

Abstract
The calcium-sensing receptor (CaSR) is a family C G-protein-coupled receptor1 (GPCR) that has a central role in regulating systemic calcium homeostasis2,3. Here we use cryo-electron microscopy and functional assays to investigate the activation of human CaSR embedded in lipid nanodiscs and its coupling to functional Gi versus Gq proteins in the presence and absence of the calcimimetic drug cinacalcet. High-resolution structures show that both Gi and Gq drive additional conformational changes in the activated CaSR dimer to stabilize a more extensive asymmetric interface of the seven-transmembrane domain (7TM) that involves key protein–lipid interactions. Selective Gi and Gq coupling by the receptor is achieved through substantial rearrangements of intracellular loop 2 and the C terminus, which contribute differentially towards the binding of the two G-protein subtypes, resulting in distinct CaSR–G-protein interfaces. The structures also reveal that natural polyamines target multiple sites on CaSR to enhance receptor activation by zipping negatively charged regions between two protomers. Furthermore, we find that the amino acid l-tryptophan, a well-known ligand of CaSR extracellular domains, occupies the 7TM bundle of the G-protein-coupled protomer at the same location as cinacalcet and other allosteric modulators. Together, these results provide a framework for G-protein activation and selectivity by CaSR, as well as its allosteric modulation by endogenous and exogenous ligands.
Similar content being viewed by others


Main
Strict regulation of circulating Ca2+ levels, a crucial component of human physiology, relies heavily on CaSR. This receptor detects increases in serum calcium and activates G-protein signalling to restrict the secretion of parathyroid hormone (PTH) and promote renal calcium excretion2,3. Hundreds of naturally occurring mutations in human CaSR lead to various diseases, including familial hypocalciuric hypercalcaemia type 1, severe neonatal hyperparathyroidism, autosomal dominant hypocalcaemia type 1 (ADH1) and type 5 Bartter syndrome3,4. Reduced expression and activation of CaSR are observed in patients with chronic kidney diseases, which affect nearly 10% of the global population5,6.
CaSR belongs to family C GPCRs, which also include the γ-aminobutyric acid B (GABAB) and metabotropic glutamate (mGlu) receptors (mGlu1–mGlu8) that modulate neurotransmission. Family C GPCRs function as obligate homo- or heterodimers with endogenous ligands engaging the extracellular domain (ECD) that resides away from the membrane plane and typically encompasses a ligand-binding Venus flytrap domain (VFT) connected to the signature 7TM bundle through, in most cases, a cysteine-rich domain (CRD). Members of this family exhibit distinct G-protein coupling selectivity. mGlu1 and mGlu5 activate multiple G proteins including Gq/11, whereas the other mGlu receptors preferentially activate Gi/o (refs. 7,8). On the other hand, CaSR predominantly activates both Gq/11 and Gi/o subtypes2,3,9. Earlier structural studies with detergent-solubilized mGlu2, mGlu4 and GABAB (refs. 10,11,12) have revealed that family C GPCRs employ a unique mechanism to couple to Gi that involves a reorganization of intracellular loops without the opening of the 7TM bundle. However, it has been unclear whether this mechanism applies to other G-protein subtypes, and how G-protein selectivity is achieved by family C receptors in general.
A notable feature of CaSR is that it is engaged by various natural ligands, including polyvalent cations such as Ca2+, anions such as phosphate, l-amino acids, peptides and polyamines13,14. Changes in the relative concentrations of these ligands under different physiological conditions can either promote or inhibit receptor activation, and thus CaSR appears to be a fine-tuned signalling platform that integrates various chemical inputs to elicit its physiological effects. CaSR is targeted therapeutically by synthetic positive and negative allosteric modulators (PAMs and NAMs) that potentiate receptor activation or inactivation, respectively15. For example, cinacalcet is an FDA-approved PAM drug targeting CaSR that is used for the treatment of secondary hyperparathyroidism and parathyroid carcinoma. Although cryo-electron microscopy (cryo-EM) structures of human CaSR bound to different ligands including Ca2+, l-tryptophan (Trp) and PAMs or NAMs have been reported16,17,18, the binding modes of other ligands such as polyamines have not been characterized.
Here we sought to establish an enriched mechanistic framework for CaSR activation and G-protein subtype coupling. Cryo-EM snapshots of human CaSR reconstituted in lipid nanodiscs, complemented by functional data, reveal how CaSR uses an asymmetric interface stabilized by lipids and spermine molecules to activate G protein in one protomer; how Gi and Gq engage the same receptor elements that adopt different conformations; how the 7TM PAM cinacalcet induces side-chain rearrangements to promote G-protein coupling; and that the natural amino acid Trp can engage the 7TM bundle in the absence of exogenous PAMs.
Reconstitution of CaSR–Gi and CaSR–Gq complexes
To evaluate the G-protein selectivity landscape of CaSR, we implemented a cell-based bioluminescence resonance energy transfer (BRET) assay19 to monitor receptor-stimulated G-protein activation within the four major classes (Gs, Gi/o, Gq/11 and G12/13) upon the addition of Ca2+. Levels of activation by CaSR were compared against the activation of G-protein subtypes by neurotensin receptor 1 (NTSR1), a promiscuous GPCR that activates most G proteins, or the β2 adrenergic receptor (β2AR), which strongly activates Gs. The results showed that CaSR mainly activates the Gi/o and Gq/11 subtypes (Extended Data Fig. 1a,b), consistent with the physiological importance of Gi/o and Gq/11 signalling pathways for systemic CaSR function2,3. We further confirmed the G-protein preference of CaSR in a complementary cellular G-protein activation assay that monitors the binding of released βγ–Venus fusion protein to a membrane-associated NanoLuc-tagged GRK3 C-terminal fragment20 (Extended Data Fig. 1c,d). Accordingly, we sought to reconstitute complexes of CaSR with Gi3 (hereafter referred to as Gi) and with Gq for structural studies.
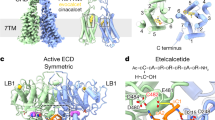
Although a complex between Gi and CaSR in lauryl maltose neopentyl glycol (LMNG) detergent micelles could be isolated in the presence of Ca2+, Trp and cinacalcet (Extended Data Fig. 1f), a low-resolution cryo-EM reconstruction revealed that the receptor assumes an active-like conformation but Gi remains in an inactive state (Extended Data Fig. 2a–d). This is evident from the ordered α-helical domain (AHD) packed against the Ras domain of Gαi, similar to the crystal structure of inactive, GDP-bound Gi (ref. 21) (Extended Data Fig. 2e,f), and suggests that CaSR is deficient in activating Gi in LMNG detergent micelles. Of note, structure docking shows that an extended C terminus of the Gαi α5 helix is positioned right below the receptor (Extended Data Fig. 2f), indicating that we trapped an intermediate state that precedes the ejection of GDP. Unlike our studies with the mGlu2–Gi complex10, the detergent micelles surrounding the 7TMs of activated CaSR appear to provide a limited planar surface for the proper landing of Gβγ, whereas a lipidic environment may be necessary for activating G proteins. Indeed, reconstitution of the CaSR–Gi complex into the relatively large MSP1E3D1 lipid nanodiscs (Extended Data Fig. 1g) enabled the release of GDP, as evidenced by the disordered Gαi AHD in high-resolution cryo-EM maps (Extended Data Fig. 3a,b). The global map of the nucleotide-free CaSR–Gi complex in nanodiscs was determined at a resolution of 3.1 Å with locally refined maps at resolutions of 2.8 Å (VFT–CRD), 3.2 Å (CRD–7TM) and 3.2 Å (Gαiβγ) (Fig. 1b and Extended Data Fig. 3a,c,d).

a–c, Cryo-EM maps of the cinacalcet (CINA)-bound active-state CaSR (a), CaSR–Gi (b) and CaSR–Gq (c) complexes in lipid nanodiscs. d,e, Asymmetric 7TM configuration and dimer interface in the CINA-bound CaSR–Gi structure. Two diametrically opposite views parallel to the membrane plane show the upper phospholipid (d) and the lower phospholipid and cholesterol (e). Cinacalcet, phospholipids (assigned as dioleoylphosphatidylcholine; DOPC), cholesterol and spermine are shown as spheres. Key residues that are involved in the 7TM dimer interface are shown as sticks. f, Comparison of the structures of the CaSR–Gi complex and the active-state CaSR (CaSR*) shows further compaction of the two 7TM bundles.
On the other hand, extensive cryo-EM trials to visualize nanodisc-embedded wild-type CaSR in complex with Gq failed. To enhance complex stability, we used a NanoBiT tethering strategy22 on an engineered CaSR dimer17 (with fused C1 and C2 tails from the GABAB heterodimer) that behaves similarly to wild-type CaSR in functional assays17 (Extended Data Fig. 1e). This approach, coupled with an optimized protocol (Methods), improved the reconstitution of the CaSR–Gq complex into nanodiscs (Extended Data Fig. 1i). Nevertheless, a relatively large cryo-EM dataset and extensive classifications were necessary to isolate the particle population representing a stable complex (Extended Data Fig. 4a,b). The global map of the CaSR–Gq complex in nanodiscs was determined at a resolution of 3.6 Å with local refinement maps at resolutions of 3.1 Å (VFT–CRD), 3.7 Å (CRD–7TM), and 3.9 Å (Gαqβγ) (Fig. 1c and Extended Data Fig. 4a,c,d). From the same dataset, we also isolated a population of particles representing active-state CaSR in the absence of Gq, enabling a global map at a resolution of 2.8 Å, and local refinement maps at resolutions of 2.6 Å (VFT–CRD) and 3.2 Å (CRD–7TM) (Fig. 1a and Extended Data Fig. 4a,e,f). Our CaSR–Gi and CaSR–Gq maps (Fig. 1b,c) both show that the CaSR dimer engages one G protein at a time, similar to mGlu2, mGlu4 and GABAB (refs. 10,11,12). Notably, CaSR is positioned off-centre in nanodiscs that extend in one direction to allow the insertion of the lipidated Gα N terminus and Gγ C terminus into the lipid bilayer (Extended Data Fig. 4g). This observation further emphasizes the importance of a sufficient membrane surface for assembling CaSR–G-protein complexes (Extended Data Fig. 2g).
The asymmetric 7TM dimer interface
To better understand the conformational changes during CaSR activation in a lipid bilayer, we also obtained a cryo-EM map of the nanodisc-embedded inactive receptor in the absence of Ca2+, albeit with limited resolution owing to receptor flexibility (Extended Data Fig. 2h–j). This inactive-state CaSR adopts a conformation with open-closed VFTs, well-separated CRDs and flexible 7TMs (Extended Data Fig. 2k). By contrast, nanodisc-embedded CaSR in the active state—as well as in the Gi- and Gq-coupled states—exhibits closed-closed VFTs bound to Ca2+, Trp and phosphate, with the CRDs in close proximity, and well-resolved 7TMs (Fig. 1a–c, and Extended Data Figs. 5 and 6a,b). The active-state CaSR shows an asymmetric 7TM configuration similar to that seen in detergent micelles17 (Extended Data Fig. 6a), whereas the two 7TMs of Gi- and Gq-coupled CaSR are substantially more compacted compared with the active state (Fig. 1a–c,f). This observation suggests that ligand binding to the ECDs alone is not sufficient to induce the fully active 7TM configuration of CaSR in the absence of G proteins.
The overall similarity between the Gi- and the Gq-coupled structures indicates that G-protein activation by CaSR depends on a common dimer arrangement (Extended Data Fig. 6b). However, in contrast to the TM6–TM6 dimer interface in the active-state structure (Extended Data Fig. 6d), the two G-protein-coupled structures reveal a distinct asymmetric 7TM dimer interface mediated by TM6 of the G-protein-coupled 7TM (7TMGC) and TM6 and TM7 of the non-G-protein-coupled 7TM (7TMNGC) (Fig. 1d,e and Extended Data Fig. 6e,f). The interface is characterized by a markedly higher sitting position of TM6 of 7TMGC relative to the opposing TM6, as is also evident when aligning the two protomers on the VFTs (Extended Data Fig. 6c). Of note, multiple disease-causing CaSR mutations are localized to the 7TM dimer interface, including F809L, I816T, S820F, F821L, A824P, F832S and V836L (refs. 23,24,25,26,27).
Another notable observation in the G-protein-coupled structures is the asymmetric binding of lipids at the 7TM dimer interface. On one side, a phospholipid (referred to as the lower phospholipid) and a cholesterol molecule are bound to the hydrophobic cleft between TM5 and TM6 of 7TMGC and TM6 and TM7 of 7TMNGC (Fig. 1e). The two phospholipid tails mediate multiple hydrophobic interactions with F7885.55 and F8096.41 along the 7TM dimer interface, and the negatively charged head group closely contacts K8056.37 in both protomers (Extended Data Fig. 6e,f). The mutations K805A and F809A significantly impaired the activation of both Gi and Gq by CaSR, and the combined F788A/K805A and K805A/F809A mutations almost abolished CaSR-mediated signalling (Extended Data Fig. 6g), highlighting an important role of protein–lipid interactions in the activation of G proteins by CaSR. On the opposite side, better resolved in the CaSR–Gi map is a phospholipid sitting on top of TM6 of 7TMNGC (referred to as the upper phospholipid) (Figs. 1d and 2a). The head group of this phospholipid inserts into a cleft formed by Y8256.57, Y8296.57 and V8337.28 in 7TMNGC, whereas the phospholipid tails appear to form limited interactions with both protomers and are not resolved in the CaSR–Gq map (Extended Data Fig. 7b). A weaker structural role for this lipid might be suggested by the inappreciable effect of CaSR mutations Y825A, Y829A and V833A in G-protein activation (Extended Data Fig. 6g). Nevertheless, in this arrangement, the upper phospholipid might help to keep TM6 of 7TMNGC in the downward position while the lower phospholipid supports the upward shift of TM6 of 7TMGC.

a, Two spermine molecules are bound to the ECL–CRD regions in the CINA-bound CaSR–Gi complex. The EM densities of the spermine (at a map threshold of 1.4) and the upper phospholipid DOPC (using the unsharpened map at a threshold of 0.45) are shown. A water molecule (red sphere) is observed between E604 of the CRD and the lower spermine molecule. b, The two spermine-binding sites located between the LB2 lobes of the two protomers in the CINA-bound CaSR–Gi complex. The EM densities of the two spermine molecules are shown, using the unsharpened map at a threshold of 0.3. c, Analysis of the spermine-binding sites by mutagenesis. IP1-accumulation assays monitoring the responses of wild-type (WT) CaSR and various CaSR mutants to Ca2+ or spermine suggest that functional responses to spermine are blunted by the mutations. The sample sizes and descriptions of error bars for all figures are described in ‘Statistics and reproducibility’ in the Methods.
Spermine targets multiple sites on CaSR
In both the cinacalcet-bound CaSR–Gi and the cinacalcet-bound CaSR–Gq maps, strong densities were observed in a narrow hydrophilic cleft between the ECL–CRD regions of the receptor dimer (Fig. 2a and Extended Data Fig. 7b). Given the shape and the surrounding negatively charged residues, we hypothesized that these densities correspond to either spermine or spermidine—two positively charged polyamines that have been previously reported to act as CaSR agonists but with unknown binding sites28. Mass spectrometry confirmed the presence of spermine but not spermidine in our sample (Extended Data Fig. 7a), consistent with the higher binding affinity of spermine to CaSR (ref. 28). The bound spermine molecules probably originated from the insect cells used for receptor expression, as polyamines are ubiquitously present in all living cells to stabilize negative charges and regulate diverse biological processes29.
The two spermine molecules bound to the ECL–CRD site are stabilized by a network of electronegative and hydrophobic residues including E604, E757 and Y829 (Fig. 2a and Extended Data Fig. 7b). E757 in ECL2 forms salt bridges with the amine nitrogen of the upper spermine, and there is a water-mediated ionic interaction between E604 of the CRD and the last amine nitrogen of the lower spermine. Nevertheless, the mutation E604A/E757A/Y829A, as well as a mutation that also includes adjacent negatively charged residues in ECL2 (E604A/E755A/E757A/D758A/E759A/Y829A), still resulted in substantial spermine-induced inositol monophosphate (IP1) responses (Fig. 2c), suggesting the presence of alternative interactions between spermine and CaSR. Indeed, closer examination of the CaSR–Gi map identified additional spermine-like densities located at two negatively charged clefts between the lower VFT lobes (LB2) of CaSR (Fig. 2b and Extended Data Fig. 7c). Although mutating the two VFT sites alone (E228A/D238A/E241A) had a limited effect on the spermine responses, mutating all three spermine-binding sites in the VFT and ECL–CRD regions (E228A/D238A/E241A/E604A/E757A/Y829A and E228A/D238A/E241A/E604A/E755A/E757A/D758A/E759A/Y829A) blunted spermine-induced CaSR activation (Fig. 2c). The latter mutation that removes additional negative charges in ECL2 resulted in high CaSR basal activity (Fig. 2c); this is likely to be a result of decreased electrostatic repulsions between the dimers, which could increase the occupancy of the active conformation in the absence of ligand30. Of note, E228K, E604K and E757K are gain-of-function ADH1 mutations31,32,33.
The above observations suggest that positively charged polyamines agonize CaSR by stabilizing the negatively charged patches at the dimer interface while ‘zipping’ the ECDs and 7TMs. This activation mechanism is different from that elicited by Ca2+, which mainly induces domain reorientation in each individual protomer to promote homodimer interactions16,17,18. These findings explain why spermine can function like a PAM to enhance Ca2+-induced CaSR activation28 but also as a CaSR agonist that is potentiated by the ECD PAM etelcalcetide17 (Extended Data Fig. 7d,e). The spermine responses seem to be less cooperative compared with the Ca2+ responses, as indicated by the shallower Hill slope in the IP1 assays (Fig. 2c), owing possibly to the lower number of spermine-binding sites on the receptor. We also note that the spermine-binding sites are distinct from those of amino acids, Ca2+ and PAMs, highlighting a complex regulatory mechanism for CaSR and potential implications for its nutrient-sensing role34.
Common features of Gi- and Gq-binding pockets
In the CaSR–Gi and CaSR–Gq structures, the cytoplasmic regions of 7TMGC differ substantially from those of 7TMNGC, which retains the conformation observed in the active-state structure (Extended Data Fig. 8a–f). After coupling to Gi or Gq, the cytoplasmic ends of TM3 and TM4, as well as intracellular loop 2 (ICL2) in 7TMGC, become ordered and extend downwards towards the G protein. This movement is accompanied by an outward tilt of TM3 and a displacement of TM5, with ICL3 moving towards the α5 helix of Gα, a downward shift of helix 8 (H8) and the ordering of the C terminus (Fig. 3a). These structural changes create an intracellular pocket involving TM3, ICL1, ICL2, ICL3, H8 and the C terminus to engage the α5 helix of Gαi or Gαq (Fig. 3a,c,d). We monitored the effects of mutating residues that are involved in forming the G-protein-binding pockets through Gi3 and Gq activation assays, as well as IP1 assays that reflect mainly Gαq activation, but also Gαi to a minor degree—as shown by the effect of pertussis toxin (PTX), which is known to abolish Gαi/o activity35.

a, A comparison of the cytoplasmic regions of the CaSR 7TM bundles in the active state and in the Gi- and Gq-bound states shows the displacement of ICL3, TM3, TM4, TM5 and H8 (indicated by the arrows) and the ordering of ICL2 and the C terminus after G-protein binding. Residues F801 in ICL3 and V7023.54 and F7063.58 in TM3 form a hydrophobic patch to engage the conserved hydrophobic residues in the α5 helix of both Gαi and Gαq. b, BRET-based G-protein-activation assays identify residues in ICL2 and H8 that are important for Gi activation versus Gq activation or for both (F801 in ICL3). AUC, area under the curve; NLuc, NanoLuc. c, Interactions between ICL1, ICL2, TM3 and H8 of the Gi-coupled protomer and the α5 helix of Gαi in the CINA-bound CaSR–Gi complex. ICL2 (residues 707–722) is highlighted in turquoise. d, Interactions between ICL1, ICL2, TM3 and H8 of the Gq-coupled protomer and the α5 helix of Gαq in the CINA-bound CaSR–Gq complex. The cytoplasmic tip of TM4 and ICL2 (residues 707–722) are highlighted in turquoise.
The movement of ICL3 seems to coordinate the ordering of the cytoplasmic ends of the receptor and the engagement of G proteins. F801 in ICL3 undergoes a substantial displacement (around 7 Å, as measured by its Cα) and forms a hydrophobic patch with V7023.54 and F7063.58 in TM3 to clamp onto the hydrophobic side of the α5 helix (I344H5.16, L348H5.20 and L353H5.25 in Gαi or L349H5.16, L353H5.20 and L358H5.25 in Gαq), whereas R7013.53 interacts with the backbone carbonyl of C351H5.23 in Gαi or Y356H5.23 in Gαq to further stabilize the C-cap of the α5 helix (Fig. 3a,c,d). This agrees with IP1 data showing that alanine mutations of F801, R7013.53 and F7063.58 markedly reduced G-protein signalling by CaSR (Extended Data Fig. 9a). The ICL3–TM3 patch might be a conserved element for G-protein activation by family C GPCRs, because similar residues in mGlu2 and GABAB are also crucial for Gi coupling10,11,12. Notably, the α5-helix binding pocket of CaSR contains a well-ordered H8, which was not observed in the Gi-coupled mGlu2, mGlu4 or GABAB structures. R8738.55 in H8 forms hydrogen bonds with D346H5.13 and Q350H5.17 in the α5 helix of Gαq, and contacts the last residue Y354H5.26 in Gαi (Fig. 3c,d). In line with this observation, the R873A mutation impaired mainly Gq but also Gi signalling when assessed in G-protein activation (Fig. 3b) and IP1 assays (Extended Data Fig. 9a), which could rationalize the loss-of-function phenotypes associated with the disease-causing mutations R873H and R873P (ref. 36). Furthermore, mutating T876 in H8 and K644 in ICL1 also impaired G-protein signalling (Extended Data Fig. 9a).
Distinct conformations of ICL2 for Gi and Gq coupling
CaSR adopts distinct ICL2 conformations to interact with Gi and Gq (Fig. 3c,d). In the Gi-coupled protomer, the ICL2 residues H715–W719 form a short α-helix that orients downwards to interact with the α5 helix of Gαi. R716 forms several salt bridges with E350H5.22, and W719 makes extensive contacts with E350H5.22 and C351H5.23 (Fig. 3c). Thus, the disease-associated CaSR inactivating mutation R716H (ref. 37) might act by weakening the ICL2–Gi interactions. By contrast, when bound to Gq, ICL2 adopts a loop conformation and shifts upwards, accompanied by a helical extension of TM4 (Fig. 3d). This rearrangement of ICL2 is due mainly to the bulky residue Y356H5.23 in the α5 helix of Gαq, which would clash with the helical conformation of ICL2 observed in the Gi-coupled protomer. As a result, ICL2 is positioned further away from Gαq, with F714 and T712 making limited contacts with Y356H5.23 (Fig. 3d). The marked conformational changes of ICL2 enable different sets of residues to engage with Gi and Gq, suggesting that it has a crucial role in determining G-protein specificity. In agreement with this interpretation, the R716A/W719A mutation in ICL2 substantially impaired Gi but not Gq signalling, whereas the T712A/F714A mutation reduced Gq but not Gi signalling (Fig. 3b). These findings highlight the importance of ICL2 in mediating G-protein selectivity by CaSR, and in particular for Gi coupling.
We also note that ICL2 establishes different interactions with the N terminus of Gαi and Gαq. The ICL2 of the Gi-coupled protomer fits into a hydrophobic groove formed by the αN–β1 junction, the β2–β3 loop and the α5 helix of Gαi, whereas the ICL2 of the Gq-coupled protomer is positioned further away from the N terminus of Gαq, with only K709 interacting with S198s2s3.2 in the β2–β3 loop of Gαq (Extended Data Fig. 8g,h). Differences in this region might be related to the 20° rotation of the αN helix between Gαi and Gαq (Extended Data Fig. 8i), and the mutations K709A, I710A and P711A had limited effects in our IP1 assays (Extended Data Fig. 9a), suggesting that this interface is less important for complex formation.
Opposing orientations of the CaSR C terminus
Although it is disordered in the NGC protomers, the CaSR C terminus is resolved up to residue 887 in the Gi-coupled protomer and up to residue 890 in the Gq-coupled protomer, in line with the previous finding that residues between 874 and 888 are crucial for CaSR signalling38. The most notable observation is that the CaSR C terminus adopts opposing orientations for the two distinct classes of G proteins (Fig. 4a,b). In the Gi-coupled protomer, the C terminus (residues 878–887) adopts a loop conformation and inverts its orientation after F881 towards the C-terminal lobe of the Gαi Ras domain. By contrast, when bound to Gq, the C terminus (residues 878–890) forms a H8 extension and further extends towards the N-terminal lobe of the Gα Ras domain (Extended Data Fig. 8j). The overall EM density for the C terminus in the Gi-coupled protomer is substantially weaker than that in the Gq-coupled protomer (Extended Data Fig. 5c), indicating that the C terminus is less critical for Gi coupling. In addition, the distinct orientations of the CaSR C terminus could be linked to the different positions of the Gα α5 helix and Ras domain in the two G-protein-coupled complexes (Extended Data Fig. 8j).

a, The interaction interface between the C terminus of the Gi-coupled protomer and Gαi in the CINA-bound CaSR–Gi complex. The resolved C terminus (residues 878–887) is highlighted in turquoise and its orientation is indicated by the arrow. b, The interaction interface between the C terminus of the Gq-coupled protomer and Gαq in the CINA-bound CaSR–Gq complex. The resolved C terminus (residues 878–890) is highlighted in turquoise and its orientation is indicated by the arrow. c, Functional BRET-based G-protein-activation assays show that key residues in the CaSR C terminus interacting with Gαq are crucial for the activation of Gq but not that of Gi.
A series of observations explain how the CaSR C terminus engages Gi and Gq with distinct orientations. First, H879 in the Gq-coupled protomer is stabilized by K345H5.12 in the α5 helix of Gαq and P798 in ICL3 (Fig. 4b), whereas H879 in the Gi-coupled protomer is positioned differently and makes no contact with Gαi (Fig. 4a). In line with these findings, the H879A mutation affected Gq coupling but not Gi coupling (Fig. 4c), which is also recapitulated in the IP1 assay (Extended Data Fig. 9a,b). Second, the side chain of F881 in the Gq-coupled protomer points towards and is stabilized by a hydrophobic cleft formed by Y325S6.2, F339H5.6 and A342H5.9 in Gαq (Fig. 4b). By contrast, the side chain of F881 in the Gi-coupled protomer faces away from Gαi to avoid a steric clash with the negatively charged residue D337H5.9 in the α5 helix (Fig. 4a and Extended Data Fig. 8k,l), facilitating the disruption of H8 and inverting the orientation of the C terminus. Furthermore, T888 in the Gq-coupled protomer is situated between the side chains of R338H5.5 and F341H5.8 in the α5 helix (Fig. 4b) whereas T888 is not resolved in the Gi-coupled protomer. We note that residues RH5.5 and FH5.8 are highly conserved in the Gαq/11 class (Extended Data Fig. 8m), and FH5.8L is a gain-of-function mutation identified in Gα11 that causes hypocalcaemia39. Moreover, superposition of the Gi- and Gq-coupled protomers on the 7TM shows that the C terminus in the Gq-bound conformation would clash with residues K192s2s3.1 and Q333H5.5 in Gαi (Extended Data Fig. 8k), and that the C terminus in the Gi-bound conformation would not be compatible with the α5 helix of Gαq (Extended Data Fig. 8l).
The CaSR C terminus confers Gq selectivity
The above analysis suggests that G-protein recognition by CaSR is determined mainly by ICL2 and the C terminus of the receptor, which undergo substantial conformational changes to accommodate the differences in the α5 helix and core region of Gαi and Gαq. Compared with Gi, the more stable interactions observed between the CaSR C terminus and Gq could partially compensate for the lack of strong interactions between ICL2 and Gq.
We performed further mutagenesis experiments to examine the importance of the CaSR C terminus in Gq coupling. IP1 data showed that two truncation mutants terminating at residues 882 (Δ882) and 886 (Δ886) almost entirely abolished Gq signalling (Extended Data Fig. 9b). Consistent with the observation that A884 in the C terminus closely contacts the α5 helix of Gαq (Extended Data Fig. 8k), the A884W mutation substantially impaired Gq-mediated signalling in IP1 assays, whereas the mutations A885W, R886A and A887W, pointing away from the α5 helix of Gq, had a minimal effect on Gq signalling (Extended Data Fig. 9b).
CaSR activation has been shown to stimulate the phosphorylation of residue T888 in the C terminus by the primary protein kinase C (PKC) enzyme40,41,42, which in turn downregulates CaSR signalling. On the basis of our structures, phosphorylation of T888 might impede its binding to Gq but not to Gi, thereby leading to biased G-protein signalling. In agreement with this hypothesis, the truncation mutant Δ887, which abolishes the binding of T888 to Gq, severely attenuated Gq signalling without affecting Gi signalling (Fig. 4c). Therefore, the physiological function of T888 phosphorylation is likely to selectively uncouple Gq signalling from CaSR activation. The structural and functional data also suggest that the gain-of-function T888M mutation43 could act by disrupting phosphorylation and increasing hydrophobic interactions with Gq. In addition, we found that the single-site mutations F881A and T888D (the T888A mutant did not express well) in the CaSR C terminus had a minimal effect on Gq and Gi signalling (Fig. 4c and Extended Data Fig. 9b), whereas the combined F881A/T888D mutation considerably attenuated Gq coupling but not Gi coupling (Fig. 4c), suggesting that these two key residues in the interaction interface (Fig. 4b) complement each other for recognizing Gq.
Collectively, our structural and functional data highlight the crucial role of the CaSR C terminus in Gq coupling, which might be further regulated by T888 phosphorylation. By contrast, the CaSR C terminus seems to contribute less to Gi coupling, because strong interactions with Gi are conferred by ICL2. The differential binding contributions of ICL2 and the C terminus for Gi and Gq further illustrate how these receptor elements mediate selective G-protein coupling.
The 7TMs in the presence and absence of cinacalcet
The active-state CaSR in nanodiscs shows different poses of cinacalcet in the two 7TMs, analogous to the active-state receptor in detergent micelles17 (Extended Data Fig. 6a). In both Gi- and Gq-coupled CaSR, 7TMGC exhibits a bent cinacalcet, whereas 7TMNGC contains an extended cinacalcet (Fig. 1d,e and Extended Data Fig. 10a,b), further suggesting key structural differences between the two 7TMs in activated CaSR. Superposition reveals that F8216.53 in the TM6 W6.50XXF6.53XP6.55 motif (in which ‘X’ denotes any amino acid) retains the inward-pointing inactive conformation17,18 in 7TMNGC, whereas it flips outwards towards the dimer interface, leading to a TM6 kink in 7TMGC (Fig. 5c and Supplementary Video 1). The outward flipping of F8216.53 appears to be promoted and stabilized by the folding down of cinacalcet’s phenyl group, which would clash with the inward-pointing conformation of F8216.53. In addition, G-protein binding appears to be associated with a slight upward displacement of TM6, including W8186.50 and P8236.55 (Fig. 5c).

a, Cryo-EM map of the PAM-free CaSR–Gi complex in lipid nanodiscs. b, Comparison of 7TMGC and 7TMNGC in the PAM-free CaSR–Gi complex. Conformational differences between 7TMGC and 7TMNGC are indicated with arrows. The EM density of the 7TM Trp is shown (at a map threshold of 0.41). c, Comparison of 7TMGC and 7TMNGC in the CINA-bound CaSR–Gi complex. Conformational differences between 7TMGC and 7TMNGC including the bound cinacalcet are indicated with arrows.
We note that our earlier functional analysis with CaSR mutants suggested that 7TMGC favours the extended PAM, including cinacalcet17. However, the unambiguous EM densities of cinacalcet in the CaSR–Gi and CaSR–Gq maps (Extended Data Fig. 5b,c) indicate that the C781W/I822W (favouring the extended pose) or the L773W/V833W (favouring the bent pose) mutant in our previous experiments did not efficiently occlude one pose of PAM while favouring the other pose. Indeed, our modelling reveals that the C781W/I822W mutation might have no effect on the binding of either pose, whereas the L773W/V833W mutation might impede PAM binding regardless of the conformations (Extended Data Fig. 10j,k).
To decipher PAM-induced conformational changes and the native 7TM configuration of G-protein-coupled CaSR, we further determined the cryo-EM structure of the nanodisc-embedded CaSR–Gi complex in the absence of exogenous PAMs at a global resolution of 3.5 Å, with local refinement maps at resolutions of 2.9 Å (VFT–CRD), 3.5 Å (CRD–7TM) and 3.5 Å (Gαiβγ) (Fig. 5a and Extended Data Figs. 1h and 3e–h). This PAM-free complex reveals an additional density in 7TMGC but not in 7TMNGC. This density is a good fit for Trp, a co-factor we added during protein purification. The indole ring of Trp occupies a similar position to that of the naphthylethylamine group of cinacalcet (Extended Data Fig. 10f) and interacts with several PAM-binding residues, including I7775.44, F6843.36 and E8377.32 (Fig. 5b and Extended Data Fig. 10d). However, Trp lacks a phenyl group like cinacalcet to facilitate the rearrangement of F8216.53, suggesting that this amino acid could function as a weak 7TM PAM. Supporting this notion, the PAM-free map reveals a weak density corresponding to a different rotamer of W8186.50 in 7TMGC (Extended Data Fig. 10g), positioned similarly to the inactive conformation of W8186.50 in the NAM-bound inactive structures17,18. Nevertheless, the hydrophobic indole ring of this alternative rotamer appears to be incompatible with the polar carboxyl group of Trp (Extended Data Fig. 10g). This suggests that some of the complexes used in the final three-dimensional (3D) reconstruction may not have a bound Trp in 7TMGC to stabilize W8186.50, potentially explaining why the W8186.50A mutation had no measurable effect on normal Ca2+-mediated CaSR activation15,18. Except for these differences, 7TMGC in the PAM-free and cinacalcet-bound CaSR–Gi complexes exhibits high similarity (Extended Data Fig. 10c,f).
By contrast, 7TMNGC in the PAM-free complex exhibits a modulatory site without a bound ligand (Extended Data Fig. 10e), which suggests that Trp has a substantially lower binding affinity for 7TMNGC than does cinacalcet. Superposition of the two 7TMs in the PAM-free complex reveals marked conformational differences in TM5 and TM6 (Supplementary Video 2), further highlighting the 7TM asymmetry of G-protein-coupled CaSR. 7TMGC shows an outward movement of TM5 away from the 7TM core, which is probably due to Trp binding (Fig. 5b). Furthermore, 7TMGC shows an upward movement of TM6 relative to 7TMNGC, which is more pronounced than that in the cinacalcet-bound CaSR–Gi complex (Fig. 5b,c). Superposition of 7TMNGC from the PAM-free and cinacalcet-bound CaSR–Gi structures also reveals a cinacalcet-induced outward repositioning of TM5 (Extended Data Fig. 10h), an aspect that could be useful for designing improved 7TM modulators.
Discussion
More than 30 years since the discovery of CaSR44, the mechanisms by which this GPCR instigates signalling have remained enigmatic. The structures presented here, supported by mutagenesis and functional data, show how the CaSR homodimer activates distinct heterotrimeric G proteins with mechanisms that are highly relevant for other family C GPCRs. Although the C1 and C2 tails (with and without NanoBiT) were used in the constructs to reconstitute the Gq-coupled and PAM-free complexes (Extended Data Fig. 1h,i), these modifications showed high flexibility and allowed the engineered CaSR to activate Gq or Gi similarly to the wild-type receptor without affecting its structural properties (Supplementary Fig. 1). Our results also provide insights into how CaSR function is regulated by diverse ligands, including polyamines, Trp and PAMs, as well as by C-terminal phosphorylation, and how known mutations might interfere with CaSR activation to cause diseases. We hope that these findings will support future strategies targeting the modulation of receptor activity within specific therapeutic windows.
The question of which CaSR protomer is primed to engage the G protein remains intriguing and is relevant to the development of therapeutics. Several lines of evidence lead us to postulate that the CaSR protomer with the extended PAM in the active state couples to the G protein and that this coupling is accompanied by PAM rearrangements in both protomers. First, the higher 7TM sitting position of this protomer relative to the opposing 7TM (Extended Data Fig. 6d) is consistent with that of 7TMGC in our G-protein-coupled structures. Second, the 7TM of this protomer is less tilted and positioned further towards the cytoplasm compared with the other 7TM (Extended Data Fig. 10i), rendering it more accessible to G proteins. Moreover, if the protomer with the bent PAM couples to the G protein, its 7TM would need to undergo a marked upward shift to establish the dimer interface observed in the CaSR–G complexes (Extended Data Fig. 10l), an unlikely transition in the lipid bilayer unless other unknown factors exert forces. Of note, the toggle switch residue F8216.53 in both protomers of the active-state CaSR shows the outward-facing active conformation (Extended Data Fig. 6d), suggesting that the additional 7TM compaction after G-protein coupling causes F8216.53 in 7TMNGC to flip back to the inactive conformation. This side-chain movement can also drive the PAM rearrangements, as an inward-pointing F8216.53 would clash with the bent pose (Fig. 5c).
The unexpected observation of a Trp molecule occupying 7TMGC suggests that this aromatic amino acid has a role as an allosteric modulator beyond its well-described binding at the VFTs. Our IP1 and GTPγS assays did not detect effects in Trp-induced CaSR activation using an ECD-truncated CaSR (ref. 15) (Extended Data Fig. 9c,d), indicating that—at least under our experimental conditions—allosteric modulation by Trp at the 7TM bundle would be weak. Nevertheless, the finding suggests that therapeutic 7TM PAM drugs have exploited the same binding site used by endogenous ligands of CaSR. It is also possible that the 7TM bundle of CaSR accommodates additional aromatic amino acids such as Phe (Supplementary Fig. 2). Collectively, these results improve our understanding of the variety of integrated signals driving CaSR activation; these range from Ca2+ and phosphate to positively charged polyamines and aromatic amino acids, and engage multiple sites across the VFT, CRD and 7TM domains of the receptor. The fine-tuning of various receptor sites for sensing environmental cues explains the relative high number of disease-causing mutations in CaSR.
Methods
Construct generation
For expression in insect cells, human CaSR (UniProt: P41180, residues 20–894) was cloned into a pFastBac1 vector with an N-terminal haemagglutinin (HA) signal sequence followed by a Flag tag and a 3×Ala linker as described previously17. This wild-type (WT) CaSR construct (Flag-CaSR) was used as the template for generating human CaSR-C1 and CaSR-C2 constructs. To make CaSR-C1, the DNA sequence of the GABAB1 C-terminal tail as well as an endoplasmic reticulum retention motif17 was inserted at the C terminus and the Flag tag was replaced with a Twin-Strep tag (WSHPQFEKGGGSGGGSGGSAWSHPQFEK). To make CaSR-C2, the sequence of the GABAB2 C-terminal tail with the endoplasmic reticulum retention motif was inserted at the C terminus. We have previously shown that only the CaSR-C1–CaSR-C2 heterodimer could reach the cell membrane; this has G-protein signalling profiles that are comparable with those of CaSR homodimers17. The LgBiT (ref. 22) sequence was fused to the C terminus of CaSR-C2 after a SGGS linker for NanoBiT tethering with Gq. For expression in HEK293 cells, human CaSR (WT and mutants), CaSR-C1–CaSR-C2 and the ECD-truncated CaSR (residues 599–894) were cloned into a pcDNA3.1(+) vector as described previously17.
Wild-type human Gαi3, a dominant-negative (DN) human Gαi3 (ref. 45) and a modified human Gαq (the N-terminal 28 residues were replaced with the N-terminal 22 residues of Gαi1 for improving the expression46) were cloned into a pFastBac1 vector. The human Gβ1 or Gβ1-SmBiT containing a C-terminal SmBiT connected with a 15-residue linker22, together with human Gγ2, were cloned into the pFastBac Dual vector with a 10×His tag at the N terminus of Gβ1.
Mutants used in this study were generated with the Q5 Site-Directed Mutagenesis kit (New England Biolabs) or with the standard QuikChange method using PrimeSTAR Max DNA Polymerase (Takara).
Expression and purification of CaSR
Wild-type CaSR was expressed in Spodoptera frugiperda Sf9 cells (Expression Systems, authenticated by supplier, not tested for mycoplasma) as described previously17. CaSR-C1–CaSR-C2 and CaSR-C1–CaSR-C2–LgBiT were expressed by infecting Sf9 cells at a density of 3.5 million–4 million cells per ml with 1% culture volume of P2 baculoviruses for each protomer. Cells were collected 48 h after infection and the pellets were snap-frozen in liquid nitrogen and stored at −80 °C until use.
For purification of CaSR that formed the cinacalcet-bound complex with Gi or Gq, cell pellets were thawed in 20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM CaCl2, 10% glycerol, 10 mM Trp, protease inhibitors, benzonase and 20 μM cinacalcet, and lysed by nitrogen cavitation. The lysate was centrifuged at 1,000g for 10 min to remove unbroken cells and nuclei, and membranes were collected by centrifugation at 100,000g for 30 min and solubilized in the same buffer with the addition of 1% n-dodecyl-β-maltoside (DDM) and 0.2% cholesteryl hemisuccinate (CHS) for 2–3 h, followed by centrifugation at 100,000g for 30 min. The resulting supernatant was incubated with anti-DYKDDDDK G1 affinity resin (GenScript) for WT CaSR or with Strep-Tactin XT 4Flow resin (IBA) for CaSR-C1–CaSR-C2–LgBiT for 3 h or overnight. The resin was then washed in the buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM CaCl2, 5% glycerol, 40 μM Trp, 20 μM cinacalcet, 0.02% DDM and 0.004% CHS and transferred to a gravity flow column. The column was washed with ten column volumes of the same buffer with 0.02% DDM and 0.004% CHS. Protein was eluted from the column using Flag peptide or Strep-Tactin XT elution buffer. Eluted protein was concentrated and injected onto a Superose 6 column with a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl (for WT CaSR) or 120 mM NaCl (CaSR-C1–CaSR-C2–LgBiT), 10 mM CaCl2, 40 μM Trp, 20 μM cinacalcet, 0.02% DDM and 0.004% CHS. The peak fractions were pooled and concentrated for later use.
For purification of the inactive-state CaSR or CaSR-C1–CaSR-C2 that formed the PAM-free complex with DN Gi, the procedures were similar to those described above, with the only difference being that no CaCl2 and cinacalcet (for the inactive-state CaSR) or no cinacalcet (for the PAM-free complex) were used throughout all steps including the lysis, solubilization and purification.
Expression and purification of heterotrimeric Gi and Gq
Heterotrimeric WT or DN Gi was expressed by infecting Trichoplusia ni Hi5 or Sf9 insect cells (Expression Systems, authenticated by supplier, not tested for mycoplasma) at a density of 3 million cells per ml, with baculoviruses encoding Gαi or DN Gαi, Gβ1, Gγ2 and Ric8A. Heterotrimeric Gq was expressed by infecting Sf9 cells at a density of 3.5 million–4 million cells per ml with baculoviruses encoding Gαq, Gβ1-SmBiT, Gγ2 and Ric8A. Cells were collected 48 h after infection and the pellets were snap-frozen in liquid nitrogen and stored at −80 °C until use.
Cell pellets containing heterotrimeric Gi or Gq were thawed in 20 mM HEPES pH 7.5, 150 mM NaCl, 10% glycerol, 5 mM MgCl2, 5 mM β-mercaptoethanol, 50 μM GDP, protease inhibitors and benzonase, and lysed by nitrogen cavitation. The lysate was centrifuged at 1,000g for 10 min to remove unbroken cells and nuclei, and membranes were collected by centrifugation at 100,000g for 30 min and solubilized in the same buffer with the addition of 1% sodium cholate, 0.05% DDM and 0.01% CHS for 2–3 h, followed by centrifugation at 100,000g for 30 min. The resulting supernatant was supplemented with 30 mM imidazole and gravity loaded over Ni-NTA resin. The resin was washed with ten column volumes with increasing concentrations of DDM and CHS and decreasing concentrations of cholate until a final wash in 20 mM HEPES pH 7.5, 150 mM NaCl, 0.02% DDM, 0.004% CHS, 5% glycerol, 1 mM MgCl2, 5 mM β-mercaptoethanol, 50 μM GDP and 30 mM imidazole, before being eluted into the same buffer containing 250 mM imidazole. Eluted G-protein heterotrimer was then supplemented with HRV-3C protease to remove the His tag and incubated overnight at 4 °C with dialysis against low-imidazole buffer. The dialysed protein was flowed through a Ni-NTA gravity column to remove HRV-3C and uncleaved heterotrimer, concentrated and injected onto an Enrich SEC 650 column. The final SEC buffer contains 20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% DDM, 0.004% CHS, 1 mM MgCl2, 100 μM TCEP and 50 μM GDP. The peak fractions were pooled and concentrated for later use.
Expression and purification of MSP1E3D1
The expression plasmid with the gene encoding MSP1E3D1 was purchased from Addgene (20066). MSP1E3D1 was expressed and purified as described previously47 with slight modifications. In brief, the plasmid was transformed into E. coli BL21 (DE3). The culture, grown to an optical density at 600 nm (OD600 nm) of 0.9, was induced with 1 mM IPTG at 23 °C for 16 h. The cells were collected and resuspended in lysis buffer comprising 20 mM sodium phosphate buffer 7.4, 1% Triton X-100 and protease inhibitors. The cells were lysed by sonication, and the soluble fraction of cell lysates after centrifugation was loaded to a Ni-NTA column three times. The column was washed with a buffer containing 40 mM Tris-HCl 8.0, 300 mM NaCl and 50 mM imidazole. MSP1E3D1 was eluted using a buffer containing 40 mM Tris-HCl 8.0, 300 mM NaCl and 300 mM imidazole, and was further purified by gel filtration chromatography (Superdex 200, Cytiva) using a buffer containing 25 mM HEPES pH 7.5 and 150 mM NaCl. The peak fractions were pooled and concentrated for later use.
Assembling CaSR and Gi in detergent micelles
For assembling CaSR and Gi in detergents, WT CaSR was mixed with WT Gi protein at a ratio of 1:1.2 CaSR to Gi, and further incubated for 2 h on ice before the addition of apyrase, and the reaction was allowed to proceed overnight at 4 °C. The next day, the reconstitution mixture was supplemented with 0.5% LMNG and further incubated on ice for 1 h. The sample was then applied onto a Superose 6 column with a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 0.001% LMNG, 0.0002% CHS, 10 mM CaCl2, 40 μM Trp and 20 μM cinacalcet. The peak fractions were screened by negative-stain EM (ref. 48), and the fractions containing the complex were collected and concentrated for preparing cryo-EM grids.
Reconstitution of CaSR and CaSR–G-protein complexes in lipid nanodiscs
Dioleoylphosphatidylcholine (DOPC; Avanti Polar Lipids) was mixed with dioleoylphosphatidylglycerol (DOPG; Avanti Polar Lipids) at a molar ratio of 3:2, dried and kept under vacuum overnight. The lipid mixture was dissolved by the addition of 20 mM HEPES pH 7.5, 150 mM NaCl and 50 mM sodium cholate.
For inactive CaSR, purified WT CaSR was mixed with the lipid mixture and the scaffold protein MSP1E3D1 at a molar ratio of 1:180:2 and incubated for 1 h on ice. Bio-Beads SM2 (Bio-Rad) were then added into the mixture (0.4 g of wet beads per ml) and incubated with gentle rocking for 6 h at 4 °C. The reconstitution mixture was spun down and the supernatant was concentrated and applied onto a Superose 6 column with a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl and 100 μM Trp. The peak fractions were screened by negative-stain EM (ref. 48), and the fractions containing inactive CaSR were collected and concentrated for preparing cryo-EM grids.
For the formation of the cinacalcet-bound CaSR–Gi complex, purified WT CaSR was mixed with purified WT Gi at a ratio of 1:1.1 with the addition of apyrase for 15 min on ice. The proteins were further mixed with the lipid mixture and the scaffold protein MSP1E3D1 at a molar ratio of 1:180:2 and incubated for 1 h on ice. Bio-Beads SM2 (Bio-Rad) were then added into the mixture (0.4 g of wet beads per ml) and incubated with gentle rocking for 6 h at 4 °C. The reconstitution mixture was spun down and the supernatant was concentrated and applied onto a Superose 6 column with a buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 10 mM CaCl2, 40 μM Trp and 20 μM cinacalcet. The peak fractions were screened by negative-stain EM (ref. 48), and the fractions containing the complex were collected and concentrated for preparing cryo-EM grids. For the formation of the PAM-free CaSR–Gi complex, the procedures were similar to those described above except that the CaSR-C1–CaSR-C2 and DN Gi proteins were used for reconstituting the complex, and no PAM was added throughout all steps.
For the formation of the cinacalcet-bound CaSR–Gq complex, purified CaSR (CaSR-C1–CaSR-C2-LgBiT) was mixed with purified Gq protein at a ratio of 1:1.3 with the addition of apyrase for 20 min at 4 °C. The proteins were further mixed with the lipid mixture and the scaffold protein MSP1E3D1 at a molar ratio of 1:150:2 and incubated for 1.5 h on ice. Bio-Beads SM2 (Bio-Rad) were then added into the mixture (0.2 g of wet beads per ml) and incubated with gentle rocking for 2 h at 4 °C. The same amount of Bio-Beads was added into the mixture again with the reconstitution proceeding for another 4 h at 4 °C. The mixture was spun down and the supernatant was directly injected onto a Superose 6 column with a buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 15 mM CaCl2, 100 μM Trp and 30 μM cinacalcet. The peak fractions were screened by negative-stain EM (ref. 48), and the fractions containing the complex were collected and concentrated for preparing cryo-EM grids.
Cryo-EM data collection and processing
A 3-μl volume of sample at around 7 mg ml−1 supplemented with 0.02% (w/v) β-octyl glucoside detergent was applied to freshly glow-discharged 300-mesh R1.2/R1.3 UltrAuFoil holey gold grids (Quantifoil) under 100% humidity at 4 °C. Excess sample was blotted away, and the grids were subsequently plunge-frozen into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific) and stored in liquid nitrogen. For CaSR–Gi or CaSR–Gq complexes in nanodiscs, frozen grids were imaged at cryogenic temperatures with a Titan Krios G2 (Thermo Fisher Scientific) transmission electron microscope with a post-column energy filter operated at 300 kV on a Gatan K3 direct electron camera at a pixel size of 0.8677 Å. Micrographs, dose-fractionated over 50 frames, were recorded in super-resolution mode with defocus values ranging from −0.5 μm to −1.5 μm using SerialEM (ref. 49). Micrographs were recorded for about 2.5 s with a total exposure dose of approximately 50 electrons per Å2. For CaSR–Gi in detergents, frozen grids were imaged at cryogenic temperatures with a Titan Krios G2 (Thermo Fisher Scientific) transmission electron microscope without a post-column energy filter operated at 300 kV on a Gatan K3 direct electron camera at a pixel size of 0.8521 Å. Micrographs, dose-fractionated over 50 frames, were recorded in super-resolution mode with defocus values ranging from −0.5 μm to −1.5 μm using SerialEM. Micrographs were recorded for 2.49 s with a total exposure dose of 68.31 electrons per Å2. For inactive CaSR in nanodiscs, frozen grids were imaged at cryogenic temperatures with a Titan Krios G2 (Thermo Fisher Scientific) transmission electron microscope with a Selectris X post-column energy filter operated at 300 kV on a Falcon 4i direct electron camera at a pixel size of 0.75 Å. Micrographs were recorded with defocus values ranging from −0.5 μm to −1.5 μm using EPU (Thermo Fisher Scientific). Micrographs were recorded for 3.78 s with a total exposure dose of 50 electrons per Å2.
For a detailed breakdown of data processing and exact particle numbers, see Extended Data Figs. 2–4. Micrographs were imported into cryoSPARC (ref. 50) and dose-fractionated image stacks were subjected to patch motion correction and patch CTF refinement. A total of 7,666 micrographs for CaSR–Gi in detergents, 12,758 micrographs for inactive CaSR in nanodiscs, 20,528 micrographs for the cinacalcet-bound CaSR–Gi complex in nanodiscs, 31,047 micrographs for the PAM-free CaSR–Gi complex in nanodiscs and 47,434 micrographs for the CaSR–Gq complex in nanodiscs were selected for further processing after manual curation. Auto-picked particles were extracted and subjected to several rounds of reference-free two-dimensional 2D classification, followed by iterative rounds of 3D ab initio reconstruction with multiple classes and 3D heterogeneous refinement to remove particles from poorly defined 3D classes. In early 3D classification rounds, particles from poor 3D classes were subjected to additional 2D classification and particles from better resolved classes were reincluded for further heterogeneous refinement.
For the dataset of CaSR–Gi in detergents, heterogenous refinement yielded maps corresponding to active-state CaSR, Gi bound to detergent micelles and CaSR–Gi in detergent micelles, consistent with the 2D averages. The particles of Gi bound to detergent micelles were further cleaned by 3D classification and the resulting particles were subjected to nonuniform refinement followed by local refinement with a soft mask around the G proteins. The particles of CaSR–Gi in detergent micelles were further cleaned by 3D and 2D classifications and the resulting particles were subjected to nonuniform refinement followed by local refinement with a soft mask around either the receptor or the G proteins.
For the dataset of inactive CaSR in nanodiscs, heterogenous refinement yielded a particle group corresponding to inactive CaSR, consistent with the 2D averages. The particles were further cleaned by 3D and 2D classifications and the resulting particles were subjected to nonuniform refinement followed by local refinement with a soft mask around either VFT–CRD or the 7TMs.
For the dataset of the cinacalcet-bound CaSR–Gi complex in nanodiscs, heterogenous refinement yielded a conformationally homogeneous group corresponding to Gi-coupled CaSR. This group of particles was subjected to local motion correction and further cleaned by 3D and 2D classifications. The resulting particles were then subjected to nonuniform refinement followed by CTF refinement and local refinement with a soft mask around VFT–CRD, CRD–7TM, or Gi.
For the dataset of the PAM-free CaSR–Gi complex in nanodiscs, heterogenous refinement yielded two particle groups corresponding to Gi-coupled CaSR and active-state CaSR. The particles of Gi-coupled CaSR were subjected to local motion correction, and further cleaned by 3D and 2D classifications. The selected particles were then subjected to nonuniform refinement followed by CTF refinement and local refinement with a soft mask around VFT–CRD, CRD–7TM or Gi. The particles of active-state CaSR were imported into RELION (ref. 51) for further cleaning by focused 3D classification without alignment on the two 7TMs. The selected particles were then transferred back to cryoSPARC for nonuniform refinement followed by CTF refinement and local refinement with a soft mask around either VFT–CRD or CRD–7TM. Most side chains of this 7TM domain are unresolved in the local refinement map (Extended Data Fig. 3i,j), suggesting conformational flexibility.
For the dataset of the cinacalcet-bound CaSR–Gq complex in nanodiscs, heterogenous refinement yielded two major particle groups corresponding to active-state CaSR and the Gq-coupled CaSR. The particles of active-state CaSR were imported into RELION for further cleaning by focused 3D classification without alignment on the two 7TMs, followed by C2 symmetry expansion and 7TM focused classification without alignment. Two good 3D classes with C1 symmetry were obtained, and the particles from one class were selected and then transferred back to cryoSPARC for local refinement with a soft mask around either VFT–CRD or CRD–7TM. The particles of Gq-coupled CaSR were subjected to local motion correction, and further cleaned by 3D and 2D classifications. The selected particles were then subjected to nonuniform refinement followed by CTF refinement and local refinement with a soft mask around VFT–CRD, CRD–7TM or Gq.
Model building and refinement
For model building and refinement, composite maps were generated in Chimera (ref. 52) by merging local refinement maps using the ‘vop maximum’ command. The active-state CaSR structure in detergents (Protein Data Bank (PDB) ID: 7M3F) and the Gi from the mGlu2–Gi structure10 (PDB ID: 7MTS) and the Gq from the CCKBR–Gq structure53 (PDB ID: 7F8W) were used as the initial models for docking into the cryo-EM maps in Chimera. The models were then subjected to iterative rounds of manual adjustment in Coot54 and real-space refinement in PHENIX (ref. 55). The model statistics were validated in MolProbity (ref. 56). The refinement statistics are provided in Extended Data Table 1. Chimera and ChimeraX (ref. 57) were used for structure visualization and figure preparation.
Mass spectrometry
Standards for spermine and spermidine were prepared in a buffer containing 20 mM HEPES pH 7.5 and 150 mM NaCl. A 100-μl sample of the cinacalcet-bound CaSR–Gi complex in nanodiscs at 1 mg ml−1 was alkalized by using 100 μl 10 M NaOH solution and vortexed for 1 min. Then, 200 μl ethyl acetate was added to extract alkalized polyamines from the aqueous layer. The ethyl acetate layer was collected and dried in a vacuum. The samples were then dissolved in methanol and subjected to direct infusion electrospray ionization–mass spectrometry (ESI–MS) analysis on an Orbitrap Elite mass spectrometer. Mass spectrometry data were processed using Thermo Xcalibur software (Thermo Fisher Scientific).
BRET assay using TRUPATH biosensors
BRET assays were performed and analysed as previously described19 with specific modifications. HEK293S cells (Thermo Fisher Scientific, authenticated by supplier, not tested for mycoplasma) grown in FreeStyle suspension medium (Thermo Fisher Scientific) were transfected at a density of 1 million cells per ml using 1,200 ng total DNA at a 1:1:1:1 ratio of receptor:Gα–RLuc8:Gβ:Gγ–GFP and a DNA:polyethylineimine ratio of 1:5. After 48 hours, cells were collected by centrifugation, incubated in assay buffer (Hank’s balanced salt solution with 25 mM HEPES pH 7.5) supplemented with 0.5 mM EGTA for 5 min, and subsequently washed with the assay buffer. The cells were then resuspended in assay buffer containing 5 μg ml−1 of freshly prepared coelenterazine 400a (GoldBio) and dispensed into white 96-well plates at a volume of 60 μl per well. Thirty microlitres of ligand solutions, prepared in assay buffer with 0.1% bovine serum albumin (BSA), were immediately added to plated cells. Five minutes after the addition of ligand, plates were read using a SpectraMax iD5 plate reader using 410-nm and 515-nm emission filters with a 1-s integration time per well. The computed BRET ratios (GFP2/RLuc8 emission) were normalized to ligand-free control (net BRET) before analysis using a four-parameter logistic equation in GraphPad Prism v.9.
Enzyme-linked immunosorbent assay (ELISA)
The cell-surface expression of Flag–CaSR WT and mutant constructs, as well as Flag–CaSR-C1 and HA–CaSR-C2 constructs, were evaluated as previously described17. In brief, cells were seeded in poly-d-lysine-coated white (white- or clear-bottomed) 96-well plates and incubated for 24–32 h (BRET assays) or 48 h (IP1 assays) at 37 °C and 5% CO2. Cells were washed twice with phosphate-buffered saline (PBS) (with 1 mM CaCl2) and fixated with 4% paraformaldehyde (PFA) in PBS for 5 min. After fixation, cells were incubated in blocking solution (ddH2O, 4% skimmed milk, 1 mM CaCl2, 1 mM MgCl2 and 50 mM Trizma hydrochloride solution, pH 7.4) for 30 min, followed by incubation with HRP-conjugated Flag (Sigma Aldrich, A8592) or HA (R&D systems, HAM0601) antibodies diluted 1:2,000 in blocking solution for 60 min. Subsequently, cells were washed four times in blocking solution and then four times with PBS. The detection signal was measured by chemiluminescence using the SuperSignal ELISA Femto solution (Thermo Fisher Scientific) or HRP substrate (Bio-Rad, 170-5060), according to the manufacturer’s instructions. The surface expression levels of CaSR WT and mutants used in the study are shown in Extended Data Fig. 9e,f.
Steady-state readout of BRET-based G-protein-activation assay
HEK293 cells (ATCC CRL-1573, authenticated by the supplier, routinely tested for mycoplasma) were transfected in suspension with DNA encoding the Flag–CaSRΔ894 WT or mutants (100–200 ng, to reach similar surface expression levels determined by ELISA) together with Venus(1–155)-Gγ2 (7 ng), Venus(159–239)-Gβ1 (7 ng), masGRK3ctNanoLuc (7 ng), human Gαi3 or Gαq proteins (40 ng) and empty pcDNA3.1(+) vector to a total of 1 µg per 1 ml of cell suspension transfected. The constructs for the BRET-based G-protein-activation assay were provided by the laboratory of K. A. Martemyanov20. In brief, DNA mixtures (in OptiMEM) were mixed with FuGENE6 transfection reagent (in OptiMEM), and incubated for 20 min at room temperature, as previously described17. After incubation, the DNA-FuGENE6 mixture was added to the HEK293 cell suspension that was diluted to 0.5 million cells per ml. Cells were seeded at 50,000 cells per well in poly-d-lysine-coated, white-bottomed 96-well plates and incubated for 24–32 h at 37 °C and 5% CO2. To initiate the assay, cells were washed once with 150 µl assay buffer (HBSS, no Ca, Mg, Gibco 14175 supplemented with 20 mM HEPES, pH 7.4), exchanged to 40 µl per well assay buffer and pre-incubated with 10 µl per well NanoBRET substrate (Promega N1572) diluted 1:250 for 5 min. Then, 50 µl per well assay buffer containing Ca2+ at increasing concentrations to give a final concentration range of 0.63–10 mM was added to separate wells, and the change in BRET ratio was recorded for 60 min using an Envision plate reader, equipped with 470/24 nm (NanoLuc donor bioluminescence) and 535/30 nm (Venus acceptor fluorescence) emission filters. To generate concentration response curves, the AUC for each curve was calculated, and the AUC was plotted as a function of the Ca2+ concentration by nonlinear regression using the ‘log agonist vs. response’ model with variable slope using GraphPad Prism v.9.
Kinetic readout of BRET-based G-protein-activation assay
HEK293 cells (ATCC CRL-1573) were transfected in suspension (50,000 per well) with receptor plasmid constructs (7–35 ng), Venus(1–155)-Gγ2 (6.3 ng), Venus(159–239)-Gβ1 (6.3 ng), masGRK3ctNanoLuc (6.3 ng), human Gα subunits (6.3 – 31.5 ng) Ric8A (6.3 ng; Gα chaperone, only co-transfected for Gα14 and Gα15 activation), Ric8B (6.3 ng; Gα chaperone, only co-transfected for Gαolf activation) and empty pcDNA3.1(+) vector (supplemented to in total 150 ng per well) using Lipofectamine 2000 reagent (Invitrogen; 1:2.5 DNA: lipofectamine ratio) in OptiMEM (Gibco). After transfection, cells were seeded in poly-d-lysine-coated, white-bottomed 96-well plates and incubated for 24–32 h at 37 °C and 5% CO2. Before the experiment, cells were washed twice with 100 µl per well HBSS (no Ca, Mg, Gibco 14175) supplemented with 20 mM HEPES, pH 7.4, and 25 µl per well HBSS was added to the cells. Subsequently, cells were incubated with the NanoGlo luciferase assay substrate (Promega; 1:250 dilution; 25 µl per well), followed by ligand stimulation prepared in the same buffer (50 µl per well).
The BRET signal was measured on a LUMIstar Omega plate reader at 37 °C, and quantified by calculating the ratio between the Venus acceptor (520–550 nm) and the NanoLuc donor (465–490 nm) emissions. To map the G-protein profiles of CaSR and other GPCRs, including the μ-opioid receptor (MOR), the histamine receptor 1 (H1R) and β2AR, the kinetic traces of G-protein activation were measured (45 s, 0.04 s per datapoint), after automated injection with a single saturating concentration of a ligand or a vehicle condition. From individual kinetic measurements, the maximum BRET signal (max. ΔBRET) was determined from the peak ligand responses and compared to corresponding measurements with a vehicle condition to calculate a fold/vehicle (basal) ratio.
To investigate the pharmacological mode of action of spermine at CaSR, concentration response curves of spermine, in the presence or absence of Ca2+ and/or EDTA, were generated for Gαi3 and Gαq activation 20 s after ligand stimulation. Concentration response curves were corrected for the basal condition (vehicle), normalized against the maximal response of the reference ligand Ca2+ and fitted by nonlinear regression using the ‘log agonist vs. response’ model with variable slope using GraphPad Prism v.9.
IP1 assays for the etelcalcetide competition and spermine mechanism studies
For competition experiments of etelcalcetide with spermine, Flp-In HEK293 cells stably expressing HA–CaSR WT (Flp-In HEK293, RRID: CVCL_U421) were generated using Flp-In technology as previously described58, and cultured in Dulbecco’s modified Eagle’s medium (Gibco, Thermo Fisher Scientific) supplemented with hygromycin B (250 mg ml−1), 10% v/v fetal bovine serum and 1% v/v penicillin–streptomycin, at 37 °C, and 5% CO2. The IP1 experiments for ligand profiling were performed using the IP-One assay (Cisbio) as previously described17. In brief, Flp-In HEK293 HA–CaSR cells (50,000 per well) were seeded into poly-d-lysine-coated, transparent 96-well plates and incubated overnight at 37 °C and 5% CO2. Subsequently, cells were rinsed with HBSS (no Ca, Mg, Gibco 14175) supplemented with 20 mM HEPES, pH 7.4, and incubated with etelcalcetide in the same solution for 5 h at 37 °C. To this end, time-course experiments indicated that the potency of etelcalcetide (at 3 mM Ca2+) reached a maximum at a 5-h pre-incubation time. After pre-incubation with etelcalcetide, cells were incubated with (2×) ligand solutions prepared in HBSS (20 mM HEPES pH 7.4) supplemented with (2×) 20 mM LiCl for 40 min at 37 °C.
To investigate the pharmacological mode of action of spermine, HEK293 (ATCC CRL-1573) cells (50,000 per well) were transfected in suspension with DNA encoding CaSR (7 ng per well) and empty PcDNA3.1(+) vector (53 ng per well) using Lipofectamine 2000 reagent (Invitrogen; 1:2.5 DNA:lipofectamine ratio) in OptiMEM (Gibco). After transfection, cells were seeded into poly-d-lysine-coated, transparent 96-well plates and incubated overnight at 37 °C and 5% CO2. Subsequently, the cells were rinsed with HBSS (20 mM HEPES pH 7.4) and incubated with ligand solutions prepared in HBSS (20 mM HEPES pH 7.4) with 20 mM LiCl for 40 min at 37 °C. After incubation with ligands, cells were then rinsed with HBSS and lysed using the IP-One Conjugate & Lysis buffer (30 μl per well) (Cisbio). Next, the lysate solution (10 μl per well) was transferred to a 384-well Optiplate (Perkin Elmer), after which (10 μl per well) detection solution (HBSS + 2.5% v/v IP1–d2 conjugate and 2.5% v/v anti-IP1 antibody–cryptate terbium conjugate) was added. The solutions were incubated in the dark for 1 h at room temperature, after which the IP1 signal (ratio) was detected using a Envision plate reader (Perkin Elmer). The data were transformed using a standard curve to obtain absolute IP1 values (nM). Subsequently, the data from individual experiments were corrected for the basal condition (vehicle), normalized against the maximal response of a reference ligand and grouped. The concentration response curves were fitted to a nonlinear regression ‘log agonist vs. response’ model with variable slope using GraphPad Prism v.9.
IP1 assay for mutant characterization
HEK293 cells (ATCC CRL-1573) were transfected in suspension with DNA encoding WT and mutant CaSR in concentrations ranging from 25 ng to 100 ng per ml of cell suspension to obtain similar surface expression levels of WT and mutant receptors. Receptor DNA diluted in OptiMEM was supplemented with empty vector pcDNA3.1(+) to a total of 1 µg DNA per ml of cell suspension transfected using FuGENE6, as described previously17. Cells were seeded at a density of 25,000 cells per well in clear, poly-d-lysine-coated 96-well plates, and incubated for 48 h before the assay. To assess the contribution of Gi signalling to IP1 accumulation, cells transfected with WT CaSR were incubated with 200 ng ml−1 PTX for 18 h before the assay. The assay was performed as described above, with the exception of incubation time with Ca2+ for 45 min at 37 °C, and cell lysis with 40 μl per well IP-One Conjugate & Lysis buffer (Cisbio).
Assays of [35S]-GTPγS binding activity
ECD-truncated human CaSR was expressed for 48 h in 50 ml HEK293S cells (Thermo Fisher Scientific, authenticated by supplier, not tested for mycoplasma) after transfection at 2 million cells per ml. The collected cells were lysed by nitrogen cavitation in lysis buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA and protease inhibitors. Unbroken cells were cleared by centrifugation at 1,000g and membranes were collected by centrifugation at 100,000g. The membranes were Dounce-homogenized into lysis buffer, reprecipitated at 100,000g, Dounce-homogenized again, snap-frozen in storage buffer (lysis buffer with 12% w/v sucrose) and then stored at −80 °C. Membrane homogenates (3.6 μg per μl) were reconstituted with 500 nM purified Gαi3β1γ2 heterotrimer in binding buffer (50 mM HEPES pH 7.4, 1 mM dithiothreitol (DTT), 1 mM EDTA and 3 µg ml−1 purified BSA) augmented with Trp or cinacalcet, in the absence and presence of 10 mM CaCl2, then pre-incubated for 10 min at 25 °C. GTPγS binding reactions were initiated by the addition of an equal volume of binding buffer supplemented with 50 mM NaCl, 10 mM MgCl2, 20 µM GDP and 4 µM [35S]-GTPγS. After a 10-min incubation at 25 °C, reactions were quenched with 20 mM Tris pH 7.7, 100 mM NaCl, 10 mM MgCl2, 1 mM GTP and 0.08% w/v C12E10 and filtered through Protran BA85 nitrocellulose filters (GE Healthcare) using a Millipore vacuum manifold. The filters were washed, dried and subjected to liquid scintillation counting.
Statistics and reproducibility
For reproducibility, sample sizes and descriptions of error bars for all figures are described in this section. In Fig. 2c, data represent mean ± s.e.m. of at least five independent experiments; for the Ca2+ concentration response curves (CRCs), data for CaSR WT represent n = 8 independent experiments, data for the mutants E604A/E757A/Y829A and E228A/D238A/E241A represent n = 6 independent experiments and data for the mutants E228A/D238A/E241A/E604A/E757A/Y829A, E604A/E755A/E757A/D758A/E759A/Y829A and E228A/D238A/E241A/E604A/E755A/E757A/D758A/E759A/Y829A represent n = 5 independent experiments; for the spermine CRCs, data for CaSR WT represent n = 8 independent experiments, data for the mutants E228A/D238A/E241A, E228A/D238A/E241A/E604A/E757A/Y829A and E604A/E755A/E757A/D758A/E759A/Y829A represent n = 6 independent experiments and data for the mutants E604A/E757A/Y829A and E228A/D238A/E241A/E604A/E755A/E757A/D758A/E759A/Y829A represent n = 5 independent experiments. In Fig. 3b, data represent mean ± s.e.m. of at least seven independent experiments; data for CaSR WT represent n = 9 independent experiments, data for the mutants R873A, T712A/F714A and R716A/W719A represent n = 8 independent experiments and data for the mutant F801A represent n = 7 independent experiments. In Fig. 4c, data represent mean ± s.e.m. of at least seven independent experiments; data for CaSR WT and Δ887 represent n = 9 independent experiments, data for the mutants H879A and F881A/T888D represent n = 8 independent experiments and data for the mutants F881A and T888D represent n = 7 independent experiments. In Extended Data Fig. 1a, data and error bars represent mean ± s.e.m. of n = 4 independent experiments. In Extended Data Fig. 1b, data represent mean ± s.e.m. of n = 3 independent experiments. In Extended Data Fig. 1c, data represent mean ± s.e.m. of n = 6 independent experiments. In Extended Data Fig. 1d, data and error bars represent mean ± s.e.m. of at least three independent experiments; data for CaSR WT, CaSR Δ894, MOR and H1R represent n = 3 independent experiments and data for β2AR represent n = 5 independent experiments. In Extended Data Fig. 1e, data represent mean ± s.e.m. of n = 4 independent experiments performed in duplicate. In Extended Data Fig. 6g, data represent mean ± s.e.m. of at least seven independent experiments; data for CaSR WT and the mutants F806A, F809A and K805A/F806A represent n = 9 independent experiments, data for the mutants K805A and I822A represent n = 8 independent experiments and data for the mutants F788A, Y825A, Y829A, V833A, F788A/K805A, K805A/F809A, Y825A/Y829A, Y825A/V833A and Y825A/Y829A/V833A represent n = 7 independent experiments. In Extended Data Fig. 7d, data represent mean ± s.e.m. of at least three independent experiments performed in duplicate; data for the Ca2+- and spermine-only CRCs represent n = 5 independent experiments, data for the spermine CRCs with 0.62 μM, 1.25 μM, 2.5 μM or 5.0 μM etelcalcetide represent n = 4 independent experiments and data for the spermine CRCs with 100 μM etelcalcetide or 10 μM NPS 2143 represent n = 3 independent experiments. In Extended Data Fig. 7e, data for the Gi3 and Gq activation assays represent mean ± s.e.m. of n = 5 independent experiments performed in duplicate, and data for the IP1 accumulation represent mean ± s.e.m. of n = 7 independent experiments performed in duplicate. In Extended Data Fig. 9a,b, data represent mean ± s.e.m. of at least five independent experiments; data for CaSR WT, WT + PTX and the mutants A884W and R886A represent n = 9 independent experiments, data for the mutants P711A, R716A, I869A, T876A, H879A, F881A and A887W represent n = 8 independent experiments, data for the mutants V705A, K709A, T712A, K805A, V872A and R873A represent n = 7 independent experiments, data for the mutants F706A, I710A, F714A, W719A, F801A, T888D, Δ886 and Δ882 represent n = 6 independent experiments and data for the mutants K644A, R701A and A885W represent n = 5 independent experiments. In Extended Data Fig. 9c,d, data and error bars represent mean ± s.e.m. of n = 3 independent experiments. In Extended Data Fig. 9e, data represent mean ± s.e.m. of at least seven independent experiments; data for CaSR WT and the mutants F806A, F809A, Δ887 and K805A/F806A represent n = 9 independent experiments, data for the mutants K805A, I822A, R873A, H879A, T712A/F714A, R716A/W719A and F881A/T888D represent n = 8 independent experiments and data for the mutants K709A, F788A, F801A, Y825A, Y829A, V833A, F881A, T888D, F788A/K805A, K805A/F809A, Y825A/Y829A, Y825A/V833A and Y825A/Y829A/V833A represent n = 7 independent experiments. In Extended Data Fig. 9f, data represent mean ± s.e.m. of at least five independent experiments; data for CaSR WT, WT + PTX and the mutant A884W represent n = 9 independent experiments, data for the mutants P711A, R716A, I869A, T876A, H879A, R886A and A887W represent n = 8 independent experiments, data for the mutants V705A, K709A, T712A, W719A, V872A, R873A and F881A represent n = 7 independent experiments, data for the mutants F706A, I710A, F714A, F801A, K805A, Δ886 and Δ882 represent n = 6 independent experiments and data for the mutants K644A, R701A, A885W and T888D represent n = 5 independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps and corresponding coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and the PDB, respectively, under the following accession codes: EMD-40914 and 8SZF (cinacalcet-bound active-state CaSR); EMD-40915 and 8SZG (cinacalcet-bound CaSR–Gq complex); EMD-40916 and 8SZH (cinacalcet-bound CaSR–Gi complex); and EMD-40917 and 8SZI (PAM-free CaSR–Gi complex). Source data are provided with this paper.
References
Kniazeff, J., Prézeau, L., Rondard, P., Pin, J.-P. & Goudet, C. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol. Ther. 130, 9–25 (2011).
Hofer, A. M. & Brown, E. M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 4, 530–538 (2003).
Hannan, F. M., Kallay, E., Chang, W., Brandi, M. L. & Thakker, R. V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 15, 33–51 (2019).
Vahe, C. et al. Diseases associated with calcium-sensing receptor. Orphanet J. Rare Dis. 12, 19 (2017).
Massy, Z. A., Henaut, L., Larsson, T. E. & Vervloet, M. G. Calcium-sensing receptor activation in chronic kidney disease: effects beyond parathyroid hormone control. Semin. Nephrol. 34, 648–659 (2014).
Eckardt, K.-U. et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382, 158–169 (2013).
Niswender, C. M. & Conn, P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 (2010).
Gregory, K. J. & Goudet, C. International union of basic and clinical pharmacology. CXI. Pharmacology, signaling, and physiology of metabotropic glutamate receptors. Pharmacol. Rev. 73, 521–569 (2021).
Leach, K. et al. International Union of Basic and Clinical Pharmacology. CVIII. Calcium-sensing receptor nomenclature, pharmacology, and function. Pharmacol. Rev. 72, 558–604 (2020).
Seven, A. B. et al. G-protein activation by a metabotropic glutamate receptor. Nature 595, 450–454 (2021).
Lin, S. et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594, 583–588 (2021).
Shen, C. et al. Structural basis of GABAB receptor–Gi protein coupling. Nature 594, 594–598 (2021).
Magno, A. L., Ward, B. K. & Ratajczak, T. The calcium-sensing receptor: a molecular perspective. Endocr. Rev. 32, 3–30 (2011).
Centeno, P. P. et al. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 10, 4693 (2019).
Leach, K. et al. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res. 26, 574–592 (2016).
Ling, S. et al. Structural mechanism of cooperative activation of the human calcium-sensing receptor by Ca2+ ions and l-tryptophan. Cell Res. 31, 383–394 (2021).
Gao, Y. et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 595, 455–459 (2021).
Park, J. et al. Symmetric activation and modulation of the human calcium-sensing receptor. Proc. Natl Acad. Sci. USA 118, e2115849118 (2021).
Olsen, R. H. et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 16, 841–849 (2020).
Masuho, I. et al. Distinct profiles of functional discrimination among G proteins determine the actions of G protein–coupled receptors. Sci. Signal. 8, ra123 (2015).
Wall, M. A. et al. The structure of the G-protein heterotrimer Giα1β1γ2. Cell 83, 1047–1058 (1995).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 4121 (2020).
Timmers, H., Karperien, M., Hamdy, N., De Boer, H. & Hermus, A. Normalization of serum calcium by cinacalcet in a patient with hypercalcaemia due to a de novo inactivating mutation of the calcium-sensing receptor. J. Intern. Med. 260, 177–182 (2006).
Hannan, F. M. et al. Identification of 70 calcium-sensing receptor mutations in hyper-and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum. Mol. Genet. 21, 2768–2778 (2012).
Nagase, T. et al. A family of autosomal dominant hypocalcemia with a positive correlation between serum calcium and magnesium: identification of a novel gain of function mutation (Ser820Phe) in the calcium-sensing receptor. J. Clin. Endocrinol. Metab. 87, 2681–2687 (2002).
Shiohara, M. et al. A novel gain-of-function mutation (F821L) in the transmembrane domain of calcium-sensing receptor is a cause of severe sporadic hypoparathyroidism. Eur. J. Pediatr. 163, 94–98 (2004).
Hu, J. et al. A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+. J. Biol. Chem. 280, 5113–5120 (2005).
Quinn, S. J. et al. The Ca2+-sensing receptor: a target for polyamines. Am. J. Physiol. 273, C1315–C1323 (1997).
Michael, A. J. Polyamines in eukaryotes, bacteria, and archaea. J. Biol. Chem. 291, 14896–14903 (2016).
Schamber, M. R. & Vafabakhsh, R. Mechanism of sensitivity modulation in the calcium-sensing receptor via electrostatic tuning. Nat. Commun. 13, 2194 (2022).
Cole, D. E. et al. Calcium-sensing receptor mutations and denaturing high performance liquid chromatography. J. Mol. Endocrinol. 42, 331–339 (2009).
Tan, Y. et al. Autosomal dominant hypocalcemia: a novel activating mutation (E604K) in the cysteine-rich domain of the calcium-sensing receptor. J. Clin. Endocrinol. Metab. 88, 605–610 (2003).
Kwan, B. et al. A novel CASR mutation (p. Glu757Lys) causing autosomal dominant hypocalcaemia type 1. Endocrinol. Diabetes Metab. Case Rep. 2018, 18-0107 (2018).
Conigrave, A. D., Quinn, S. J. & Brown, E. M. l-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc. Natl Acad. Sci. USA 97, 4814–4819 (2000).
Mangmool, S. & Kurose, H. Gi/o protein-dependent and-independent actions of pertussis toxin (PTX). Toxins 3, 884–899 (2011).
Vargas-Poussou, R. et al. Familial hypocalciuric hypercalcemia types 1 and 3 and primary hyperparathyroidism: similarities and differences. J. Clin. Endocrinol. Metab. 101, 2185–2195 (2016).
Koltin, D. et al. Mild infantile hypercalcemia: diagnostic tests and outcomes. J. Pediatr. 159, 215–221 (2011).
Ray, K., Fan, G.-F., Goldsmith, P. K. & Spiegel, A. M. The carboxyl terminus of the human calcium receptor: requirements for cell-surface expression and signal transduction. J. Biol. Chem. 272, 31355–31361 (1997).
Nesbit, M. A. et al. Mutations affecting G-protein subunit α11 in hypercalcemia and hypocalcemia. N. Engl. J. Med. 368, 2476–2486 (2013).
Bai, M. et al. Protein kinase C phosphorylation of threonine at position 888 in Ca2+o-sensing receptor (CaR) inhibits coupling to Ca2+ store release. J. Biol. Chem. 273, 21267–21275 (1998).
Davies, S. L., Ozawa, A., McCormick, W. D., Dvorak, M. M. & Ward, D. T. Protein kinase C-mediated phosphorylation of the calcium-sensing receptor is stimulated by receptor activation and attenuated by calyculin-sensitive phosphatase activity. J. Biol. Chem. 282, 15048–15056 (2007).
Jiang, Y. F. et al. Protein kinase C (PKC) phosphorylation of the Ca2+o-sensing receptor (CaR) modulates functional interaction of G proteins with the CaR cytoplasmic tail. J. Biol. Chem. 277, 50543–50549 (2002).
Lazarus, S. et al. A novel mutation of the primary protein kinase C phosphorylation site in the calcium-sensing receptor causes autosomal dominant hypocalcemia. Eur. J. Endocrinol. 164, 429–435 (2011).
Brown, E. M. et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366, 575–580 (1993).
Robertson, M. J., Meyerowitz, J. G., Panova, O., Borrelli, K. & Skiniotis, G. Plasticity in ligand recognition at somatostatin receptors. Nat. Struct. Mol. Biol. 29, 210–217 (2022).
Maeda, S. et al. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, 3712 (2018).
Bayburt, T. H., Grinkova, Y. V. & Sligar, S. G. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 (2002).
Peisley, A. & Skiniotis, G. 2D projection analysis of GPCR complexes by negative stain electron microscopy. Methods Mol. Biol. 1335, 29–38 (2015).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Zhang, X. et al. Structures of the human cholecystokinin receptors bound to agonists and antagonists. Nat. Chem. Biol. 17, 1230–1237 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Falk‐Petersen, C. B. et al. Development of a robust mammalian cell‐based assay for studying recombinant α4β1/3δ GABAA receptor subtypes. Basic Clin. Pharmacol. Toxicol. 121, 119–129 (2017).
Acknowledgements
We thank E. Montabana at the Stanford Cryo-EM Center (cEMc) for support with data collection. This work was supported by R01 DK132902 (G.S.), R01 NS122394 (G.S. and J.M.M.), R01 DK071801 (L.L.), S10 RR029531 (L.L.), the Lundbeck Foundation Experiment R346-2020-1829 (S.N.R. and J.M.M.) and the Independent Research Fund Denmark (Medical Sciences, 1030-00050A) (S.N.R. and J.M.M.).
Author information
Authors and Affiliations
Contributions
F.H. and Y.G. generated expression constructs for structural studies. F.H., C.-G.W. and Y.G. expressed and purified CaSR and G proteins. F.H. designed and generated the CaSR mutants. F.H. reconstituted and purified the inactive-state CaSR and the CINA-bound active-state CaSR and CaSR–Gq complexes in nanodiscs. C.-G.W. and F.H. reconstituted and purified the PAM-free CaSR–Gi complex in nanodiscs. Y.G. reconstituted and purified CaSR–Gi in detergents and the CINA-bound CaSR–Gi complex in nanodiscs. C.-G.W., F.H. and Y.G. prepared cryo-EM grids. M.Z., F.H., C.-G.W. and Y.G. collected cryo-EM data. F.H., Y.G. and C.-G.W. processed cryo-EM data. F.H. and Y.G. built and refined the structural models. C.-G.W. and Y.G. expressed and purified MSP1E3D1. S.N.R. optimized and performed in vitro cellular G-protein activation and signalling assays and assisted in figure preparation under the supervision of J.M.M. C.-G.W. and F.H. performed TRUPATH assays. J.M.M. conceived, optimized and performed in vitro cellular G-protein activation and signalling mutagenesis assays and interpreted results. M.M.P.-S. performed the GTPγS activity assay and analysed the data. T.-J.G. collected and analysed mass spectrometry data under the supervision of L.L. M.J.R. provided constructs for Gi expression and assisted in modelling. A.B.S. assisted in data collection and processing. F.H. and G.S. analysed data and wrote the manuscript with input from all authors. G.S. supervised the project.
Corresponding author
Ethics declarations
Competing interests
G.S. is a co-founder of and consultant for Deep Apple Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Qiuyan Chen, Reza Vafabakhsh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 G-protein signalling of CaSR and reconstitution of human CaSR–G-protein complexes.
a, Survey of Emax of Ca2+-dependent CaSRΔ894 activation of 11 different G-protein BRET-based TRUPATH biosensors using benchmark receptors NTSR1 and β2AR. The BRET signal monitors dissociation of the heterotrimeric Gαβγ complex induced by receptor-mediated guanine nucleotide exchange at the Gα subunit19. nd, no coupling detected. b, Concentration response curves of Gi3 and Gq activation pathways of CaSRΔ894 by Ca2+ using the BRET-based TRUPATH assay. c, Ca2+ concentration response curves of Gi3, Gq and Gs activation by CaSRΔ894 using the BRET-based G-protein-activation assay. d, G-protein-activation assays measuring the functional responses of 15 Gα subtypes by full-length CaSR (WT) and CaSRΔ894 after stimulation with 10 mM Ca2+, by MOR after stimulation with 10 mM morphine, by H1R after stimulation of 100 mM histamine and by β2AR after stimulation with 10 mM isoproterenol, respectively. The BRET signal monitors the binding of the released Gβγ from receptor-mediated Gαβγ dissociation to a membrane-associated reporter20. e, G-protein-activation assays monitoring functional responses of the CaSRΔ894 dimer fused to C1(WT-C1)/C2(WT-C2) tails to Ca2+ alone or in combination with cinacalcet. f–i, Size-exclusion chromatography profiles of purified CINA-bound CaSR/Gi in LMNG detergent (f), CINA-bound CaSR–Gi complex in nanodiscs (g), PAM-free CaSR–Gi complex in nanodiscs (h), CINA-bound CaSR–Gq complex in nanodiscs (i). Schematic diagram of the constructs used for CaSR and G protein is shown for each complex. The sample sizes and descriptions of error bars for all Extended Data Figures are described in the Methods under the section ‘Statistics and reproducibility’.
Extended Data Fig. 2 Cryo-EM processing and reconstruction of CaSR–Gi in LMNG detergent and the inactive-state CaSR in nanodiscs.
a, Cryo-EM data processing workflow for CaSR/Gi in detergents. b, Representative 2D averages. c, Local refinement maps of the class of Gi alone bound to detergent micelles. d, Low-resolution local refinement maps of the class of CaSR–Gi in detergents with a soft mask on receptor or G protein. e, Map of Gi alone with docked structure of inactive Gi (PDB ID: 1GP2). f, Low-resolution composite map of CaSR–Gi in LMNG detergent with docked structures of active-state CaSR (PDB ID: 7M3F) and inactive Gi (PDB ID: 1GP2). g, Model of the CINA-bound CaSR–Gi structure in nanodiscs docked into the density of receptor in the map of CaSR–Gi in LMNG detergent. h, Cryo-EM data processing workflow for inactive CaSR in nanodiscs. i, Representative 2D averages of inactive CaSR in nanodiscs. j, Local refinement maps of 7TM and VFT–CRD for inactive CaSR in nanodiscs. k, Low-resolution composite map of inactive CaSR in nanodiscs with open-closed VFTs and flexible 7TMs.
Extended Data Fig. 3 Cryo-EM processing and reconstruction of the CINA-bound and PAM-free CaSR–Gi complexes in lipid nanodiscs.
a, Cryo-EM data processing workflow for the CINA-bound CaSR–Gi complex in nanodiscs. b, Representative cryo-EM micrograph (out of 20528 similar micrographs) and 2D averages for the CINA-bound CaSR–Gi complex in nanodiscs. c, Local refinement maps of VFT–CRD, CRD–7TM and Gαiβγ for the CINA-bound CaSR–Gi complex in nanodiscs with corresponding Fourier shell correlation (FSC) curves indicating nominal resolutions using the FSC = 0.143 criterion. d, Angular distribution heat map of particle projections for the global reconstruction of the CINA-bound CaSR–Gi complex in nanodiscs. e, Cryo-EM data processing workflow for the PAM-free CaSR–Gi complex in nanodiscs. f, Representative cryo-EM micrograph (out of 31047 similar micrographs) and 2D averages for the PAM-free CaSR–Gi complex in nanodiscs. g, Local refinement maps of VFT–CRD, CRD–7TM and Gαiβγ for the PAM-free CaSR–Gi complex in nanodiscs with corresponding Fourier shell correlation (FSC) curves indicating nominal resolutions using the FSC = 0.143 criterion. h, Angular distribution heat map of particle projections for the global reconstruction of the PAM-free CaSR–Gi complex in nanodiscs. i, Local refinement maps of VFT–CRD, CRD–7TM for the PAM-free active-state CaSR in nanodiscs with corresponding FSC curves indicating nominal resolutions using the FSC = 0.143 criterion. j, Angular distribution heat map of particle projections for global reconstruction of the PAM-free active-state CaSR in nanodiscs.
Extended Data Fig. 4 Cryo-EM processing and reconstruction of the CINA-bound CaSR–Gq complex in lipid nanodiscs.
a, Cryo-EM data processing workflow for the CINA-bound CaSR–Gq complex in nanodiscs. b, Representative cryo-EM micrograph (out of 47434 similar micrographs) and 2D averages for the CINA-bound CaSR–Gq complex in nanodiscs. c, Local refinement maps of VFT–CRD, CRD–7TM and Gαqβγ for the CINA-bound CaSR–Gq complex in nanodiscs with corresponding FSC curves indicating nominal resolutions using the FSC = 0.143 criterion. d, Angular distribution heat map of particle projections for global reconstruction of the CINA-bound CaSR–Gq complex in nanodiscs. e, Local refinement maps of VFT–CRD, CRD–7TM for the CINA-bound active-state CaSR in nanodiscs with corresponding FSC curves indicating nominal resolutions using the FSC = 0.143 criterion. f, Angular distribution heat map of particle projections for reconstruction of the CINA-bound active-state CaSR in nanodiscs. g, Comparison of the maps of the CINA-bound CaSR–Gq and CaSR–Gi complexes in nanodiscs. The angles of the Gα αN helix relative to the lipid membrane are highlighted by dashed lines.
Extended Data Fig. 5 Agreement between cryo-EM density and model.
a, EM density and model for the 7TM with the extended CINA (at map thresholds of 0.3–0.35), and cinacalcet (at a map threshold of 0.3) in the CINA-bound active-state CaSR. b, EM density and model for the 7TM and H8 (residues 869 to 877) of the Gi-bound protomer (at map thresholds of 1.1–1.7), cinacalcet (at a map threshold of 1.35), and lipids (using the unsharpened map at thresholds of 0.4–0.6) in the CINA-bound CaSR–Gi complex. c, EM density and model for the 7TM and the extended H8 (residues 869 to 885) of the Gq-bound protomer (at map thresholds of 0.9–1.0), cinacalcet (at a map threshold of 0.85), and lipids (at a map threshold of 0.82) in the CINA-bound CaSR–Gq complex. Residues 878 to 885 in the C terminus are highlighted in turquoise as in Fig. 4b. d, EM density and model for the 7TM and H8 (residues 869 to 877) of the Gi-bound protomer (at map thresholds of 0.41–0.46), the 7TM Trp (at a map threshold of 0.4), and lipids (at map thresholds of 0.31–0.4) in the PAM-free CaSR–Gi complex.
Extended Data Fig. 6 Asymmetric dimer interface of CaSR in the active state and in G-protein-coupled states.
a, Structural comparison between active-state CaSR in detergent (PDB ID: 7M3F) and lipid nanodiscs. The ECDs in the two structures show different tilting angles relative to the 7TMs. b, Structural comparison between the CINA-bound CaSR–Gi and CaSR–Gq complexes in nanodiscs. The 7TMs (except the cytoplasmic regions) and the bound ligands are well aligned. c, The VFT of the NGC protomer of the CINA-bound CaSR–Gi or CaSR–Gq complex is aligned to the VFT of the GC protomer. d, The asymmetric TM6–TM6 interface in the structure of CINA-bound active-state CaSR in nanodiscs. e,f, The asymmetric 7TM interface in the structure of the CINA-bound CaSR–Gi in nanodiscs shown in two views, highlighting the interface between TM6 of 7TMNGC and TM6 of 7TMGC (e), and the interface between TM5 and TM6 of 7TMGC and TM7 of 7TMNGC (f). The residues that are involved in dimer interactions, and the models of phospholipids (DOPC) and cholesterol are shown. g, Functional responses of CaSR WT and mutants to Ca2+, for Gi3 and Gq activation, categorized based on interactions with the lower (F776A, K805A, F806A and F809A) and upper (I822A, Y825A, Y829A and V833A) phospholipids.
Extended Data Fig. 7 Polyamine-binding sites on CaSR.
a, Mass spectrometry for spermidine and spermine standards, and sample of purified CINA-bound CaSR–Gi complex in lipid nanodiscs. No significant amount of spermidine was detected. b, The spermine-binding site in the CRD–ECL regions of the CINA-bound CaSR–Gq complex in nanodiscs. The model and cryo-EM density of the bound spermine (at a map threshold of 1.13), a water molecule (in red sphere), the upper phospholipid (using the unsharpened map at a map threshold of 0.51) are shown. c, Electrostatic surface distribution of the modelled surface of CaSR in the CINA-bound CaSR–Gi complex in nanodiscs. The three spermine-binding sites are highlighted with dashed boxes. d, IP1-accumulation assays monitoring the functional responses of full-length CaSR to Ca2+, spermine or spermine in presence of NAM NPS 2143. Pre-incubated NPS 2143 inhibited the spermine response, whereas increasing concentrations of the PAM etelcalcetide potentiated the spermine response. e, G-protein-activation and IP1-accumulation assays monitoring the functional responses of full-length CaSR to Ca2+, spermine or spermine with 3 mM Ca2+, in the presence/absence of 20 mM (G-protein-activation assay) or 10 mM (IP1 accumulation) EDTA. Functional responses by Gi3 and Gq activation were measured 20 seconds after ligand stimulation to capture the initial activation by Gi3 and Gq, as well as to circumvent any potential long-term cellular effects of EDTA, albeit responses were similar in the IP1-accumulation assay. Pharmacological analysis across G-protein-activation and IP1-accumulation assays indicate that spermine (dark blue) may function like an ago-PAM, which is potentiated by Ca2+ as evident from its left-shifted potency with 3 mM Ca2+ (light green), and right-shifted potency in the presence of EDTA (light blue, dark green). Similar data were obtained for CaSR Δ894 WT (data not shown).
Extended Data Fig. 8 Conformational changes of CaSR associated with Gi and Gq coupling, and distinct Gi and Gq binding interfaces.
a,b, Superposition of the structures of the CINA-bound active-state CaSR and CaSR–Gi complex (a) or CaSR–Gq complex (b) based on VFT alignment. Conformational differences between the superimposed structures are indicated by arrows. c, Comparison of the two 7TMs in the CINA-bound active-state CaSR structure. d, Comparison between 7TMNGC in the CINA-bound CaSR–Gi complex and the 7TM in the active-state CaSR. e,f, Comparison of 7TMGC and 7TMNGC in the CINA-bound CaSR–Gi (e) and CaSR–Gq (f) complexes. g,h, Interactions between ICL2 of CaSR and the N-terminal regions of Gi (g) and Gq (h). i,j, Superposition of the structures of the CINA-bound CaSR–Gi and CaSR–Gq complexes by aligning the two 7TMs in top (i) and side (j) views. Conformational differences of Gi and Gq are indicated by arrows. k,l, The CaSR C terminus selectively recognizes Gi and Gq. The structures of the CINA-bound CaSR–Gi and CaSR–Gq complexes are overlaid based on the alignment of 7TMGC showing only the Gq-bound protomer (k) or the Gi-bound protomer (l). Potential clashes are represented by yellow stars. m, Sequence alignment of the α5 helix in the Gαq/11 class. The strictly conserved residues are highlighted in red.
Extended Data Fig. 9 Functional characterization and relative expression levels of wild-type CaSR and CaSR mutants.
a, IP1-accumulation assays monitoring functional responses of CaSR WT (with or without pretreatment with PTX) and CaSR mutants to Ca2+, categorized based on different structural domains/regions. b, Selected mutants identical or complementary to the mutants tested in the G-protein-activation assays in Fig. 4. c, [35S]-GTPγS binding activity assays monitoring functional responses of an extracellular domain truncated CaSR mutant ΔECD CaSR (CaSR residues 599–894) to Trp and cinacalcet in the presence and absence of 10 mM Ca2+. d, IP1-accumulation assay monitoring the functional response of ΔECD CaSR to cinacalcet and Trp. e,f, Surface expression levels of CaSR WT and mutants for Gi3 and Gq activation assays (e), and surface expression levels of CaSR WT and mutants for IP1-accumulation assays (f).
Extended Data Fig. 10 The 7TM modulatory sites in the presence and absence of cinacalcet.
a, The PAM-binding pocket of 7TMGC in the CINA-bound CaSR–Gi complex. b, The PAM-binding pocket of 7TMNGC in the CINA-bound CaSR–Gi complex. c, Overall structure of the PAM-free CaSR–Gi complex. The 7TM Trp, phospholipids (DOPC), and cholesterol are shown as spheres. d, The PAM-binding pocket of 7TMGC in the PAM-free CaSR–Gi complex. e, The PAM-binding pocket of 7TMNGC in the PAM-free CaSR–Gi complex. f, Trp and cinacalcet occupy a similar position in 7TMGC in the CINA-bound and in the PAM-free CaSR–Gi complexes. The binding-pocket residues share almost identical conformations in the two structures. g, The model and density of the two W818 rotamers in 7TMGC in the PAM-free CaSR–Gi complex. h, Comparison of 7TMNGC in the PAM-free and CINA-bound CaSR–Gi complexes. i, The two 7TMs in the CINA-bound active-state CaSR show different tilt angles in the lipid bilayer, and the protomer with the extended CINA has a better resolved C terminus. j,k, Modelling based on the structure of the CINA-bound CaSR–Gi complex shows how the C781W/I822W mutations would not efficiently occlude the bent PAM conformation (j), and how the L773W mutation would occlude the bent PAM conformation (k). A potential clash is represented by the yellow star. l, Superposition of the 7TMs in the CINA-bound CaSR–Gi complex with those in the active-state CaSR (CaSR*) based on the poses (bent or extended) of cinacalcet.
Supplementary information
Supplementary Information
Supplementary Figs 1 and 2.
Supplementary Video 1
Conformational dynamics of 7TMGC relative to 7TMNGC in the structure of the CINA-bound CaSR–Gi complex. Morph between the CINA-bound 7TMNGC and the CINA-bound 7TMGC. The models are aligned as shown in Fig. 5c.
Supplementary Video 2
Conformational dynamics of 7TMGC relative to 7TMNGC in the structure of the PAM-free CaSR–Gi complex. Morph between the PAM-free 7TMNGC and the Trp-bound 7TMGC. The models are aligned as shown in Fig. 5b.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, F., Wu, CG., Gao, Y. et al. Allosteric modulation and G-protein selectivity of the Ca2+-sensing receptor. Nature 626, 1141–1148 (2024). https://doi.org/10.1038/s41586-024-07055-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-024-07055-2
- Springer Nature Limited
This article is cited by
-
Delineating the stepwise millisecond allosteric activation mechanism of the class C GPCR dimer mGlu5
Nature Communications (2024)
-
The Ca2+-sensing receptor and the pocketome: comparing nature’s complexity with human intervention in receptor modulation
Signal Transduction and Targeted Therapy (2024)