extract in basal medium with 10 mM BES added in each culture. The 20 amino acids include alanine, arginine, aspartate, cysteine, methionine, glutamate, glycine, histidine, lysine, l-asparagine, l-glutamine, l-isoleucine, l-leucine, l-phenylalanine, l-proline, l-serine, l-threonine, l-tryptophan, l-tyrosine and l-valine. The stereoisomer not given is the mixtures of l and d configurations. All of the incubations were performed in triplicate at 55 °C in the dark without shaking.
Methane production and qPCR were used to determine the growth of strain LWZ-6. The specific growth or methane production rate (µ) of strain LWZ-6 at the log phase was calculated according to µ = (ln[X2] − ln[X1])/(t2−t1), in which X is the copy numbers of strain LWZ-6 or methane production at incubation time t. The methanogenic rate was calculated according to the equation previously described49.
Chemical analysis
The concentration of CH4 and CO2 in the headspace was measured using the Agilent GC 7820A gas chromatography (GC) system equipped with a Porapak Q column (length, 3 m; inner diameter, 0.32 mm) and a thermal conductivity detector50 using H2 (99.999%) as carrier gas at a flow rate of 27 ml min−1. The GC temperature of injection, column and the thermal conductivity detector was 65 °C, 120 °C and 130 °C, respectively. The concentration of H2 in the headspace was determined using the same Agilent GC 7820A parameters with nitrogen as the carrier gas at a flow rate of 38 ml min−1. The gas pressure in Hungate tubes or serum bottles was measured using a barometer (Ashcroft) at room temperature. Each measurement was carried out by injection of 0.1 ml gas using a pressure lock syringe (Vici) at room temperature.
Methanol was measured using the Agilent GC 7890A GC system equipped with a DB-WAX column (length, 30 m; inner diameter, 0.32 mm) and a flame ionization detector using nitrogen (99.999%) as a carrier gas at a flow rate of 27 ml min−1. The initial oven temperature was 50 °C, held for 1 min, and increased to a temperature of 220 °C at a rate of 60 °C min−1; the flame ionization detector temperature remained at 250 °C. Acetate was determined by high-performance liquid chromatography (Agilent HPLC 1200)51.
The carbon isotopic compositions of CH4 and CO2 in the headspace were determined by coupling GC and a mass spectrometer (IsoPrime100)52. In brief, CH4 and CO2 in gas samples were first separated by GC (Agilent GC 7890B) using the HP-PLOT/Q column (30 m; inner diameter, 0.32 mm; film thickness, 20 μm). The carrier gas was helium (99.999%), with a flow rate of 2.514 ml min−1. The oven and injector temperatures were 60 °C and 105 °C, respectively. The CH4 was then converted to CO2 in a combustion furnace (IsoPrime) at a temperature of 1,050 °C. The carbon isotopic composition of CO2 was measured by the IsoPrime100 isotope ratio mass spectrometer. The carbon isotope composition of methanol was determined by stable isotope mass spectrometer (Delta V advantage; Thermo Fisher Scientific) coupled with TraceGC 3000 and equipped with the HP-INNOWAX (30 m × 0.25 mm × 0.25 μm) column using helium (99.999%) as the carrier gas at a flow rate of 1.5 ml min−1. BCR-653 WINE (ethanol, low level) (JRC-IRMM) was used as a standard.
The labelled methanol in the medium and labelled CH4 in the headspace was calculated using the following equations:
Labelled methanol (mol) = total methanol × 13C-methanol content − total methanol × 13C-methanol in control group
Labelled CH4 (mol) = total CH4 × 13C-CH4 content − total CH4 × 13C-CH4 in control group
The control group refers to the culture without addition of 13C-labelled methanol.
DAPI staining
Cells were collected and centrifuged at 16,000g for 10 min. The cells were then washed twice with PBS (1 M, pH 6.8) and fixed in 4% formaldehyde for 3 h. The fixed cells were washed again and mixed with Mini-Q water. Cells were dropped on the slide and dyed with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) staining solution (Beyotime Biotechnology). The dyed cells were dried and mounted with antifade mounting medium (Beyotime Biotechnology). The mounted slide was imaged under the laser-scanning confocal microscope (ZEISS).
CARD-FISH
Samples were fixed in 4% formaldehyde for 3 h and then washed with PBS (1 M, pH 6.8). The cells were collected on a 0.22 μm filter. Embedding was performed with 0.2% agar on the filter. The embedded filter was then dehydrated with 96% pure ethanol. Permeabilization of the samples was performed with lysozyme solution (0.05 M EDTA pH 8.0, 0.1 M Tris/HCl pH 8.0, 10 mg ml−1 lysozyme, Sigma-Aldrich) at 37 °C for 60 min and proteinase K solution (0.05 M EDTA pH 8.0, 0.1 M Tris/HCl pH 8.0, 15 μg ml−1 proteinase K, Merck) at room temperature for 5 min. The samples were hybridized with 16S rRNA gene-specific oligonucleotide probes of EUB-338 targeting bacteria and ARCH-91553,54 for Archaea (Takara Bio). The optimal formamide concentration in hybridization buffer for the stringency of probes was tested with increasing formamide concentrations (20–45%). 20% and 35% formamide were chosen for EUB-338 and ARCH-915 for hybridization, respectively. Double hybridization was performed after inactivation of peroxidases from the first hybridization. Inactivation of cells was conducted by incubating with endogenous peroxidases (10 ml methanol, 50 ml 30% hydrogen peroxidase) at room temperature for 30 min in the dark. The signal amplification was continued using tyramide in dark conditions. Alexa Fluor 488 and Alexa Fluor 594 (AAT Bioquest) were used for laser-scanning confocal microscopy observation (Zeiss).
SEM
Cells were fixed with 2.5% glutaraldehyde for 1 h at room temperature. The samples were then washed with sterilized ultrapure water for 5 min twice. Dehydration of the samples was performed by washing with increasing concentrations of ethanol (30%, 50%, 70%, 80%, 90%, 95% and 100%, each for 10 min). Cells were finally sputter-coated with osmium (Hitachi) and observed under SEM (FEI).
TEM
The samples were fixed with 3% glutaraldehyde and then with 1% osmium tetroxide. The cells were dehydrated with acetone at concentrations of 30%, 50%, 70%, 80%, 90% and 95% once, and 100% three times. The cells were then incubated in a mixture of acetone and resin (Quetol-812: Nissin) with a ratio of 3:1, 1:1, 1:3, and polymerized with resins. The polymerized cells were made into 60–90-nm ultrathin slices using the ultramicrotome (UC&rt, Leica) and the slices were unfolded and mounted onto copper grids. The grids were stained with uranyl acetate for 15 min and then stained with lead stain solution for 2 min, after which the grids were imaged using a transmission electron microscope (JEOL).
NanoSIMS
The cells were fixed in 2.5% glutaraldehyde for 2 h. The fixed samples were then rinsed three times in PBS buffer to remove excess glutaraldehyde. The rinsed samples were dropped onto silicon wafers, and then dehydrated sequentially with 25%, 50%, 80% and 100% solutions of ethanol. SEM imaging was used to localize regions of interest (ROI) with cocci cells of LWZ-6 for further NanoSIMS analysis. Before SEM imaging, the wafers were coated with 5 nm platinum. SEM was conducted on the Zeiss EVO18 system at a working distance of 6 mm and an electron high tension of 2.0 kV. The wafer samples were further analysed using the NanoSIMS 50L (CAMECA) system using a Cs+ primary source55,56. Secondary ions of 12C and 13C were collected by electron multipliers. The samples were scanned in a 10 × 10 µm2 area with a 256 × 256 pixel raster. The 13C/12C ratios of the ROI were measured using the OpenMIMS plugin in ImageJ (v.1.45) (http://www.nrims.hms.harvard.edu/NRIMS_ImageJ.php). All of the images were corrected for the electron multiplier dead time (44 ns) and drift corrected.
Lipid-SIP
Lipids of the culture cell were extracted using an acid hydrolysis extraction method57. In brief, the culture cells were collected by centrifugation at 12,000g for 10 min, the cells were acid hydrolysed by 10% hydrochloric acid in methanol at 70 °C for 8 h and then phase-separated by adding dichloromethane and ultrapure water. The total lipid extracts were filtered and dried before analysis.
The total lipid extracts were analysed using the Waters ACQUITY I-Class Ultra-performance liquid chromatography (UPLC) coupled to an SYNAPT G2-Si quadrupole time-of-flight (qTOF) high-resolution mass spectrometer (HRMS) through an electrospray ionization device. The MS setting was identical to that previously described58. The mass acquisition mode was Fast-DDA (digital differential analyser) with a mass range for MS m/z of 100–2,000 and MS2 m/z of 50–2,000.
The raw data were processed with Masslynx software (v.4.1). The unlabelled samples were used for lipid structure identification, according to exact molecular mass and MS2 fragment spectra. The degree of 13C stable isotope labelling into lipids was estimated based on their MS1 mass spectra following calculations described previously59. Two representative archaeal lipids60, archaeol (C43H88O3) and Me-GMGT (C87H160O6) were selected for the calculation of labelling extent. The mass range of isotoplogs was narrowed to m/z 650–710 for archaeol and m/z 1,300–1,400 for Me-GMGT. The base peak intensity of an isotoplog with i13C atoms (BPIi) was calculated by normalizing it to the highest intensity of all isotoplogs, which was further normalized to the sum BPI of all isotoplogs, resulting in the BPIi(norm). The degree of 13C labelling of an isotopolog with i13C atoms (DoLi) and the total degree of 13C labelling of a lipid (DoL) was calculated with the following equations:
i refers to the number of 13C atoms; n refers to the number of carbon atoms in a molecule.
DNA and RNA extraction
DNA was extracted using bead-beating (Sigma-Aldrich, ≤106 μm) methods combined with an Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech) according to the manufacturer’s protocol. DNA concentration was measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and DNA was stored at −80 °C until further processing.
RNA extraction was performed using bead-beating for releasement and extracted by using the RNAprep Pure Cell/Bacteria Kit (TIANGEN Biotech) according to the manufacturer’s protocol.
PCR
PCR was performed using the universal primers 27F/1492R61 or 109F/915R62 to determine the bacteria and archaea as previously described47. PCR products were sequenced by Sanger sequencing (Sangon Biotech).
qPCR
qPCR was performed to quantify the bacteria and Methanoculleus using primers targeting their 16S rRNA gene (519F/907R61 and ZC2F/ZC2R, respectively; Supplementary Table 6), and primers targeting the 16S rRNA and mcrA genes for strain LWZ-6 (MSR4F/MSR4R and mcrA4F/mcrA4R; Supplementary Table 6). The qPCR primers of strain LWZ-6 and Methanoculleus were designed using NCBI/Primer-BLAST61. The qPCR process was performed as described previously16. The standard DNA for qPCR of bacteria, Methanoculleus and strain LWZ-6 were obtained from the 16S rRNA gene of Escherichia coli DH5α and Methanoculleus recpetaculi ZC-2T, and the 16S rRNA gene and mcrA gene of strain LWZ-6. The qPCR for each targeted gene was determined in triplicate. PCR and qPCR products were sequenced by Sanger sequencing (Sangon Biotech).
16S rRNA gene amplicon sequencing
A total of 1 ml of culture was taken for DNA extraction and amplicon sequencing. The 16S rRNA gene was amplified using the primer set for bacteria with barcode (341F/806R)63,64, primer set for archaea Arch519F/Arch915R65 (Supplementary Table 6), and universal primers targeting both bacteria and archaea with barcode 515FmodF/806RmodR66 (Supplementary Table 6). The amplicon product was then sequenced using the NovaSeq 6000 sequencer (Illumina) with paired-end 250 bp mode (PE250) at Novogene Bioinformatics Technology (Novogene) or Shanghai Majorbio Bio-pharm Technology (Majorbio). All of the sequencing data were first filtered to remove the low-quality reads67,68. Further reads were analysed according to the Qiime2 (v.2021.2) pipeline69. The sequences were denoised to amplicon sequence variants (100% similarity). The taxonomy was determined using the sklearn method in Qiime2 (v.2021.2) and the Silva NR99 database (release138) as the reference70,71.
Metagenome sequencing, assembly, genome binning and annotation
Genomic DNA was extracted and sent to Novogene for library sequencing using the NovaSeq 6000 platform with PE150, generating raw metagenomic sequencing data. The raw reads were processed as follows: qualified trimmed reads were obtained using Trimmomatic (v.0.38)72. De novo assembly was performed using metaSPAdes (v.3.12.0) with k-mer sizes of 21, 33, 55 and 7773. Genome binning of the assemblies was performed using BBMap (v.38.24) and MetaBAT (v.2.12.1). The ambiguous contigs and redundant contigs were removed. Completeness, contamination and strain heterogeneity were identified using CheckM (v.1.0.8) to evaluate the estimated quality and completeness of each recovered MAGs74. Taxonomic classification of MAGs was performed according to the GTDB database (release 95.0, July 2020)75. In total, 23 MAGs grouping with Methanosuratincolia were retrieved from the enrichments. Similarities of each MAG to the other MAGs and publicly available Methanosuratincolia MAGs were determined using the 16S rRNA gene sequence identity, the AAI, the ANI and the percentage of conserved proteins (POCP) with other published Methanosuratincolia MAGs. Barrnap was used to obtain the 16S rRNA gene sequences (https://github.com/tseemann/barrnap). The ANI of the MAGs was calculated using the OrthoANIu (Orthologous ANI using USEARCH) tool76. The AAI was calculated using CompareM (v.0.1.2; https://github.com/dparks1134/CompareM). POCP calculation was performed as described previously77. The 23 MAGs could be dereplicated78 into five species-level clusters using an ANI cut-off of 97% (Supplementary Table 3). One MAG was chosen per cluster for downstream analyses. These MAGs were first analysed by prodigal (v.2.6.3)79, and then annotation of open reading frames was predicted using the KEGG server (BlastKOALA)80 and eggnog-mapper (v.2)81. The predicted genes ascribing to methanogenesis were further verified using UniProt82 and CD-search (conserved domain) in NCBI83.
Nanopore for closed genome binning and analysis
Metagenome sequencing of the simple culture (Fig. 1 (stage 3)) was conducted as described above. The Illumina metagenome was assembled using metaSPAdes (v.3.13.0)73 under the default parameters. Subsequent binning of the contigs using MetaWRAP (v.1.3)84 gave a Methanosuratincolia bin (1.54 Mb, 2 contigs). Nanopore data were generated to close the genome: the library was prepared with 60 ng genomic DNA using the ligation sequencing gDNA kit (ONT) according to the manufacturer’s instructions and minor modifications. In brief, DNA repair and end-prep were performed using the NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End repair/dA-tailing Module (New England Biolabs), followed by adapter ligation using Quick T4 ligase (New England Biolabs). Subsequently, a clean-up using AMPure beads (Beckman Coulter) and the short fragment buffer (SFB) was performed to retain DNA fragments of all sizes with a final incubation at 37 °C for 10 min. The library was then directly loaded onto a primed SpotON R9.4.1 flow cell in a MinION Mk1C (1,366 pores available). Sequencing was performed for 44 h resulting in 1.57 million reads and 2.73 Gb raw data. Raw Nanopore reads were corrected using Canu (v.2.2)85 and used to perform a metaSPAdes hybrid assembly with the Illumina data. After binning the contigs with MetaWRAP, this again resulted in a Methanosuratincolia bin (1.54 Mb, 2 contigs). Moreover, corrected Nanopore reads were assembled using metaFlye (v.2.9.1)86. This resulted in a Methanosuratincolia single contig of 1.43 Mb. This contig was corrected with the raw Illumina reads, in three rounds/iterations, using Pilon (v.1.23)87. Analysis showed that the overlapping areas of the five contigs of the three Methanosuratincolia bins were identical and could be combined into a single contig. To this end, raw Illumina reads were mapped onto the five contigs using Burrows-Wheeler Aligner (bwa, https://github.com/lh3/bwa), after which mapped reads were extracted from the original Illumina dataset. These reads were used in a SPAdes v.3.14.0 assembly to which the five contigs were added under the flag -trusted-contigs. This gave a single circular contig (1,538,194 bp) of strain LWZ-6, as confirmed using Bandage (v.0.8.1)88.
Phylogenomic and phylogenetic tree construction
To phylogenomically place the five representative MAGs, publicly available MAGs classified as ‘Ca. Verstraetearchaeota’, ‘Candidatus Culexarchaeia’, ‘Ca. Nezhaarchaeota’ and ‘Ca. Methanomethylicia’ were downloaded from the GTDB, JGI and NCBI repositories, together with a set of 92 high-quality genomes spanning archaeal diversity. All MAGs were first analysed using checkM2 (https://github.com/chklovski/CheckM2). MAGs with a completeness of below 60% and contamination above 10% were removed from the dataset. Subsequently, the proteomes of all of the remaining MAGs were predicted using Prodigal79. The phylogenomic tree was constructed using a set of 76 archaeal markers (Archaea76)89. To retrieve orthologues from the proteomes and create single-marker alignments, GtoTree was run with the standard parameters. Maximum likelihood (ML) individual protein phylogenies were generated using IQ-TREE (v.2.0.3)90 under the LG+C20+F+G substitution model with 1,000 ultrafast bootstraps. Phylogenetic trees were manually inspected for erroneous inclusion of paralogous or contaminated sequences. If present, such sequences were removed from the dataset, after which the remaining sequences were realigned, and phylogenies were re-estimated, as described above. The final single-marker alignments were then concatenated into one. Columns with gaps in more than 90% of the sequences were removed using trimAl (v.1.4.rev22)91, resulting in an alignment of 12,084 positions. A first ML phylogenetic tree was generated using IQ-TREE (v.2.0.3)90 (-bb 1000 -alrt 1000) with the model LG+C60+F+G. The resulting ML tree was then used to generate a posterior mean site frequency ML tree (-tbe -b 100 flags).
Furthermore, a mcrA gene phylogeny tree was constructed using 115 mcrA sequences retrieved from the six representative Methanosuratincolia MAGs and draft genomes from publicly available (potential) methanogenic taxa of the TACK superphylum, Euryarchaeota and Helarchaeota. The sequences were first aligned using MAFFT (v.7.310)92. Columns with gaps in more than 10% of the sequences were removed using trimAl (v.1.4.rev22)91. The ML phylogenetic tree was then generated using IQ-TREE (v.2.0.3)90 (-bb 1000 -alrt 1000) with the model Q.pfam+C40+F+G8, as chosen by the Bayesian information criterion using ModelFinder Plus93. The resulting ML tree was then used to generate a posterior mean site frequency ML tree (-tbe -b 100 flags).
Comparative genomics of Methanosuratincolia
The dataset containing all publicly available MAGs classified as ‘Ca. Verstraetearchaeota, ‘Ca. Nezhaarchaeota’, ‘Ca. Culexarchaeia’ and ‘Ca. Methanomethylicia’ was also used for a comparative genomics analysis of Methanosuratincolia. By combining the CheckM2 completeness of the MAGs and the phylogenomic tree of the class Methanosuratincolia, 38 genomes were selected (that is, 19 Methanosuratincolales, 2 ‘Ca. Aukarchaeales’, 8 ‘Ca. Nezhaarchaeales’ and 9 ‘Ca. Culexarchaeales’). These all had a CheckM2 completeness of at least 70% and spanned the entire phylogenomic diversity of Methanosuratincolia. Moreover, the five representative Methanosuratincolia genomes of this study were added to the dataset, together with a Methanosuratincolia genome of an enrichment culture of LCB070 from hot springs in Yellowstone National Park (Kohtz, A. J. et al., unpublished data) and four near-complete euryarchaeotal type strain genomes of known hydrogenotrophic methanogens. The proteome of each genome was predicted using Prodigal (v.2.6.3)79, after which the entire proteome dataset was functionally annotated with eggNOG-mapper (v.2)81 with the minimum percentage identity set to 25%. The EggNOG-mapper results were inspected for the presence of proteins involved in methanogenesis, electron cycling and energy conservation. Moreover, the proteomes were analysed for the presence of these proteins using BLASTp94.
Transcriptome sequencing and data analysis
Total RNA was extracted at the exponential methane production stage of strain LWZ-6 when Methanosuratincolia culture was incubated with methanol and hydrogen in fresh medium. RNA was sent to Novogene for sequencing. The RNA-seq data were produced using the NovaSeq 6000 instrument with PE150 at Novogene (https://en.novogene.com). The raw data were first trimmed by removing the adaptors and low-quality sequences using Trimmomatic72, and the mRNA was retrieved with SortMeRNA (v4.3.6)95 using the default settings after removal of tRNA and rRNA.
Evaluation of the activity of strain LWZ-6 was conducted using the complete circular LWZ-6 genome for analysis. The transcription of genes was calculated using bwa (v.0.7.17-r1188)96. The SAM mapping file was transformed into BAM files using SAMtools (v.1.13)97. The read coverage was calculated using BEDTools (v.2.30.0)98. The number of reads was normalized to the length of the genome. FPKM was used to normalize the expression level. The transcribed rank of genes was calculated on the basis of log2[FPKM].
Screening of the 16S rRNA gene of Methanosuratincolia in public databases
The 16S rRNA gene reference sequences extracted from the Methanosuratincolia genomes were submitted to the Integrated Microbial Next Generation Sequencing (IMNGS) platform (https://www.imngs.org/). SRA sequences were retrieved using a similarity cut-off of 90% at a minimum length of 200 bp. In total, 10,774 sequences were obtained from IMNGS, in which 5,187 were present at a relative abundance of at least 0.1% in the dataset. These were grouped into 692 clusters with a 95% similarity cut-off using CD-HIT99. The Methanosuratincolia reference sequences and the representative sequence of each CD-HIT cluster together with near-complete 16S rRNA gene sequences from other Archaea and Bacteria were aligned using MAFFT (v.7.310)92. This alignment was used to construct a phylogenetic tree using IQ-TREE v.2.0.3 (-bb 1000 -alrt 1000)90. From this tree, CD-HIT cluster representative sequences robustly grouping with Methanosuratincolales were linked back to the unclustered dataset of 5,187 sequences. This resulted in 1,091 sequences clustering with Methanosuratincolales. The metadata from the sequencing datasets in which these sequences were found were used to analyse the global distribution of Methanosuratincolales. World map shape with geographical coordinates of Methanosuratincolales 16S rRNA gene sequences and Methanosuratincolia genomes were created using the maps100 and ggplot2101 packages in R.
Description of Methanosuratincola gen. nov
Methanosuratincola (Me.tha.no.su.rat.in’co.la. N.L. pref. methano- pertaining to methane; L. masc./fem. n. incola inhabitant, dweller; N.L. masc. n. Methanosuratincola methane organism inhabiting the Surat Basin).
Anaerobic, non-motile, coccid, neutrophilic, thermophilic methanogen. The DNA G+C content of the type species is 54.42 mol%. The type species is M. petrocarbonis.
Description of M. petrocarbonis sp. nov
petrocarbonis (pe.tro.car.bo’nis. Gr. fem. n. petra rock; L. masc. n. carbo coal; N.L. gen. n. petrocarbonis of coal from a rock). Strain LWZ-6 is the type strain (CCAM 1872).
Cells are non-motile and coccoid with a diameter of approximately 0.5 μm. Methanol and monomethylamine can be used as catabolic substrates, with hydrogen as an electron donor to produce methane. Yeast extract or casamino acids are essential growth factors. Strain LWZ-6 was isolated from oily sludge of the Shengli oil field in China. The complete circular genome size is 1.54 Mb, has a DNA G+C content of 54.42 mol%, 1 copy of the 16S rRNA gene, 23S rRNA gene and 5S rRNA gene, and 45 tRNAs. The GenBank accession numbers of the 16S rRNA and genome are OR243905 and GCA_030522375.1, respectively.
Besides strain LWZ-6, 18 other MAGs belonging to M. petrocarbonis (cluster 1; Supplementary Table 3) were obtained from oily sludge of the Shengli oilfield. The estimated completeness of these 18 MAGs ranged from 88.47% to 100% and contamination ranged from 0.00% to 1.87%. The genome sizes of 18 MAGs are 0.95–1.75 Mb, and the DNA G+C contents are 54.29–56.37 mol% (detailed information is provided in Supplementary Table 3).
Description of Methanosuratincolaceae fam. nov
Methanosuratincolaceae (Me.tha.no.su.rat.in.co.la’ce.ae N.L. masc. n. Methanosuratincola, type genus of the family; -aceae, ending to designate a family; N.L. fem. pl. n. Methanosuratincolaceae, the family of Methanosuratincola). The type genus is Methanosuratincola gen. nov.
Description of Methanosuratincolales ord. nov
Methanosuratincolales (Me.tha.no.su.rat.in.co.la’les. N.L. masc. n. Methanosuratincola, type genus of the order; -ales, ending to designate an order; N.L. fem. pl. n. Methanosuratincolales, the order of Methanosuratincola). The type genus is Methanosuratincola gen. nov.
Description of Methanosuratincolia classis nov
Methanosuratincolia (Me.tha.no.su.rat.in.co’li.a. N.L. masc. n. Methanosuratincola, type genus of the class; -ia, ending to designate a class; N.L. neut. pl. n. Methanosuratincolia, the class of Methanosuratincola). The type genus is Methanosuratincola gen. nov.
Description of ‘Ca. Methanosuratincola thermotaenarum’ sp. nov
thermotaenarum (ther.mo.tae.na’rum. Gr. masc. adj. thermos, hot; Gr. neut. N. taenarum, referring to Taenarum, a town named after Taenarus, a son of Zeus. Taenarum was believed to be the gate to the Underworld; N.L. neut. adj. thermotaenarum, referring to a hot underground site).
This uncultivated organism is potentially capable of methyl-reducing methanogenesis and is represented by MAG XY_C20_T55_P1_bin.49, the representative of cluster 2 (Supplementary Table 3). The MAG has an estimated completeness of 88.21% and an estimated contamination of 0%. No ribosomal RNA genes were present in the MAG. A total of 19 tRNAs could be detected. As derived from the draft genome, the DNA G+C content is 54.64 mol%. The draft genome sequence of MAG XY_C20_T55_P1_bin.49 has an estimated size of 1.09 Mb. The two cluster 2 MAGs were retrieved from an enrichment culture of oily sludge of the Shengli oilfield that was incubated with alkanes anaerobically at 55 °C (detailed information is provided in Supplementary Table 3).
Description of ‘Ca. Methanosuratincola mesotaenarum’ sp. nov
mesotaenarum (me.so.tae.na’rum. Gr. masc. adj. mesos, middle; Gr. neut. N. taenarum, referring to Taenarum, a town named after Taenarus, a son of Zeus. Taenarum was believed to be the gate to the Underworld; N.L. neut. adj. mesotaenarum, referring to a moderately warm underground site).
This uncultivated organism is potentially capable of methyl-reducing methanogenesis and is represented by MAG GD_Cm_T35_P0_bin.90, the sole representative of cluster 3 (Supplementary Table 3). The draft genome has an estimated completeness of 95.33% and an estimated contamination of 0%. The draft genome contained 37 tRNAs but no rRNA genes. Its estimated DNA G+C content is 53.47 mol%. The MAG was retrieved from oily sludge of the Shengli oilfield incubated with alkanes anaerobically at 35 °C (detailed information is listed in Supplementary Table 3).
Description of ‘Ca. Methanoluiimicrobium’ gen. nov
Methanoluiimicrobium (Me.tha.no.lu.i.i.mi.cro’bi.um. N.L. neut. n. methanum, methane; N.L. pref. methano-, referring to the methane group; N.L. neut. n. microbium, a microorganism; N.L. neut. n. Methanoluiimicrobium, a methane-producing microorganism named to honour Yahai Lu for his contribution on methane metabolism in rice paddy soil). The type species is ‘Ca. Methanoluiimicrobium subterraneum’.
Description of ‘Ca. Methanoluiimicrobium subterraneum’ sp. nov
subterraneum (sub.ter.ra.ne’um. L. neut. adj. subterraneum, under the Earth, indicating the environment of isolation). This organism is a potential methyl-reducing methanogen and not cultivated, represented by a single MAG GD_Cm_T35_P0_bin.15 of cluster 4 (Supplementary Table 3) with an estimated completeness of 81.31%, an estimated contamination of 0.93% and 30 tRNAs. No ribosomal RNA genes were present in the MAG. The DNA G+C content, as derived from the draft genome, is 58.76 mol%. Its approximate genome size is 1.49 Mb. The MAG was retrieved from oily sludge of the Shengli oilfield which was incubated with alkanes anaerobically at 35 °C (detailed information is provided in Supplementary Table 3).
Description of ‘Candidatus Methanostamsia’ gen. nov
Methanostamsia (Me.tha.no.stams’i.a. N.L. neut. n. methanum, methane; N.L. pref. methano-, pertaining to methane; N.L. fem. n. Methanostamsia, a methane [producing] microorganism named in honour of Alfons J. M. Stams for his work on syntrophic methanogenesis). The type species is ‘Ca. Methanostamsia’ subterranea.
Description of ‘Ca. Methanostamsia’ subterranea sp. nov
subterranea (sub.ter.ra’ne.a. L. fem. adj. subterranea, underground, subterranean). An uncultivated and potential methyl-reducing methanogen, represented by a single MAG XY_C20_T55_P2_bin.56 (Supplementary Table 3). The genome has an estimated completeness and contamination of 91.12% and 0.93%, respectively. One partial 16S rRNA gene copy and 44 tRNAs were present in the draft genome. The DNA G+C content, as derived from the genome, was 47.00 mol%. The draft genome sequence of XY_C20_T55_P2_bin.56 has an estimated genome size of 1.40 Mb. The MAG was retrieved from an enrichment culture of oily sludge of the Shengli oilfield that was incubated with alkanes anaerobically at 55 °C (detailed information is listed in Supplementary Table 3).
Description of Acetomicrobium sp. strain CY-2
Strain CY-2 was isolated from oily sludge of Shengli oil field using the roll tube method. Cells are rod-shaped, 0.5–1.3 μm in width and 1.4–4.0 μm in length (Supplementary Fig. 1a–c). The fermentation products were hydrogen, acetate and carbon dioxide when strain CY-2 was incubated with 0.5 g l−1 yeast extract at 55 °C (Supplementary Fig. 1d). No methanol was detected when strain CY-2 was incubated with 0.5 g l−1 yeast extract whereas methanol was detected in 5 g l−1 yeast extract (data not shown).
CY-2 has a 16S rRNA gene similarity of 98.75 and 99.31% to Acetomicrobium thermoterrenum DSM 13490T and Acetomicrobium hydrogeniformans OS1T, respectively. The ANI with A. thermoterrenum DSM 13490T and A. hydrogeniformans OS1T is 96.85% and 96.76%, respectively. Here we propose CY-2 as Acetomicrobium sp. strain CY-2. Its estimated genome size is 1.97 Mb (DNA G+C content of 46.64%). The GenBank accession numbers of the 16S rRNA gene and genome are PP732077 and JBCLWM000000000, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The 16S rRNA gene amplicon sequences, metagenomic, genomic and transcriptome data generated in the current study are publicly available under BioProject accession number PRJNA992765 and at the NODE database (https://www.biosino.org/node/project/detail/OEP003742). Further details are provided in Supplementary Table 7. All other data are presented in the Article and its Supplementary Information. Source data are provided with this paper.
Code availability
The codes and programs used for analyses are mentioned in the Methods, and they are available at GitHub (https://github.com/zhuozhou1993/Methanosuratus/blob/main/code).
Change history
02 September 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-08000-z
References
Evans, P. N. et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438 (2015).
Vanwonterghem, I. et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1, 16170 (2016).
McKay, L. J. et al. Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat. Microbiol. 4, 614–622 (2019).
Wang, Y., Wegener, G., Hou, J., Wang, F. & Xiao, X. Expanding anaerobic alkane metabolism in the domain of Archaea. Nat. Microbiol. 4, 595–602 (2019).
Martin, W. & Russell, M. J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. Lond. B 362, 1887–1925 (2007).
Saunois, M. et al. The global methane budget 2000-2017. Earth Syst. Sci. Data 12, 1561–1623 (2020).
Ferry, J. G. Methanogenesis: Ecology, Physiology, Biochemistry & Genetics (Springer, 2012).
Woese, C. R., Kandler, O. & Wheelis, M. L. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl Acad. Sci. USA 87, 4576–4579 (1990).
Ermler, U., Grabarse, W., Shima, S., Goubeaud, M. & Thauer, R. K. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462 (1997).
Wang, J. et al. Evidence for nontraditional mcr-containing archaea contributing to biological methanogenesis in geothermal springs. Sci. Adv. 9, eadg6004 (2023).
Berghuis, B. A. et al. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc. Natl Acad. Sci. USA 116, 5037–5044 (2019).
Borrel, G. et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 4, 603–613 (2019).
Ou, Y.-F. et al. Expanding the phylogenetic distribution of cytochrome b-containing methanogenic archaea sheds light on the evolution of methanogenesis. ISME J. 16, 2373–2387 (2022).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Mand, T. D. & Metcalf, W. W. Energy conservation and hydrogenase function in methanogenic archaea, in particular the genus Methanosarcina. Microbiol. Mol. Biol. Rev. 83, e00020-19 (2019).
Zhou, Z. et al. Non-syntrophic methanogenic hydrocarbon degradation by an archaeal species. Nature 601, 257–262 (2022).
Mayumi, D. et al. Methane production from coal by a single methanogen. Science 354, 222–225 (2016).
Garcia, P. S., Gribaldo, S. & Borrel, G. Diversity and evolution of methane-related pathways in archaea. Annu. Rev. Microbiol. 76, 727–755 (2022).
Liu, Y.-F. et al. Long-term cultivation and meta-omics reveal methylotrophic methanogenesis in hydrocarbon-impacted habitats. Engineering 24, 265–274 (2023).
Hua, Z.-S. et al. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat. Commun. 10, 4574 (2019).
Evans, P. N. et al. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 17, 219–232 (2019).
Cheng, L. et al. Isolation and characterization of Methanoculleus receptaculi sp. nov. from Shengli oil field, China. FEMS Microbiol. Lett. 285, 65–71 (2008).
Oren, A., Garrity, G. M., Parker, C. T., Chuvochina, M. & Trujillo, M. E. Lists of names of prokaryotic Candidatus taxa. Int. J. Syst. Evol. Microbiol. 70, 3956–4042 (2020).
Lang, K. et al. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of Candidatus Methanoplasma termitum. Appl. Environ. Microbiol. 81, 1338–1352 (2015).
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W. & Hedderich, R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591 (2008).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645 (2014).
Konstantinidis, K. T., Rosselló-Móra, R. & Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406 (2017).
Speth, D. R. et al. Microbial communities of Auka hydrothermal sediments shed light on vent biogeography and the evolutionary history of thermophily. ISME J. 16, 1750–1764 (2022).
Wang, Y. et al. A methylotrophic origin of methanogenesis and early divergence of anaerobic multicarbon alkane metabolism. Sci. Adv. 7, eabj1453 (2021).
Liu, Y.-F. et al. Anaerobic degradation of paraffins by thermophilic Actinobacteria under methanogenic conditions. Environ. Sci. Technol. 54, 10610–10620 (2020).
Feldewert, C., Lang, K. & Brune, A. The hydrogen threshold of obligately methyl-reducing methanogens. FEMS Microbiol. Lett. 367, fnaa137 (2020).
Zhuang, G.-C., Lin, Y.-S., Elvert, M., Heuer, V. B. & Hinrichs, K.-U. Gas chromatographic analysis of methanol and ethanol in marine sediment pore waters: validation and implementation of three pretreatment techniques. Mar. Chem. 160, 82–90 (2014).
Zhuang, G.-C., Peña-Montenegro, T. D., Montgomery, A., Hunter, K. S. & Joye, S. B. Microbial metabolism of methanol and methylamine in the Gulf of Mexico: insight into marine carbon and nitrogen cycling. Environ. Microbiol. 20, 4543–4554 (2018).
Miller, T. L. & Wolin, M. J. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 141, 116–122 (1985).
Sprenger, W. W., van Belzen, M. C., Rosenberg, J., Hackstein, J. H. & Keltjens, J. T. Methanomicrococcus blatticola gen. nov., sp. nov., a methanol- and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int. J. Syst. Evol. Microbiol. 50, 1989–1999 (2000).
Dridi, B., Fardeau, M.-L., Ollivier, B., Raoult, D. & Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62, 1902–1907 (2012).
Sorokin, D. Y. et al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2, 17081 (2017).
Verhees, C. H. et al. The unique features of glycolytic pathways in Archaea. Biochem. J 375, 231–246 (2003).
Tersteegen, A., Linder, D., Thauer, R. K. & Hedderich, R. Structures and functions of four anabolic 2‐oxoacid oxidoreductases in Methanobacterium thermoautotrophicum. Eur. J. Biochem. 244, 862–868 (1997).
Glasemacher, J., Bock, A. K., Schmid, R. & Schönheit, P. Purification and properties of acetyl‐CoA synthetase (ADP‐forming), an archaeal enzyme of acetate formation and ATP synthesis, from the hyperthermophile Pyrococcus furiosus. Eur. J. Biochem. 244, 561–567 (1997).
Adam, P. S., Kolyfetis, G. E., Bornemann, T. L., Vorgias, C. E. & Probst, A. J. Genomic remnants of ancestral methanogenesis and hydrogenotrophy in Archaea drive anaerobic carbon cycling. Sci. Adv. 8, eabm9651 (2022).
Beulig, F., Røy, H., McGlynn, S. E. & Jørgensen, B. B. Cryptic CH4 cycling in the sulfate-methane transition of marine sediments apparently mediated by ANME-1 archaea. ISME J. 13, 250–262 (2019).
Kohtz, A. J. et al. Cultivation and visualization of a methanogen of the phylum Thermoproteota. Nature https://doi.org/10.1038/s41586-024-07631-6 (2024).
Rabus, R., Hansen, T., Widdel, F. & Dworkin, M. The Prokaryotes: Ecophysiology and Biochemistry (Springer, 2006).
Wolin, E. A., Wolin, M. J. & Wolfe, R. S. Formation of methane by bacterial extracts. J. Biol. Chem. 238, 2882–2886 (1963).
Plugge, C. M. Anoxic media design, preparation, and considerations. Methods Enzymol. 397, 3–16 (2005).
Wu, K. et al. Gudongella oleilytica gen. nov., sp. nov., an aerotorelant bacterium isolated from Shengli oilfield and validation of family Tissierellaceae. Int. J. Syst. Evol. Microbiol. 70, 951–957 (2020).
Neubauer, S. et al. U13C cell extract of Pichia pastoris—a powerful tool for evaluation of sample preparation in metabolomics. J. Sep. Sci. 35, 3091–3105 (2012).
Ver Eecke, H. C., Akerman, N. H., Huber, J. A., Butterfield, D. A. & Holden, J. F. Growth kinetics and energetics of a deep-sea hyperthermophilic methanogen under varying environmental conditions. Environ. Micobiol. Rep. 5, 665–671 (2013).
Cheng, L., Dai, L., Li, X., Zhang, H. & Lu, Y. Isolation and characterization of Methanothermobacter crinale sp. nov., a novel hydrogenotrophic methanogen from the Shengli oil field. Appl. Environ. Microbiol. 77, 5212–5219 (2011).
Cheng, L., Rui, J., Li, Q., Zhang, H. & Lu, Y. Enrichment and dynamics of novel syntrophs in a methanogenic hexadecane-degrading culture from a Chinese oilfield. FEMS. Microbiol. Ecol. 83, 757–766 (2013).
Cheng, L. et al. DNA-SIP reveals that Syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS ONE 8, e66784 (2013).
Stahl, D. A. & Amann, R. in Nucleic Acid Techniques in Bacterial Systematics (eds Stackebrandt, E. & Goodfellow, M.) (Wiley, 1991).
Pernthaler, A., Preston, C. M., Pernthaler, J., DeLong, E. F. & Amann, R. Comparison of fluorescently labeled oligonucleotide and polynucleotide probes for the detection of pelagic marine bacteria and archaea. Appl. Environ. Microbiol. 68, 661–667 (2002).
Chi, Z.-L., Yu, G.-H., Kappler, A., Liu, C.-Q. & Gadd, G. M. Fungal–mineral interactions modulating intrinsic peroxidase-like activity of iron nanoparticles: implications for the biogeochemical cycles of nutrient elements and attenuation of contaminants. Environ. Sci. Technol. 56, 672–680 (2021).
Yu, G.-H. et al. Fungal nanophase particles catalyze iron transformation for oxidative stress removal and iron acquisition. Curr. Biol. 30, 2943–2950 (2020).
Zeng, Z. et al. GDGT cyclization proteins identify the dominant archaeal sources of tetraether lipids in the ocean. Proc. Natl Acad. Sci. USA 116, 22505–22511 (2019).
Chen, Y. et al. The production of diverse brGDGTs by an Acidobacterium providing a physiological basis for paleoclimate proxies. Geochim. Cosmochim. Acta 337, 155–165 (2022).
Thiele, B. & Matsubara, S. in Plant and Food Carotenoids. Methods in Molecular Biology (eds Rodríguez-Concepción, M. & Welsch, R.) (263–277 (Humana, 2020).
Knappy, C. et al. Mono‐, di‐ and trimethylated homologues of isoprenoid tetraether lipid cores in archaea and environmental samples: mass spectrometric identification and significance. J. Mass Spectrom. 50, 1420–1432 (2015).
Lane, D. J. in Nucleic Acid Techniques in Bacterial Systematics (eds Stackebrandt, E. & Goodfellow, M.) 115–175 (Wiley, 1991).
Grosskopf, R., Janssen, P. H. & Liesack, W. Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl. Environ. Microbiol. 64, 960–969 (1998).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2012).
Hiergeist, A., Reischl, U. & Gessner, A. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int. J. Med. Microbiol. 306, 334–342 (2016).
Wei, S. et al. Comparative evaluation of three archaeal primer pairs for exploring archaeal communities in deep-sea sediments and permafrost soils. Extremophiles 23, 747–757 (2019).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108, 4516–4522 (2011).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648 (2013).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S. & Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286 (2017).
Qin, Q. L. et al. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215 (2014).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2020).
Marchler-Bauer, A. & Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331 (2004).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Lee, M. D. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164 (2019).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Katoh, K., Misawa, K., Kuma, K. I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arxiv.org/abs/1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 47, 11.12.11–11.12.34 (2014).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Brownrigg., R., Becker, R. A. & Wilks, A. R. maps: draw geographical maps. R package version 3.4.2 (2023); cran.r-project.org/web/packages/maps/maps.pdf
Wilkinson, L. ggplot2: elegant graphics for data analysis. Biometrics 67, 678–679 (2011).
Acknowledgements
We thank P. Geesink for performing Nanopore sequencing of strain LWZ-6; W. B. Whitman for comments on the manuscript; L.-r. Dai, M. Yang and L. Fu for assisting in cultivation and experiments; Z. Zhou for technical support; and P. Vandamme for advice on the etymology of the new names proposed. This study was supported by National Natural Science Foundation of China (92051108, 92351301, 31970066, 42203080 and 42207167), Agricultural Science and Technology Innovation Project of the Chinese Academy of Agriculture Science (CAAS-ASTIP-2021-BIOMA-01), CAAS Center for Science in Agricultural Green and Low Carbon (CAAS-CSGLCA-202301), Sichuan Science and Technology program (2024NSFTD0093), Netherlands Ministry of Education, Culture and Science (project 024.002.002: Soehngen Institute of Anaerobic Microbiology), European Research Council (grant 817834), the Dutch Research Council (grant VI.C.192.016), the Central Public-interest Scientific Institution Basal Research Fund (1610012017002_05103), the Stable Support Plan Program of Shenzhen Natural Science Fund (20200925173954005) and the Shenzhen Key Laboratory of Marine Archaea Geo-Omics, Southern University of Science and Technology (ZDSYS201802081843490). The Article is dedicated to our co-author W.-H.H.
Author information
Authors and Affiliations
Contributions
L.C. initiated the study. L.C., D.Z.S., K.-j.W., T.J.G.E. and L.Z. designed the research. L.-y.L. performed the initial cultivation. K.-j.W., L.Z., L.-y.L. and J.L. performed the isolation process and physiological experiments. G.T. and W.-h.H. performed all bioinformatics analyses. C.-p.D. performed CARD-FISH. J.-c.Z. performed NanoSIMS. F.-f.Z. and C.-l.Z. performed lipid-SIP. L.F. and L.-p.B. performed biochemical analysis. K.-j.W. and L.Z. analysed data. X.-z.D. provided constructive suggestions on the isolation process. K.-j.W., L.C., X.-z.D., T.J.G.E. and D.Z.S. wrote the manuscript with the contributions from all of the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Methanosuratincolia communities were detected in numerous enrichments.
The enrichments were initially established from oily sludge or oil-produced water and incubated with various substrates such as acetate, propionate, butyrate, alkanes, and oil at temperatures ranging from 25 °C to 75 °C. Alkanes mix includes n-docosane, n-hexadecylcyclohexane, and n-hexadecylbenzene. The blank represents no enrichments performed on these conditions. Abundance 20, 40, and 60 show the relative abundance of Methanosuratincolia populations in total archaea by 16S rRNA gene amplicon sequencing.
Extended Data Fig. 2 Consecutive subcultures amended with methanol as a substrate to eliminate Methanoculleus recptaculi (Mcl).
a, CH4 production, b, the copy numbers of LWZ-6, c, the copy numbers of Mcl. Culture-1,2,3 represent triplicate tubes used for subcultures. The arrows indicate the subculture points, the subculture dilution varied from 0.1% to 10%. Copy numbers of LWZ-6 and Mcl were determined by qPCR with the primers mcr4F/mcr4R and ZC2F/ZC2R targeting their mcrA and 16S rRNA genes.
Extended Data Fig. 3 The microbial community compositions of the co-culture during 5 consecutive transfer incubations.
The relative abundance of strain LWZ-6 and CY-2 was determined by 16S rRNA gene amplicon sequencing using the primer 515FmodF/806RmodR66. The samples in each generation were collected from the cultures grown at the exponential phase of methane production.
Extended Data Fig. 4 Consecutive subcultures amended with antibiotics to eliminate Acetomicrobium sp. CY-2.
a, CH4 production, b, CO2 change, c, the copy numbers of LWZ-6 determined at the endpoint of each generation, d, the copy numbers of CY-2 determined at the endpoint of each generation, ND means the copy numbers are under detectable level, the subculture dilution varied from 1%-10%, e, the proportion of CY-2 in the total bacteria and archaea, f, gel electrophoresis of PCR products using 27F/1492R61 targeting bacteria, PC: positive control of coculture, NC: negative control of ddH2O. Samples were run on one gel and the lanes in images are representative blot of n = 3 biological replicates.
Extended Data Fig. 5 Microscopy observation of the Methanosuratincolia co-culture (strain LWZ-6/CY-2).
a, d, CARD-FISH showing cells hybridized with nucleotide probes that target archaea ARCH-915 (green) and bacteria EUB-338 (red). b, e, Fluorescence images hybridized with nucleotide probes that target archaea ARCH-915 (green), representative images are from 18 recorded images of n = 3 biological replicates. c, f, fluorescence images hybridized with nucleotide probes that target bacteria EUB-338 (red). g, h, TEM showing ultrathin section of the co-culture, the black arrows are strain LWZ-6, representative images are from 8 recorded images of n = 2 biological replicates. Scale bars: 10 μm in (a-c), 2 μm (d-f), 200 nm (g-h).
Extended Data Fig. 6 Methane activity and growth dynamics of the Methanosuratincolia co-culture (strain LWZ-6/CY-2).
a, the CH4 production, b, methanol consumption, c, H2 change, d, copies numbers of strain LWZ-6, e, copies numbers of bacteria in the 50 ml fresh medium. Three groups with different substrates were set up: 0.5 g l−1 yeast extract (YE); 0.5 g l−1 YE, 10 mM methanol and 10 kPa H2 (YE + methanol + H2); without substrates addition, YE (-). All symbols represent means of three individual incubations; error bars represent SD of triplicates; the invisible error bars are smaller than symbols.
Extended Data Fig. 7 The changes of the carbon isotope composition of CH4 (a) and CO2 (b).
The Methanosuratincolia co-culture was incubated with initial different ratios of (1.76 ± 0.01)%, (2.3 ± 0.10)%, (3.65 ± 0.08)%, and (5.98 ± 0.79)%.
Extended Data Fig. 8 The physiological properties of strain LWZ-6.
a, C1-methylated compounds utilization, b, growth factors, c, Temperature, d, pH, e, NaCl tolerance concentration. µ represents the maximum specific growth rate of strain LWZ-6 or the specific CH4 production. MeOH: methanol, MMA: monomethylamine, DMA: dimethylamine, TMA: trimethylamine, MeSH: methanethiol, YE: yeast extract, CA: casamino acids, ND indicates no growth of strain LWZ-6 or CH4 detected. Data in a and b are mean ± standard deviation of triplicates. All symbols represent means of three individual incubations, error bars represent SD of triplicates, the invisible error bars are smaller than symbols.
Extended Data Fig. 9 Global distribution of Methanosuratincolia in different biotopes.
The bold circles represent the sampling locations of Methanosuratincolia MAGs. The numbers of the genomes in environments are anaerobic digester: 6, groundwater: 3, freshwater/sediment: 2, hot spring: 36, hydrothermal sediment: 10, petroleum field: 3. The triangles represent Methanosuratincolales 16S rRNA gene sequence derived from IMNGS (Detailed information was listed in Supplementary Table 5).
Supplementary information
Supplementary Information
Supplementary Figs. 1–3.
Supplementary Table 1
Genome annotation of M. petrocarbonis LWZ-6.
Supplementary Table 2
Transcript of M. petrocarbonis LWZ-6 grown on methanol and H2.
Supplementary Table 3
General information on the 23 Methanosuratincolia genomes obtained in our study.
Supplementary Table 4
Methanogenetic features and energy mechanism of five Methanosuratincolia clusters in our study, other Methanosuratincolales, ‘Ca. Nezhaarchaeales’ and ‘Ca. Culexarchaeales’ genomes and other H2-dependent methylotrophic methanogens in Euryarchaeota. 1, present; 0, absent.
Supplementary Table 5
Global distribution of 16S rRNA gene sequences and MAGs of Methanosuratincolia in different biotopes.
Supplementary Table 6
Primers and probes used in this study.
Supplementary Table 7
Summary of sequencing data submitted to the NCBI and The National Omics Data Encyclopedia.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, K., Zhou, L., Tahon, G. et al. Isolation of a methyl-reducing methanogen outside the Euryarchaeota. Nature 632, 1124–1130 (2024). https://doi.org/10.1038/s41586-024-07728-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07728-y
- Springer Nature Limited