

Abstract
Antibody responses during infection and vaccination typically undergo affinity maturation to achieve high-affinity binding for efficient neutralization of pathogens1,2. Similarly, high affinity is routinely the goal for therapeutic antibody generation. However, in contrast to naturally occurring or direct-targeting therapeutic antibodies, immunomodulatory antibodies, which are designed to modulate receptor signalling, have not been widely examined for their affinity–function relationship. Here we examine three separate immunologically important receptors spanning two receptor superfamilies: CD40, 4-1BB and PD-1. We show that low rather than high affinity delivers greater activity through increased clustering. This approach delivered higher immune cell activation, in vivo T cell expansion and antitumour activity in the case of CD40. Moreover, an inert anti-4-1BB monoclonal antibody was transformed into an agonist. Low-affinity variants of the clinically important antagonistic anti-PD-1 monoclonal antibody nivolumab also mediated more potent signalling and affected T cell activation. These findings reveal a new paradigm for augmenting agonism across diverse receptor families and shed light on the mechanism of antibody-mediated receptor signalling. Such affinity engineering offers a rational, efficient and highly tuneable solution to deliver antibody-mediated receptor activity across a range of potencies suitable for translation to the treatment of human disease.
Similar content being viewed by others
Main
The humoral immune response constitutes a major component of immunity. Antibody responses during infection and vaccination typically undergo affinity maturation that leads to the selection of B cell clones that produce higher affinity antibodies2. Such antibodies are required for efficient pathogen opsonization and can be sufficient for pathogen neutralization3. This high affinity and specificity for target antigens has seen antibodies broadly adopted as the biologic of choice in the treatment of many diseases, with more than 100 monoclonal antibodies now approved for use in humans4,5. Current approaches used to generate therapeutic monoclonal antibodies, including hybridoma technology6, antibody display platforms7 and direct sequencing modalities8, have typically prioritized high affinity as a key criterion for selection. High affinity is crucial for the activity of naturally occurring and conventional direct-targeting therapeutic antibodies. Such antibodies bind to antigens on pathogens or tumour cells and commonly require crystallizable fragment (Fc)-mediated immune effector functions to elicit full therapeutic activity. A conceptually distinct class of monoclonal antibody are agonistic and target immune receptors such as the TNF receptors (TNFRs) CD40 and 4-1BB to induce intracellular signalling9. Compared with direct-targeting monoclonal antibodies, the rules governing the activity of agonistic monoclonal antibodies are more complex and multifactorial, and are determined by a combination of antibody epitope, isotype and Fcγ receptor (FcγR) requirements10,11,12. These agonistic monoclonal antibodies have entered clinical trials and offer promise for cancer immunotherapy9. However, unlike conventional direct-targeting monoclonal antibodies, no study has systematically examined the impact of affinity on their activity, and it remains unclear whether high affinity is similarly important.
Generation of anti-CD40 affinity variants
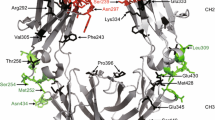
To investigate the effect of affinity on the agonistic activity of immunostimulatory receptor monoclonal antibodies, we chose the human CD40 system as a paradigm. A crystal structure of CD40 in complex with the antigen-binding fragment (Fab) of a clinically relevant anti-CD40 monoclonal antibody, ChiLob 7/4, was used as a model to design affinity mutants based on the complementary-determining regions (CDRs). This structure was analysed in PISA using the QtPISA interface to identify potential interacting residues, which were then mutated to alanine in PyMOL and subsequently re-analysed in PISA. The resulting difference in the change in Gibbs free energy (ΔG) and binding energy were used to predict the effect of mutations on binding affinity. A series of single, double and triple mutants were subsequently evaluated through the application of a confusion matrix to produce combined mutation scores. Final mutations were determined on the basis of the scores in the confusion matrix and the proximity of the residue to the binding interface. In addition, we took advantage of a series of non-CDR, framework mutants generated during the humanization of ChiLob 7/4. Thus, we produced six mutants by mutating the CDR sequence (prefixed CDR-) and six mutants by altering the variable domain framework sequence (prefixed FW-) (Fig. 1a). Surface plasmon resonance (SPR) analyses revealed that these ChiLob 7/4 mutants bound CD40 with a range of affinities (5.22–925 nM) and differed in both the on and off rates (Fig. 1b and Extended Data Fig. 1a,b). We have previously demonstrated that the antibody epitope can greatly influence the agonistic activity of anti-CD40 monoclonal antibodies11. Here results from competitive cell-binding assays showed that they could differentially inhibit wild-type (WT) ChiLob 7/4 binding to CD40 in a dose-dependent manner according to their respective CD40-binding affinities. This result indicates that anti-CD40 monoclonal antibodies bind cell surface CD40 through the same epitope (Extended Data Fig. 1c).

a, Left, surface overview of the CD40–ChiLob 7/4 Fab complex. ChiLob 7/4 heavy and light chains are shown in dark and light grey, respectively, bound to CD40 (blue). Mutated residues are coloured, showing their proximity to the interface. Middle, ChiLob 7/4 variable region surface interface showing mutated residues as coloured and translucent. Right, ChiLob 7/4 showing interacting residues (coloured with transparent surface) in relation to CD40 (blue). Below each close up, the chain (green), residue (red), change in binding energy (purple) and ΔG value (light blue) are shown for each mutated residue. Mutants are ordered from high to low affinity. Figures were created in PyMOL 2.5.2. b, SPR results (in response units (RU)) of various ChiLob 7/4 mIgG1 (m1) affinity mutants injected at 250, 50, 10, 2, 0.4 and 0 nM binding to CD40ECD. Data are representative of three independent experiments. c,d, Purified hCD40Tg mouse B cells were incubated with ChiLob 7/4 m1 mutants for 2 days, and surface CD23 (c) or CD86 (d) expression levels (in mean fluorescence intensity (MFI)) were determined. Plots on the far right show expression as a function of affinity (KD). e, Purified hCD40Tg mouse B cells were incubated with ChiLob 7/4 m1 mutants for 3 days and then 3H-thymidine was added for 18 h to measure proliferation. Plot on the far right shows proliferation as a function of KD. For all experiments, data are shown as the mean ± s.e.m., n = 3, data representative of three independent experiments. c.p.m., counts per minute; ECD, extracellular domain.
Low-affinity anti-CD40 antibodies show greater agonism
We next examined the activity of these monoclonal antibodies in the human CD40 transgenic (hCD40Tg) mouse system13. As most anti-CD40 monoclonal antibodies require FcγR2b crosslinking for agonistic activity10,14, we initially expressed the ChiLob 7/4 mutants as mouse IgG1 (mIgG1) to enable efficient engagement with mouse FcγR2b receptors10. Splenic B cells from hCD40Tg mice were treated with these anti-CD40 affinity variants, and levels of the activation markers CD23 and CD86, and B cell proliferation were assessed. Contrary to expectations from direct-targeting monoclonal antibodies, anti-CD40 antibodies with lower affinity exhibited stronger agonistic activity. This was evidenced by the upregulation of CD23 and CD86 and increased B cell proliferation and cell–cell homotypic adhesion (Fig. 1c–e and Extended Data Fig. 2a–c). The affinity–agonism relationship revealed a bell-shaped curve, whereby B cell activation increased from the parental ChiLob 7/4 mIgG1 baseline with reducing affinity until a threshold was reached at which proliferation fell (Fig. 1c–e). The most agonistic variants (FW-16 mIgG1 and CDR-Y102A/Y32A mIgG1) induced approximately 26-fold and 8-fold higher CD23 and CD86 expression, respectively (Fig. 1c,d), than the WT monoclonal antibody. The same bell-shaped response between agonism and affinity was seen for B cell proliferation, in which the most agonistic variant induced twofold higher proliferation (Fig. 1e). Although the magnitude of the effects differed between these readouts, which indicated that there is a differential threshold for activation (with CD23 expression the lowest and proliferation the highest), they all confirm that lower affinity drives increased agonism. Moreover, in each case, the off rate (kd), but not on rate (ka), of the monoclonal antibodies exhibited a similar bell-shaped trend as the equilibrium affinity (KD; Extended Data Fig. 2a–c). This result indicates that the off rate is the driving parameter.
To investigate whether such low-affinity-driven agonism could be recapitulated in vivo, we used an OTI CD8 T cell expansion model. Mice were injected with ovalbumin (OVA)-specific OTI cells followed by the administration of OVA and low-affinity anti-CD40 variants12. Consistent with the in vitro data, the low-affinity monoclonal antibodies induced significantly higher levels of antigen-specific CD8 T cell expansion. This effect was identified through SIINFEKL tetramer staining, with the most agonistic variant FW-16 mIgG1 inducing eightfold higher expansion than the WT parental monoclonal antibody (Fig. 2a,b). Notably, CDR-Y102A/Y32A mIgG1, which displayed equally strong agonistic activity as FW-16 mIgG1 in vitro, mediated a more modest enhancement in OTI expansion. This result indicated that the precise affinity range for maximal agonism differed among the experimental systems. We next examined whether enhanced agonism could be translated into therapeutic benefit in the OVA+ EG7 tumour model. Mice were subcutaneously inoculated with EG7 tumour cells and then treated with an anti-CD40 monoclonal antibody when tumours became palpable. The ability of low-affinity anti-CD40 monoclonal antibodies to drive strong tumour-specific T cell responses was retained in tumour-bearing mice (Fig. 2c). The parental monoclonal antibody conferred a modest degree of survival benefit compared with the isotype control. By contrast, the low-affinity variant CDR-Y102A/Y32A mIgG1 was able to cure most of the mice and was significantly more effective than the higher affinity parent monoclonal antibody (Fig. 2d and Extended Data Fig. 2d). In addition, cured mice were resistant to tumour rechallenge, which provides evidence of strong immunological memory (Fig. 2e). These data collectively demonstrate that low-affinity anti-hCD40 monoclonal antibodies are able to mediate agonistic activity in vitro and in vivo and exhibit potent antitumour activity compared with the high-affinity parent antibody.

a,b, OTI cells were adoptively transferred into hCD40Tg mice 1 day before treatment with the indicated ChiLob 7/4 m1 mutants with peripheral SIINFEKL+ CD8 cells identified by flow cytometry on day 5. a, Representative dot plots, gated on CD8+ cells. b, Summary of OTI expansion data. Mean ± s.e.m., n = 8, data pooled from 2 independent experiments. Each dot represents one mouse. Two-tailed, non-paired Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001. P values for WT m1 versus various mutants (left to right): P = 0.0002, P = 0.0030, P = 0.0002, P = 0.0006 and P = 0.1304. c–e, hCD40Tg mice were inoculated with EG7 cells. After 7 days, mice received OTI cells and then ChiLob 7/4 m1 mutants 24 h later. c, Per cent of SIINFEKL+ CD8 cells determined on day 5. Mean ± s.e.m., n = 17, 17, 13 (left to right), data pooled from 2–3 independent experiments. Each dot represents one mouse. Two-tailed, non-paired Student’s t-test, P < 0.0001 for isotype versus WT m1, P = 0.0044 for WT m1 versus CDR-Y102A/Y32A m1. d, Tumour size and survival. n = 13–17, data pooled from 2–3 independent experiments. Survival curves compared using log-rank test, *P < 0.05, **P < 0.01, ***P < 0.001. P = 0.0006 for isotype versus WT m1, P < 0.0001 for isotype m1 versus CDR-Y102A/Y32A m1. e, After the initial tumour challenge, tumour-free mice were re-inoculated with EG7 cells and monitored for tumour size and survival (compared using log-rank test, *P < 0.05, **P < 0.01, ***P < 0.001). P = 0.0066 for isotype versus WT m1, P < 0.0002 for isotype m1 versus CDR-Y102A/Y32A m1.
Strong agonistic activity in human systems
We next examined whether low-affinity-mediated agonism could be recapitulated in human systems. As the mIgG1 isotype effectively engages with mouse but not human FcγRs, we isotype-switched the ChiLob 7/4 mIgG1 variants to the human IgG2 (hIgG2) isotype to optimize receptor agonism11,12,15. SPR analysis showed that these hIgG2 affinity variants exhibited an affinity ranking for CD40 similar to their mIgG1 counterparts (Extended Data Fig. 3a,b). Moreover, they inhibited the binding of WT ChiLob 7/4 to Ramos cells in a dose-dependent manner as anticipated from their affinities. This result provides further support of a conserved epitope for these antibodies (Extended Data Fig. 3c). Consistent with previous data using the mIgG1 monoclonal antibodies, the low-affinity ChiLob 7/4 hIgG2 variants induced greater levels of CD23 and CD86 expression on hCD40Tg B cells and more proliferation compared with the WT ChiLob 7/4 hIgG2 monoclonal antibody (Extended Data Fig. 3d,e). Greater OTI expansion in vivo was also observed (Extended Data Fig. 3f). Purified human B cells also underwent greater proliferation and displayed higher expression of CD23 and CD86 in response to low-affinity ChiLob 7/4 hIgG2 variants (Extended Data Fig. 3g–i). Next, as CD40 stimulates immune responses primarily through dendritic cells (DCs)16, we derived immature DCs from CD14+ human monocytes. We found that low-affinity ChiLob 7/4 hIgG2 variants induced higher levels of CD86 expression than parental (higher affinity) ChiLob 7/4 hIgG2, a well-established marker for DC activation (Extended Data Fig. 4a,b). Moreover, in a mixed leukocyte reaction, DCs treated with low-affinity variants induced greater allogeneic T cell proliferation. This result demonstrates that these activated DCs are functional and have an enhanced ability to stimulate T cells (Extended Data Fig. 4c,d). Furthermore, in a human peripheral blood mononuclear cell (PBMC) antigen recall assay, in which a mixture of viral and bacterial peptide antigens were used to elicit a T cell recall response, low-affinity ChiLob 7/4 hIgG2 monoclonal antibodies induced higher levels of CD25 expression on the responding proliferating T cells (Extended Data Fig. 4e,f). These data collectively demonstrate that low-affinity anti-CD40 hIgG2 monoclonal antibodies induce higher levels of agonism in human systems than parental higher affinity ChiLob 7/4 hIgG2.
FcγR-independent clustering drives agonism
After establishing that low-affinity anti-CD40 monoclonal antibodies induced better agonism than the parental WT monoclonal antibody, we investigated the underlying mechanism of this effect. The agonistic activity of anti-CD40 monoclonal antibodies is governed by a complex interplay between epitope and isotype, which mediates differential levels of receptor clustering11,15,17. We proposed that low-affinity monoclonal antibodies that retained sufficient antigen binding induced higher agonism through increased receptor clustering, which is independent of FcγR engagement. We tested this hypothesis by using Jurkat cells expressing the reporter nuclear factor-κB (NF-κB)–GFP and hCD40 but lacking FcγR expression. We also utilized the mIgG1 isotype as it lacks intrinsic Fc-dependent, FcγR-independent agonistic activity12. Therefore, any increased agonism could be solely attributed to the affinity effect. Consistent with our hypothesis, low-affinity ChiLob 7/4 mIgG1 variants induced significantly higher levels of NF-κB activation compared with the higher affinity WT parent antibody (Fig. 3a and Extended Data Fig. 5a). In support of this result, low-affinity variants induced significantly more CD23 and CD86 expression and higher levels of B cell proliferation and cell–cell homotypic adhesion in B cells purified from hCD40Tg FcγR-null mice, which lack all FcγRs15 (Fig. 3b–d and Extended Data Fig. 5b–d). Although the presence of the inhibitory FcγR2b receptor produced greater agonism, with higher CD23 and CD86 expression and more B cell proliferation in WT versus FcγR-null B cells, its enhancing effect was less apparent with low-affinity variants. This result indicates that these monoclonal antibodies induce agonism in a distinct manner, independent of FcγR interaction (Fig. 3e–h). Nonetheless, it is clear that FcγR interaction can influence receptor agonism in vivo, and differential FcγR expression in different disease contexts may modulate activity. We also noted that low-affinity antibodies do not require saturating concentrations to sustain their agonistic activity. That is, reduced receptor engagement by low-affinity variants drove more powerful agonism (Fig. 3i,j). Low-affinity ChiLob 7/4 hIgG2 mutants also induced greater CD23 and CD86 upregulation and B cell proliferation in hCD40Tg FcγR-null B cells (Extended Data Fig. 6). These data demonstrate that low-affinity monoclonal-antibody-mediated agonism of CD40 is independent of FcγRs.

a, Jurkat NF-κB–GFP–CD40 cells were incubated with ChiLob 7/4 m1 mutants, and NF-κB activation (based on GFP) was assessed. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. b–d, Purified hCD40Tg FcγR-null mouse B cells were incubated with ChiLob 7/4 m1 mutants for 2 days and then assessed for surface expression of CD23 (b) and CD86 (c). Plots show affinity (KD) versus maximum CD23 or CD86 MFI. d, On day 3 3H-thymidine was added to assess B cell proliferation. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. Plot shows KD versus maximum proliferation. e–g, Overlay of hCD40Tg (WT) and hCD40Tg FcγR-null mouse B cell activation. e, CD23 MFI. f, CD86 MFI. g, Cell proliferation. h, hCD40Tg FcγR-null B cell proliferation expressed as the percentage of hCD40Tg B cell proliferation. Plot shows affinity (KD) versus per cent of hCD40Tg B cell proliferation. i, Jurkat NF-κB–GFP–CD40 cells were pre-incubated with ChiLob 7/4 m1 mutants. After washing, the remaining bound monoclonal antibody level was quantified. j, Same experiment as i, but with NF-κB activation (GFP) assessed. For h–j, mean ± s.e.m., n = 3, data representative of 3 independent experiments. k, Jurkat CD40–GFP cells incubated with ChiLob 7/4 m1 mutants were imaged by confocal microscopy. Green, hCD40–GFP. Scale bar, 4 μm. Images representative of >15 images from 3 independent experiments. l, Same experiment as k, showing clustering index values for ChiLob 7/4 m1 mutants. Mean ± s.e.m., n = 5, data representative of 3 independent experiments. m, Jurkat CD40–GFP cells incubated with ChiLob 7/4 m1 mutants were fixed and imaged by confocal microscopy. Z-stack images shown. Blue, nucleus; green, CD40–GFP. Scale bar, 4 μm. Images representative of >10 images from 3 independent experiments.
To further unravel the molecular mechanism of low-affinity-induced agonism, we evaluated the ability of ChiLob 7/4 variants to induce CD40 clustering. We previously demonstrated that TNFR agonists induce differential agonistic activity through distinct patterns of receptor clustering17. To better distinguish and quantify the level of receptor clustering among the low-affinity antibody variants, we used Jurkat cells expressing GFP–CD40 and lacking FcγRs and quantified the level of receptor clustering through a clustering index (Methods). According to this clustering index, higher numbers denote greater receptor clustering (Extended Data Fig. 7a). Jurkat CD40–GFP cells treated with low-affinity ChiLob 7/4 mIgG1 variants induced significantly greater levels of receptor clustering, with a higher clustering index than the WT mIgG1 parent, which did not induce appreciable clustering (Fig. 3k,l and Extended Data Fig. 7b). This demonstrated that for antibodies sharing the same isotype and epitope, lowering the affinity induced enhanced receptor clustering and downstream signalling. Augmented clustering was also qualitatively supported by z-stacked confocal images showing extensive receptor redistribution and clustering for low-affinity variants (Fig. 3m), which was not associated with changes in cell shape (Extended Data Fig. 7c). Moreover, bivalent, but not monovalent Fab engagement, was required for agonism and receptor clustering (Extended Data Fig. 7d–f). In addition, low-affinity monoclonal antibodies did not alter the extent of receptor internalization compared with higher affinity controls (Extended Data Fig. 7g,h). These data collectively demonstrate that low-affinity anti-CD40 monoclonal antibodies mediate higher levels of agonistic activity through increased receptor clustering independent of FcγRs. To further dissect the subcluster structures that underlie the differential agonism effect, we performed super-resolution direct stochastic optical reconstruction microscopy (dSTORM) of Jurkat CD40–GFP cells. Wide-field fluorescence images showed that low-affinity anti-CD40 monoclonal antibodies formed clusters predominantly at cell–cell junctions, unlike CD40 ligand (CD40L), which induced clusters that were more widely dispersed throughout the membrane (Extended Data Fig. 8a). However, at the subcluster level evaluated by dSTORM, differences were less obvious. ChiLob 7/4 hIgG2, FW-16 mIgG1 and FW-32 mIgG1, but not CD40L, displayed significantly higher density subclusters at the cell–cell junctions compared with untreated samples (Extended Data Fig. 8b). Moreover, correlation analysis of area and density showed that CD40L induced fewer medium and large subclusters of medium-high density than ChiLob 7/4 hIgG2, FW-16 mIgG1 and FW-32 mIgG1 (Extended Data Fig. 8c). These results indicate that subclusters within monoclonal antibodies and ligand-induced clusters are similar in density but differ substantially in their membrane localization.
Low-affinity anti-4-1BB antibodies elicit agonism
We next explored whether the low-affinity-mediated agonism observed with anti-CD40 monoclonal antibodies applied to other TNFRs. We chose 4-1BB, another co-stimulatory TNFR of interest in cancer immunotherapy18. To date, two anti-4-1BB monoclonal antibodies have entered clinical trials, and both had differing outcomes. Urelumab demonstrated strong pharmacodynamic effects accompanied by liver toxicity, whereas utomilumab was quiescent regarding both these aspects18. In agreement, we previously showed that urelumab induced strong receptor clustering and activity in multiple isotypes, whereas utomilumab, even as a hIgG2, did not elicit receptor clustering or activity17. Therefore, we chose to generate affinity variants for utomilumab in an attempt to induce agonism for this otherwise inert monoclonal antibody. We used the crystal structure of the utomilumab Fab–4-1BB complex19 to generate a panel of affinity mutants. As decsribed above (Fig. 1a), we mutated key residues within the CDRs (Fig. 4a). The mutants exhibited lower binding to 4-1BB immobilized on a SPR chip compared with the WT monoclonal antibody (Fig. 4b and Extended Data Fig. 9a), and less binding to 4-1BB expressed at the cell surface (Fig. 4c). Moreover, the low-affinity mutants mediated less potent macrophage-mediated antibody-dependent cellular phagocytosis (ADCP) and human PBMC-mediated antibody-dependent cellular cytotoxicity (ADCC) (Fig. 4d,e). This result is consistent with the idea that high-affinity binding is required for efficient Fc-mediated antibody effector functions. In contrast to reduced Fc-mediated effector functions, the low-affinity utomilumab variants induced significantly higher levels of NF-κB activation in Jurkat NF-κB–GFP reporter cells expressing h4-1BB, whereas the WT utomilumab was inactive, as expected (Fig. 4f). Enhanced NF-κB activity correlated with more efficient receptor clustering, as evidenced by a higher clustering index and visible clusters by confocal microscopy (Fig. 4g,h and Extended Data Fig. 9b). In addition, the lower 4-1BB binding affinity correlated positively with a higher clustering index (Fig. 4i), but not with changes in cell shape (Extended Data Fig. 9c). Similar to the anti-CD40 monoclonal antibodies, low-affinity anti-4-1BB monoclonal antibodies also required bivalent receptor engagement for agonistic activity and receptor clustering (Fig. 4j–l). Moreover, saturating monoclonal antibody concentrations were not required for sustained agonistic activity (Fig. 4m,n). These data show that as with the anti-CD40 monoclonal antibodies, low-affinity anti-4-1BB monoclonal antibodies mediate higher agonistic activity, which suggests that low-affinity antibody-induced agonism is a conserved feature among TNFRs.

a, Left, surface overview of the 4-1BB–utomilumab Fab complex. Utomilumab heavy and light chains bound to 4-1BB (blue). Middle, utomilumab variable region surface, showing mutated residues coloured and translucent. Right, utomilumab interacting residues (coloured with transparent surface) in relation to 4-1BB (blue). Figures created using PyMOL. b, SPR of utomilumab mutants binding to 4-1BBECD. Data representative of three independent experiments. c, Utomilumab mutants binding to 4-1BB Jurkat cells. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. d, Utomilumab-opsonized Ramos 4-1BB cells were incubated with human monocyte-derived macrophages to evaluate phagocytosis (ADCP). e, Utomilumab-opsonized IIA1.6 4-1BB cells were incubated with human PBMCs to assess ADCC. For d and e, mean ± s.e.m., n = 2, data representative of 3 independent experiments. f, Jurkat NF-κB–GFP–4-1BB cells were incubated with utomilumab mutants, and NF-κB activation (GFP) was determined. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. g, Jurkat 4-1BB–GFP cells were incubated with utomilumab, fixed and imaged by confocal microscopy. Z-stack images are shown. Blue, nucleus; green, 4-1BB–GFP. Scale bar, 4 μm. Images representative of >10 images taken from 2 independent experiments. h, Jurkat 4-1BB–GFP cells were incubated with utomilumab, and clustering index values were calculated. Mean ± s.e.m., n = 5, data representative of 3 independent experiments. i, Same experiment as h, showing clustering index values versus utomilumab affinity (KD). j, Binding of utomilumab mutant Fab versus IgG to Jurkat NF-κB–GFP–4-1BB cells. Data representative of three independent experiments. k, Same experiment as f, with NF-κB activation (GFP) quantified. l, Same experiment as h, showing clustering index values. For k and l, mean ± s.e.m., n = 3–5, data representative of 3 independent experiments. m, Jurkat NF-κB–GFP–4-1BB cells were incubated with utomilumab mutants. After washing, the remaining bound monoclonal antibody was quantified. n, Same experiment as f, with NF-κB activation (GFP) assessed. For m and n, mean ± s.e.m., n = 3, data representative of 3 independent experiments.
Low-affinity anti-PD-1 antibodies elicit agonism
To explore whether this low-affinity effect observed with monoclonal antibodies directed to TNFRs is applicable to other receptors that mediate receptor signalling, we chose to evaluate the most clinically important immune checkpoint receptor: PD-1. Antagonistic anti-PD-1 monoclonal antibodies that block PD-1 signalling represent a major breakthrough in cancer immunotherapy and have been approved in multiple cancer types20. Recently, agonistic anti-PD-1 monoclonal antibodies, designed to induce active PD-1 signalling, have been suggested as potential therapeutics for autoimmune diseases such as systemic lupus erythematosus and psoriasis21,22,23. We used the crystal structure of PD-1 in complex with the clinically approved antagonistic anti-PD-1 monoclonal antibody nivolumab24 to design a panel of mutants to investigate the effect of monoclonal antibody affinity on PD-1 agonism (Fig. 5a). SPR analysis showed that the nivolumab mutants exhibited a range of affinities towards immobilized PD-1 (Extended Data Fig. 9f,g). Affinities spanned a 28-fold difference from WT nivolumab and correlated with the level of binding to cell surface PD-1 (Extended Data Fig. 9h). To evaluate the ability of these monoclonal antibodies to block the PD-1–PD-L1 interaction, we developed an assay in which PD-1-transfected Jurkat NFAT–Luc cells, which exhibit luciferase activity after NFAT activation, were mixed with CHO cells expressing PD-L1 and an anti-CD3 with a single-chain fragment variable (scFv) designed to engage CD3 on the Jurkat cells to induce NFAT activation (Extended Data Fig. 10a, left). The mutants dose-dependently rescued NFAT activation from PD-L1-mediated suppression, and the blocking activity positively correlated with PD-1 binding affinity (Fig. 5b). That is, the blocking activity reduced as the affinity decreased. These data demonstrate that blockade of PD-1–PD-L1 depends on high-affinity anti-PD-1 monoclonal antibodies as expected. Next, to investigate the impact of affinity on cell intrinsic PD-1 signalling, we designed a chimeric PD-1 construct, whereby the intracellular signalling domain of PD-1 was replaced with that of CD40. Then we generated Jurkat NF-κB–GFP cells stably expressing this construct to assess PD-1 signalling directly in response to anti-PD-1 treatment (Extended Data Fig. 10a, middle). As shown in Fig. 5c, WT nivolumab and its high-affinity variant T28A were unable to induce PD-1 signalling, whereas the low-affinity variants induced significant signalling in a dose-dependent manner, similar to the anti-CD40 and anti-4-1BB affinity variants described above (Figs. 3a and 4f). As PD-1 is known to signal concurrently with the T cell receptor complex to deliver negative signals in T cells22,25,26,27, we next examined whether PD-1 signalling induced by low-affinity variants would impair CD3-mediated T cell activation. To this end, we generated CHO cells expressing a scFv specific for hIgG Fc to capture both anti-CD3 and anti-PD-1 monoclonal antibodies. These monoclonal-antibody-coated CHO cells were then incubated with PD-1-transfected Jurkat NFAT–Luc cells. Thereafter, we measured NFAT signalling and CD69 expression to assess T cell activation (Extended Data Fig. 10a, right). As expected, the CHO cells captured the anti-PD-1 variants equivalently (Extended Data Fig. 10b). Moreover, the WT anti-PD-1 monoclonal antibody suppressed anti-CD3-induced T cell activation, as evidenced by reduced NFAT signalling and CD69 downregulation compared with the isotype control (Fig. 5d,e). Low-affinity nivolumab variants exhibited stronger T cell suppression than the WT parent, with affinity correlated to reduced NFAT signalling and CD69 expression (Fig. 5d,e). Notably, lower affinity variants induced equal levels of suppression to soluble PD-L1. Moreover, the monoclonal antibody off rate, but not on rate, displayed an inverse correlation with NFAT signalling and CD69 expression (Extended Data Fig. 10c,d). These data demonstrate that low-affinity anti-PD-1 variants induce strong PD-1 signalling and T cell suppression.

a, Top left, surface overview of the PD-1–nivolumab Fab complex. Nivolumab heavy and light chains bound to PD-1 (blue). Top right, nivolumab variable region surface, showing mutated residues coloured and translucent. Bottom, nivolumab interacting residues (coloured with transparent surface) in relation to PD-1 (blue). Figures created using PyMOL. b, Jurkat NFAT–Luc–PD-1 cells were cultured with CHO OKT3–scFv–CD8α–PD-L1 cells in the presence of titrated nivolumab mutants. NFAT signalling activity was then assessed. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. c, Jurkat NF-κB–GFP–PD-1 reporter cells were incubated with nivolumab mutants and NF-κB activation (GFP) assessed. Mean ± s.e.m., n = 3, data representative of 3 independent experiments. d, CHO SB2H2–scFv–CD8α cells were opsonized with OKT3, and nivolumab affinity mutants and then cultured with Jurkat NFAT–Luc–PD-1 cells. NFAT activity was then assessed. Right, NFAT activity versus affinity (KD). Mean ± s.e.m., n = 3, data representative of 3 independent experiments. One-way analysis of variance (ANOVA) followed by Kruskal–Wallis test, P = 0.0006. e, Same experiment as d, but CD69 expression determined. Right, CD69 expression versus affinity (KD). Mean ± s.e.m., n = 3, data representative of 3 independent experiments. One-way ANOVA followed by Kruskal–Wallis test, P < 0.0001. f, Clustering index of IIA1.6 PD-1–GFP cells incubated with nivolumab mutants. Mean ± s.e.m., n = 5, data representative of 3 independent experiments. g, IIA1.6 PD-1–GFP cells were incubated with nivolumab mutants, fixed and imaged using confocal microscopy. Z-stack images shown. Blue, nucleus; green, PD-1-GFP. Scale bar, 4 μm. Images representative of >10 images from 2 independent experiments. h, Jurkat NFAT–Luc–PD-1 cells were incubated with AF488-labelled nivolumab mutants at 37 °C or 4 °C, and level of antibody internalization assessed as detailed in methods. Data representative of 3 independent experiments. i, Same experiment as g. Orthogonal images shown.
To uncover the molecular mechanism of this effect, we investigated the ability of the same variants to induce PD-1 clustering using IIA1.6 cells stably transfected with GFP–PD-1 and lacking FcγRs28. Untreated cells, cells treated with parental nivolumab or higher affinity variants did not display significant PD-1 clustering. By contrast, almost all low-affinity variants induced significant PD-1 clustering that correlated with increased clustering index values (Fig. 5f and Extended Data Fig. 10e), which was corroborated by confocal microscopy imaging (Fig. 5g and Extended Data Fig. 10f). Similar to the CD40 system, low-affinity anti-PD-1 monoclonal antibodies did not induce significant receptor internalization (Fig. 5h,i), and saturating monoclonal antibody concentrations were not required for sustained agonism (Extended Data Fig. 10g). In addition, changes in cell shape did not correlate with agonism (Extended Data Fig. 10h). These data provide direct support that stronger monoclonal-antibody-mediated agonism can also be achieved by reducing the affinity of immune receptors outside the TNFR family. Similar mechanisms of action also underpin these effects. That is, FcγRs, changes to receptor internalization or saturating concentrations are not required.
Discussion
Unlike natural and direct-targeting antibodies, agonistic immunomodulatory monoclonal antibodies differ in their mechanism of action, needing to elicit productive receptor signalling in addition to specific target engagement. Antibodies targeting TNFRs remain highly studied owing to their potential in cancer immunotherapy, with epitope, isotype and FcγR engagement known to modulate activity10,11,12.
Here we investigated the impact of affinity on immunomodulatory monoclonal antibodies directed to three different receptors. In general, therapeutic antibodies are considered high affinity at KD values of <1 nM (refs. 29,30). As epitope and isotype can affect monoclonal antibody agonism, as outlined above, we designed experiments so that these aspects were controlled. Our structure-guided mutational approach did not obviously alter specificity, as affinity mutants retained binding for their target without exhibiting binding to close homologues (Extended Data Fig. 11a–f). Low-affinity anti-CD40 monoclonal antibodies displayed augmented agonism compared with higher affinity parent molecules. The relationship between affinity and agonism was bell-shaped, with activity increasing as affinity decreased until a threshold at which activity significantly dropped, presumably owing to insufficient target engagement. For ChiLob 7/4, this occurred at >288 nM and was associated with an inability to compete with the parental monoclonal antibody for cell surface binding. Therefore, the application of low-affinity agonism through rationale design is limited by the need to sufficiently engage the receptor on the target cell. Moreover, the monoclonal antibody off rate, but not the on rate, exhibited a similar bell-shaped relationship with agonism in both FcγR-competent and FcγR-deficient systems, which suggests that faster dissociation is responsible for the increased agonistic activity of low-affinity variants. In support of this finding, and in the reverse approach to ours, anti-Fas monoclonal antibodies engineered with higher affinities demonstrated reduced activity, with lower off rates as the key determinant. It was proposed that monoclonal antibodies with slow dissociation rates ‘lock’ two receptor monomers into a non-signalling complex, thereby preventing receptor oligomerization and activation31. These data support the hypothesis that high-affinity binding is not essential for receptor agonism and is indeed detrimental.
Low-affinity anti-CD40 monoclonal antibodies induced agonistic activity and extensive receptor clustering independent of FcγRs. This result provides support for the ‘fast dissociation’ model for the mechanism of action and indicates that agonism is mediated through receptor clustering akin to conventional high-affinity monoclonal antibodies17. Low-affinity variants also displayed enhanced agonism as hIgG2 monoclonal antibodies in various human assays, which indicates that low affinity drives agonism independently of isotype. Further studies are needed to evaluate the in vivo effect of the hIgG2 monoclonal antibodies in a fully hFcγR transgenic animal model. To extend the observation beyond CD40, we investigated the anti-4-1BB monoclonal antibody utomilumab, which lacks intrinsic agonism17,32. Lowering the affinity of utomilumab led to strong 4-1BB-mediated NF-κB activation and enhanced receptor clustering. As with the anti-CD40 mutants, bivalency was required for low-affinity agonism, with Fab fragments inactive. By contrast, lower affinity provided reduced ADCC and ADCP. This divergent activity profile supports the paradigm that low-affinity targeting is conducive to receptor signalling but detrimental for Fc-mediated effector functions. To assess whether low-affinity targeting could be a general strategy to enhance receptor signalling beyond TNFRs, we generated affinity mutants of the anti-PD-1 monoclonal antibody nivolumab20. Consistent with our earlier findings, low-affinity anti-PD-1 monoclonal antibodies induced greater PD-1 signalling through enhanced receptor clustering, which in turn induced more potent T cell receptor-activated T cell suppression.
A possible mechanism that underpins the activity of low-affinity monoclonal antibodies is reduced internalization induced by higher affinity monoclonal antibodies33. This results in increased signalling through prolonged retention at the cell surface. However, for all receptors examined in this study, this was not the case. By contrast, by confocal, but not quenching assays, certain low-affinity monoclonal antibodies appeared to display some vesicular internalization, which may affect their activity (Extended Data Fig. 11g–i). Low-affinity agonism did not require constant receptor saturation. Nevertheless, it remains possible that weaker interactions conferred by lower affinity monoclonal antibodies may trigger sufficient agonism to drive activity but not negative feedback mechanisms as a rheostat, thereby triggering higher net activity. Although unlikely, it is also possible that the introduced mutations could induce a small shift in the epitope bound by the affinity-modified monoclonal antibody to enhance agonism.
Similar to ChiLob 7/4 mutants, the anti-PD-1 monoclonal antibody off rate correlated with T cell suppression. It is therefore conceivable that all low-affinity mutants use a similar mechanism that requires partial dissociation (afforded by high off rates) to induce agonism. This is supported by their ability to mediate receptor clustering and agonism independent of FcγRs. CDR-Y102A/Y32A m1, CDR-Y102A/Y50A and CDR-Y102A/Y32A/Y96A exhibited higher on rates than the WT parent. Here, the Fab regions may engage the antigen sequentially, with less steric hindrance compared with higher affinity monoclonal antibodies, which might be expected to engage the antigen with both Fab regions concurrently, with sequential engagement providing these monoclonal antibodies faster on rates. Importantly, low-affinity mutants induced stronger agonism at various receptor densities, which encompassed physiological levels (Extended Data Fig. 11j–o). We also compared receptor clusters induced by low-affinity monoclonal antibodies and natural ligands. dSTORM revealed that although low-affinity anti-CD40 monoclonal-antibody-induced clusters were predominantly located at cell–cell junctions, ligand-induced clusters were dispersed throughout the cell membrane, which was consistent with our previous findings17. However, the subcluster density showed only modest differences between monoclonal antibody and ligand, which suggests that differential receptor cluster localization could influence agonistic activity. Conventional monoclonal antibody and native TNFR ligands differ in their binding valency34, which is reflected in their distinct binding kinetics to their cognate receptors (Extended Data Fig. 11p). Whether such differences in valency influences receptor cluster localization requires further investigation. It is also possible that these properties of the native ligand have evolved to elicit optimal, smaller, receptor clustering arrangements, not fully resolved by dSTORM, to deliver maximal receptor activity without the need for the larger clusters required by low-affinity monoclonal antibodies.
In summary, our affinity approach provides an efficient alternative to generating large sets of new monoclonal antibodies to achieve a desired receptor outcome. For three different receptors expressed on antigen-presenting cells or T cells, spanning positive (immunostimulatory) and negative (checkpoint inhibitors) immune regulators, we demonstrated that low-affinity monoclonal antibodies deliver agonistic signalling. Such affinity engineering offers a broadly applicable, tractable, rational and highly tuneable solution to deliver desired antibody-mediated receptor activity, whereby a range of affinities mediate diverse activities suitable for translation in the human disease setting.
Methods
Mice
hCD40Tg mice were provided by R. Noelle (King’s College, London)13. hCD40Tg FcγR-null mice (hCD40Tg;Fcer1g−/−;FcγR2b−/−) were generated as previously described15 by first breeding Fcer1g−/− and FcγR2b−/− mice to generate homozygous FcγR-null mice (Fcer1g−/− × FcγR2b−/−). Homozygous FcγR-null mice were subsequently crossed with hCD40Tg mice15. All mice were bred in-house and maintained on a 12 h light–dark cycle, and food and water were provided ad libitum. The temperature was maintained at 20–24 °C with 55 ± 15% humidity. Mice were checked daily to ensure healthy status. Both male and female mice were used between 8–24 weeks old, mice were randomly divided into groups based on age and sex for each experiment for which blinding was not performed. All experiments were conducted under UK Home Office licence numbers PB24EEE31, P4D9C89EA, P540CBA98 and P39FE2AA7 and according to local ethics committee guidelines.
Human samples
Human PBMCs were derived from blood cones collected from healthy donors through Southampton National Blood Services with previous informed consent. The use of human blood was approved by the East of Scotland Research Ethics Service, Tayside, UK.
Cell lines
WT Ramos, CHO-k1, EG7, A20 and Jurkat cells were obtained from the American Type Culture Collection. IIA1.6 cells were used as previously described35. The WT Jurkat NF-κB–GFP reporter cell line was from System Biosciences. The WT Jurkat NFAT–Luc reporter cell line was from Promega. All cell lines were maintained in a humidified incubator at 37 °C and 5% CO2, and cultured in RPMI medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 1 mM pyruvate, 100 U ml–1 penicillin, 100 µg ml–1 streptomycin and 50 µM β-mercaptoethanol (complete RPMI medium, all from ThermoFisher), with the exception of CHO-k1 cells, which were cultured without β-mercaptoethanol.
Generation of affinity mutants
Models of Fab–receptor complexes were obtained from the Protein Data Bank (PDB) with the following accession codes: 6FAX for ChiLob 7/4–CD40; 6MI2 for utomilumab–4-1BB; and 5WT9 for nivolumab–PD-1. To identify potential interacting residues, the models were analysed in PISA using the QtPISA interface36,37. Potential mutants were generated by mutating the potential interacting residues identified by PISA to alanine in PyMOL 2.5.2. using the mutation wizard. Each mutant was subsequently analysed in PISA. The resulting difference in the ΔG and binding energy values from the WT structure were recorded and used to predict effects on binding affinity. To generate double mutants, a confusion matrix of the difference in ΔG and binding energy values from WT was used to give combined mutation scores. Decisions on which mutants to produce were based on the scores in the confusion matrix and the proximity of the residue to the binding interface. Humanization was performed, and resulting sequences were provided by Glycotope.
Antibodies and reagents
All antibodies were produced using the ExpiCHO system as previously described15. The variable domain sequence for utomilumab38, nivolumab39, varlilumab40, TGN1412 (ref. 41) and OKT3 (ref. 42) were derived from published sequences. The light and heavy chain variable domain sequences were synthesized by GeneArt and subcloned into pEE12.4 and pEE6.4 expression vectors, respectively (Lonza), encoding the constant domain of different IgG isotypes. The CDR mutations for ChiLob 7/4, utomilumab and nivolumab were achieved by site-directed mutagenesis using a QuickChange Site-Directed Mutagenesis kit (Agilent). Plasmids encoding the heavy and light chains were transiently transfected into ExpiCHO cells for 10 days before the supernatant was collected and antibodies were purified on a MabSelect SuRe column (GE Healthcare). All antibody preparations were checked by HPLC to contain <1% aggregate and by Endosafe-PTS portable tests (Charles River Laboratories) to contain <5 endotoxin units per mg antibody.
The DNA constructs containing CD40–GFP and 4-1BB–GFP were generated as previously described17. The PD-1–GFP construct was generated by subcloning human PD-1 (NCBI GenBank accession: U64863) into a pcDNA3 plasmid containing the GFP fragment at the carboxy terminus. The OKT3–scFv–CD8α DNA construct was designed as VL-(G4S)3-VH-CD8α with an EcoRI restriction site between the VH site and CD8α and cloned into a pCIpuro vector. The variable domain sequence of the anti-human IgG Fc monoclonal antibody SB2H2 was obtained from the hybridoma by sequencing the cDNA generated using the MMLV reverse transcriptase and a universal primer as previously described43. The SB2H2–scFv–CD8α DNA construct was designed as VL-(G4S)3-VH-CD8α with an EcoRI restriction site between the VH site and CD8α and cloned into a pcDNA3 vector. Human PD-L1 (NCBI GenBank accession: NM_014143) was cloned into a pcDNA3 vector.
Recombinant soluble trimeric CD40L and 4-1BBL were produced in-house as previously described17. Fab fragments of mouse IgG1 anti-CD40 monoclonal antibody was generated using a Pierce Mouse IgG1 Fab and F(ab′)2 Preparation kit (ThermoFisher). The Fab fragment of human IgG4 anti-4-1BB monoclonal antibody was generated using immobilized papain (ThermoFisher).
SPR analysis
A Biacore T200 instrument was used throughout. Recombinant extracellular domains of human CD40 (R&D Systems), 4-1BB (R&D Systems), PD-1 (R&D systems), TNFRII (R&D systems), CD27 (in-house) and CD28 (BioLegend) were immobilized onto a CM5 chip by amine-coupling chemistry. To compare the binding affinity of various target-specific monoclonal antibody mutants and natural soluble ligands towards their respective receptor or homologue, the monoclonal antibody and ligand were injected through the flow cell at 250, 50, 10, 2, 0.4 and 0 nM in HBS-EP+ running buffer at a flow rate of 30 ml min–1, allowing 300 s for association and 300 s for dissociation. Data were collected using Biacore T200 control software. The sensorgrams were fitted using the bivalent analyte model, and the ka and kd values were calculated using Biacore Bioevaluation software; the KD values were calculated as kd/ka. All SPR reagents and software were from GE Healthcare.
Assessment of antibody cell surface receptor binding
To assess the level of antibody binding to cells expressing human CD40, 4-1BB or PD-1, relevant cells were incubated with various concentrations of anti-CD40, anti-4-1BB or anti-PD-1 monoclonal antibodies as indicated in the figure legends for 30 min at 4 °C. Unbound monoclonal antibodies were then washed off using FACS wash buffer (PBS, 1% BSA and 0.01% sodium azide), and PE-conjugated polyclonal goat F(ab′)2 secondary anti-human Fc (1:200) or PE-conjugated polyclonal goat F(ab′)2 secondary anti-mouse Fc (1:200, both from Abcam) were added for 30 min at 4 °C. Unbound monoclonal antibody was then washed off using FACS wash buffer. To detect the level of bound Fab fragment, FITC-conjugated anti-mouse IgG Fab (1:100) or AF647-conjugated anti-human IgGκ light chain (1:100) was used depending on the monoclonal antibody isotype. The level of bound monoclonal antibody was quantified by flow cytometry.
Competitive cell surface receptor binding
For competitive cell-binding assays, Ramos cells were incubated with a fixed concentration of AF647-labelled ChiLob 7/4 hIgG1 (0.5 μg ml–1) and various concentrations of competing ChiLob 7/4 mIgG1 or ChiLob 7/4 hIgG2 affinity mutants for 30 min. Cells were then washed, and the level of AF647-labelled ChiLob 7/4 hIgG1 remaining bound to the cell surface was quantified by flow cytometry.
Flow cytometry
Flow cytometry experiments were conducted using FACSCalibur, FACSCanto II or FACSMelody instruments (all from BD Biosciences). Flow cytometry data were collected using BD CellQuest and BD FACSDIVA software, and data analysis was performed using FCS Express software v.3 (De Novo Software) or Flowjo (BD Biosciences).
B cell activation assay
Human B cells were purified from human PBMCs using a MojoSort Human B Cell Isolation kit (BioLegend). Mouse B cells were purified from splenocytes using a MojoSort Mouse Pan B Cell Isolation kit (BioLegend). Purified B cells were incubated with various anti-CD40 monoclonal antibodies as indicated in the figure legends for 2 days and imaged using a conventional light microscope (Olympus CKX41). CD23 (anti-CD23, 1:160) and CD86 (anti-CD86, 1:100) expression was assessed by flow cytometry. To assess B cell proliferation, 3H-thymidine (Perkin Elmer) was added at 1 µCi per well on day 3 for an additional 18 h as previously described15.
OTI expansion assay
OTI expansion assays were performed as previously described15. To assess the ability of anti-CD40 monoclonal antibodies to induce OTI T cell expansion, 1 × 105 OTI cells were intravenously injected into CD40KOTg mice 1 day before the intravenous injection of 100 µg OVA in combination with 25 µg or 500 µg of various anti-CD40 monoclonal antibodies. Mice were then bled 4–5 days later as indicated in the figure legends, and the level of OTI expansion was assessed on the basis of the proportion of CD8+ SIINFEKL tetramer-positive cells by flow cytometry.
EG7 tumour therapy
The EG7 model was generated as previously described15. In brief, mice were intravenously inoculated with 5 × 105 EG7 cells and then treated with 25 µg anti-CD40 monoclonal antibody and 100 µg OVA intravenously when the sum of tumour length and width reached approximately 10 mm. Tumour size was measured three times per week using digital calipers, and mice were culled when the sum of the tumour length and width reached 30 mm or when the general health of the mice reached humane end-point criteria. For re-challenge, tumour-free mice were intravenously inoculated with 5 × 105 EG7 cells and monitored for tumour growth as described above. Tumour volume was calculated using the following formula: V = (W2 × L)/2, where W is the tumour width and L is the tumour length.
Human DC activation and mixed leukocyte reaction
Human immature DCs were generated as previously described44. In brief, CD14+ monocytes were isolated from human PBMCs using a magnetic negative selection kit (Miltenyi Biotech) and then cultured in the presence of 500 IU ml–1 IL-4 and 1,000 IU ml–1 GM-CSF (both cytokines produced in-house) for 5–6 days. The identity of DCs was confirmed by CD11c (anti-CD11c, 1:20) and DC-SIGN (anti-CD209, 1:20) expression. For direct stimulation, immature DCs were treated with 50 µg ml–1 anti-CD40 monoclonal antibody for 2 days, and the level of CD86 expression (anti-CD86, 1:20) was quantified by flow cytometry. A mixed leukocyte reaction was performed as previously described15. In brief, varying numbers of immature DCs were first treated with 50 µg ml–1 anti-CD40 monoclonal antibody for 2 days and then washed and further incubated with 0.1 × 106 purified human allogeneic CD4+ T cells (MojoSort Human CD4 T Cell Isolation kit, BioLegend) for 4 days. 3H-thymidine was added at 1 µCi per well on day 4 for an additional 18 h to assess T cell proliferation.
Human PBMC peptide recall assay
The expansion of antigen-specific T cells within PBMCs was achieved using a CEFX Ultra SuperStim Pool (JPT Peptide Technologies), which contains a pool of 176 known peptides based on different infectious agents that have been shown to induce antigen-specific T cell expansion45,46. In brief, fresh human PBMCs were labelled with 2 µM CFSE (ThermoFisher) in PBS and then 0.2 × 106 PBMCs were incubated with 50 µg ml–1 anti-CD40 monoclonal antibody and 0.6 µM CEFX Ultra SuperStim Pool for 5 days to recall antigen-responsive CD8+ T cells. The proliferating cells expressing CD3 (anti-CD3, 1:20) and CD8 (anti-CD8, 1:20) were regarded as antigen-responsive cells, and their level of activation was measured on the basis of CD25 expression (anti-CD25, 1:20) on day 5 by flow cytometry.
NF-κB assay
A pCIpuro vector encoding CD40, 4-1BB (expressing the hCD40 intracellular signalling domain) or PD-1 (expressing the CD40 transmembrane and intracellular signalling domain) was transfected into Jurkat NF-κB–GFP cells, and stable clones were selected using 1 µg ml–1 puromycin. To examine NF-κB activation, cells stably transfected with each receptor were incubated with the relevant monoclonal antibody as indicated in each legend for 6 h at 37 °C. The level of NF-κB activation was subsequently quantified by GFP fluorescence, which was assessed by flow cytometry.
Assays to evaluate the impact of receptor density on monoclonal antibody agonism
Jurkat NF-κB–GFP cells stably transfected with CD40, 4-1BB or PD-1 were first sorted into populations expressing low, medium or high levels of the respective receptor using a FACSMelody instrument (BD). Cells were then treated with various monoclonal antibodies as indicated for 6 h at 37 °C, and the level of NF-κB activation was subsequently quantified by GFP fluorescence, which was assessed by flow cytometry. Receptor quantification was performed using a Quantum Alexa Fluor 647 MESF kit (Bangs Laboratories). To quantify the level of PD-1 expression on human primary T cells, human PBMCs were activated with Immunocult (Stem Cell Technologies) for 2 days, and then CD3+ T cells were analysed for PD-1 expression by flow cytometry. To assess the level of 4-1BB expression on human primary CD8+ T cells, CD8+ T cells were purified from human PBMCs using a MojoSort Human CD8 T Cell Isolation kit (BioLegend) and then activated with plate-bound anti-CD3 (clone OKT3) and anti-CD28 (clone TGN1412) (both produced in-house) for 24 h before 4-1BB quantification by flow cytometry.
Assays to evaluate the effect of antibody concentration on monoclonal antibody agonism
Jurkat NF-κB–GFP cells stably transfected with CD40, 4-1BB or PD-1 were treated with 50 µg ml–1 monoclonal antibody as indicated for 30 min at room temperature, and then excess unbound monoclonal antibody was washed off. Cells were then incubated at 37 °C for various periods as indicated, and the level of monoclonal antibody remaining bound to the cell surface was quantified by DL650-conjugated goat F(ab′)2 secondary anti-mouse Fc (1:200) or by DL650-conjugated goat F(ab′)2 secondary anti-human Fc (1:200, both from Abcam) using flow cytometry. The level of NF-κB activation was concurrently quantified by GFP fluorescence, which was assessed by flow cytometry.
Confocal microscopy
DNA encoding CD40ECD–GFP and 4-1BBECD–GFP were subcloned into a pCIpuro vector and transfected into Jurkat cells using Nucleofector kit V (Lonza). Stable Jurkat clones were selected using 1 µg ml–1 puromycin. IIA1.6 cells stably transfected with full length PD-1–GFP (IIA1.6 PD-1–GFP cells) were generated by transfecting IIA1.6 cells with a pCIpuro plasmid encoding PD-1–GFP using nucleofection kit V (Lonza), and stable clones were selected using 4 µg ml–1 puromycin. Confocal microscopy was performed as previously described17. Jurkat cells were incubated with 50 µg ml–1 of the indicated monoclonal antibody for 3 h at 37 °C, and then fixed with cold methanol on ice for 10 min before the nucleus was stained with DAPI (ThermoFisher). Alternatively, cells were fixed with 2% paraformaldehyde (ThermoFisher) at room temperature for 10 min before the nucleus was stained with DAPI. For live-cell imaging, cells were imaged directly without fixation. Confocal images were acquired using a Leica SP8 confocal microscope, and data were analysed using Leica Application Suite X (all from Leica). To measure receptor clustering at the cell–cell junctions in relation to the periphery of the cells, a clustering index was calculated (Extended Data Fig. 7a). Confocal images through the centre of the cells were opened in Leica Application Suite X software (Leica), and fluorescence intensity measurements were taken for regions of interest (ROIs) at the cell–cell junctions or at the periphery of the cells (non-contacting membrane). These were determined by eye, with cell periphery measurements taken for each corresponding cell–cell junction; that is, two cell periphery ROIs would be collected for a cell that contained two cell–cell cluster ROIs. A clustering index was calculated for five confocal images, with up to five cell–cell clusters per image for each treatment. An average clustering index was calculated from five confocal images, with up to five cell–cell clusters per image for each treatment. The clustering index designates the ratio of fluorescence intensity at the cell–cell junction over the fluorescence intensity at the cell periphery, which was calculated using Leica Application Suite X software (Leica). A larger clustering index denotes higher levels of receptor clustering. The circularity of the cells was measured in ImageJ, whereby a circularity value of 1.0 indicates a perfect circle (circularity = 4π(area/perimeter2). Confocal images through the centre of the cells were opened in ImageJ, a ROI was manually drawn around individual cells following the membrane and the circularity measured. Cell circularity was measured for five confocal images for each treatment, and the results of three independent experiments were pooled.
dSTORM
IBIDI glass-bottom chambers were first coated with poly-d-lysine (Sigma). Jurkat cells expressing CD40ECD–GFP were incubated with 25 µg ml–1 anti-CD40 or CD40L at 37 °C for 1 h and then washed with PBS and fixed with 4% paraformaldehyde. GFP was detected using AF647-conjugated anti-GFP nanobodies (Proteintech Europe, 1:500) following the manufacturer’s instructions. TCEP STORM buffer comprises three solutions (A, B and C). Solution A contains 1 µg ml–1 catalase, 0.2 mM TCEP, 2.5% glycerol, 1.25 mM KCl, 1 mM Tris-HCl and 50 µg ml–1 glucose oxidase. Solution B contains 40 mg ml–1 glucose and 4% glycerol. Solution C contains 0.1 M MEA-HCl. Immediately before dSTORM collection, the TCEP STORM buffer solutions A (50 μl), B (400 μl), C (100 μl) and PBS (450 μl) were mixed and then added to the well. A wide-field fluorescence reference image was acquired before dSTORM images (10,000 frames, 30 ms of exposure) were collected using an ONI Nanoimager equipped with a 640 nm laser and NimOS1.6 software (ONI). Analyses of dSTORM data were carried out using the CODI cloud analysis platform (beta version, ONI). Images were subjected to drift correction and filtering before ROIs were drawn around the cell–cell junctions. For CD40L, ROIs were also drawn around large clusters present outside the cell–cell junctions. Localizations within the ROIs were identified and grouped into subclusters using HDBSCAN81. The following features were extracted for each individual subcluster: number of localizations; density (localizations/area); and area (computed from the convex hull of the cluster).
In vitro assessment of receptor internalization
The level of CD40 and PD-1 internalization was quantified using a fluorescence quenching assay as previously described47. To assess CD40 internalization, AF488-labelled anti-CD40 monoclonal antibody or AF488-labelled anti-CD20 rituximab hIgG2 were added to Ramos cells as indicated for 10, 30, 60, 120 or 180 min at 4 °C or 37 °C. To assess PD-1 internalization, AF488-labelled anti-PD-1 monoclonal antibody or the anti-CD3 OKT3 hIgG1 pre-opsonized with AF488-labelled anti-human IgG Fc monoclonal antibody SB2H2 (produced in-house) were added to Jurkat NFAT–Luc–PD-1 cells as indicated for 10, 30, 60, 120 or 180 min at 4 °C or 37 °C. Subsequently, Ramos or Jurkat NFAT–Luc–PD-1 cells were washed, and half the cells treated with anti-AF488 antibody (ThermoFisher, 1:100) at 4 °C that quenches AF488 fluorescence. The remaining unquenched AF488 fluorescence analysed by flow cytometry correlates to internalized CD40 or PD-1. The percentage of total expression quantifies remaining cell-surface-bound receptor and was calculated as a percentage (unquenched fluorescence – quenched fluorescence)/(unquenched fluorescence).
ADCP
Ramos cells stably transfected with 4-1BBECD-Tm (Ramos 4-1BB cells) were generated by transfecting Ramos cells with pcDNA3 plasmid encoding 4-1BBECD-Tm using nucleofection kit V (Lonza), and stable clones were selected using 1 mg ml–1 geneticin (Extended Data Fig. 10c). ADCP was performed as previously described17, using Ramos 4-1BB cells as target cells and human monocyte-derived macrophages (hMDMs) as the effector cells. The hMDMs were derived by culturing monocytes in the presence of 100 ng ml–1 M-CSF (in-house) for 6 days. The day before the phagocytosis assay, 1 × 105 hMDMs were plated onto a 96-well flat-bottom plate (ThermoFisher). The next day, target Ramos 4-1BB cells were labelled with CFSE followed by opsonization with various anti-4-1BB monoclonal antibodies as indicated in the figure legends for 30 min at 4 °C. In total, 5 × 105 target cells were then added to each well and incubated at 37 °C for 30 min for phagocytosis to occur. The samples were subsequently stained with anti-CD14-APC (1:20) to identify hMDMs, and cells positive for both CFSE and CD14 as assessed by flow cytometry were classified as hMDMs that had undergone phagocytosis. The percentage of ADCP was calculated as follows: (CFSE+CD14+ cells)/(total CD14+ cells) × 100.
ADCC
IIA1.6 cells stably transfected with full-length human 4-1BB (IIA1.6-4 1BB cells) were generated by transfecting IIA1.6 cells with pCIpuro plasmid encoding human 4-1BB using nucleofection kit V (Lonza), and stable clones were selected using 4 µg ml–1 puromycin (Extended Data Fig. 10d). ADCC was performed as previously described48, using IIA1.6-4 1BB cells as targets and human PBMCs as effector cells. In brief, target cells were labelled with calcein-AM (ThermoFisher), added to wells of a 96-well round-bottom plate (ThermoFisher), at 8 × 105 per well and then incubated with various anti-4-1BB monoclonal antibodies as specified in the figure legends for 30 min at 4 °C. In total, 4 × 106 effector cells were then added to each well and incubated at 37 °C for 4 h before cells were centrifuged and the supernatant quantified for calcein-AM fluorescence using a Varioskan Flash plate reader (ThermoFisher). The control wells for maximal lysis contained 4% Triton X-100 (Sigma). The level of ADCC was expressed as the percentage of maximal lysis = (experimental fluorescence – background fluorescence)/(maximal lysis – background fluorescence) × 100.
PD-1 blockade assay
Jurkat NFAT–Luc–PD-1 cells were generated by transfecting WT Jurkat NFAT–Luc cells (Promega) with pcDNA3 plasmid encoding human PD-1 using nucleofection kit V (Lonza), and stable clones were selected using 1 mg ml–1 geneticin. CHO OKT3–scFv–CD8α–PD-L1 cells were generated by transfecting CHO-k1 cells with pCIpuro vector encoding OKT3–scFv–CD8α and pcDNA3 vector encoding human PD-L1 and selecting stable clones using 10 µg ml–1 puromycin and 1 mg ml–1 geneticin. To assess the ability of anti-PD-1 monoclonal antibodies to block the PD-1–PD-L1 interaction, 5 × 104 CHO OKT3–scFv–CD8α–PD-L1 cells were plated onto wells of sterile white opaque 96-well microplates (Perkin Elmer) overnight. The next day, 5 × 104 Jurkat NFAT–Luc–PD-1 cells were added to each well along with the various anti-PD-1 monoclonal antibodies as indicated in each figure legend for 6 h before ONE-Glo reagent (Promega) was added. Luciferase activity was read using a Varioskan Flash plate reader (ThermoFisher).
PD-1-mediated T cell suppression assay
CHO-k1 cells stably transfected with SB2H2–scFv–CD8α (CHO SB2H2–scFv–CD8α cells) were generated by transfecting WT CHO-k1 cells with pcDNA3 plasmid encoding SB2H2–scFv–CD8α using GenePorter (Amsbio). Stable clones were selected using 1 mg ml–1 geneticin. For the PD-1-mediated T cell suppression assay, CHO SB2H2–scFv–CD8α cells were first incubated with 20 µg ml–1 nivolumab variants and 5 µg ml–1 OKT3 hIgG1 for 30 min before excess unbound monoclonal antibody was washed off using complete RPMI medium. Jurkat NFAT–Luc–PD-1 cells were then added to the opsonized CHO SB2H2–scFv–CD8α cells and incubated for 6 h. CD69 expression (anti-CD69, 1:20) on Jurkat NFAT–Luc–PD-1 cells was measured by flow cytometry, and the level of NFAT activation was quantified by measuring the luciferase activity using ONE-Glo Reagent (Promega) as described above.
Statistics and reproducibility
Data analysis was performed using GraphPad Prism 9.2.0 (GraphPad Software). Two-tailed, non-paired Student t-test was used for pairwise comparisons. One-way analysis of variance followed by Kruskal–Wallis test was used for multiple comparisons as specified in the figure legends. Throughout, *P < 0.05, **P < 0.01, ***P < 0.001 and NS, not significant. Reproducibility, including technical replicates and independent biological experiments, is stated in each figure legend.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Original raw data will be provided upon request to include all supporting information. Models of Fab–receptor complexes were obtained from the PDB with the following accession codes: 6FAX for the ChiLob 7/4–CD40 complex; 6MI2 for the utomilumab–4-1BB complex; and 5WT9 for the nivolumab–PD-1 complex. Source data are provided with this paper.
References
Muramatsu, M. et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563 (2000).
Viant, C. et al. Antibody affinity shapes the choice between memory and germinal center B cell fates. Cell 183, 1298–1311.e11 (2020).
Forthal, D. N. Functions of antibodies. Microbiol. Spectr. 2, 1–17 (2014).
Singh, S. et al. Monoclonal antibodies: a review. Curr. Clin. Pharmacol. 13, 85–99 (2018).
Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 20, 491–495 (2021).
Kohler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975).
Clackson, T., Hoogenboom, H. R., Griffiths, A. D. & Winter, G. Making antibody fragments using phage display libraries. Nature 352, 624–628 (1991).
Pedrioli, A. & Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 42, 1143–1158 (2021).
Mayes, P. A., Hance, K. W. & Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 17, 509–527 (2018).
White, A. L. et al. Interaction with FcγRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J. Immunol. 187, 1754–1763 (2011).
Yu, X. et al. Complex interplay between epitope specificity and isotype dictates the biological activity of anti-human CD40 antibodies. Cancer Cell 33, 664–675.e4 (2018).
White, A. L. et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 27, 138–148 (2015).
Ahonen, C. et al. The CD40–TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol. 3, 451–456 (2002).
Dahan, R. et al. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell 29, 820–831 (2016).
Yu, X. et al. Isotype switching converts anti-CD40 antagonism to agonism to elicit potent antitumor activity. Cancer Cell 37, 850–866.e7 (2020).
Ma, D. Y. & Clark, E. A. The role of CD40 and CD154/CD40L in dendritic cells. Semin. Immunol. 21, 265–272 (2009).
Yu, X. et al. TNF receptor agonists induce distinct receptor clusters to mediate differential agonistic activity. Commun. Biol. 4, 772 (2021).
Chester, C., Sanmamed, M. F., Wang, J. & Melero, I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood 131, 49–57 (2018).
Chin, S. M. et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat. Commun. 9, 4679 (2018).
Gong, J., Chehrazi-Raffle, A., Reddi, S. & Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother. Cancer 6, 8 (2018).
Paluch, C., Santos, A. M., Anzilotti, C., Cornall, R. J. & Davis, S. J. Immune checkpoints as therapeutic targets in autoimmunity. Front. Immunol. 9, 2306 (2018).
Curnock, A. P. et al. Cell-targeted PD-1 agonists that mimic PD-L1 are potent T cell inhibitors. JCI Insight 6, e152468 (2021).
Bryan, C. M. et al. Computational design of a synthetic PD-1 agonist. Proc. Natl Acad. Sci. USA 118, e2102164118 (2021).
Lee, J. Y. et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat. Commun. 7, 13354 (2016).
Bardhan, K. et al. Phosphorylation of PD-1-Y248 is a marker of PD-1-mediated inhibitory function in human T cells. Sci. Rep. 9, 17252 (2019).
Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H. & Riley, J. L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954 (2004).
Patsoukis, N., Wang, Q., Strauss, L. & Boussiotis, V. A. Revisiting the PD-1 pathway. Sci. Adv. 6, eabd2712 (2020).
Jones, B., Tite, J. P. & Janeway, C. A. Jr Different phenotypic variants of the mouse B cell tumor A20/2J are selected by antigen- and mitogen-triggered cytotoxicity of L3T4-positive, I-A-restricted T cell clones. J. Immunol. 136, 348–356 (1986).
Lu, R. M. et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27, 1 (2020).
Tabasinezhad, M. et al. Trends in therapeutic antibody affinity maturation: from in-vitro towards next-generation sequencing approaches. Immunol. Lett. 212, 106–113 (2019).
Chodorge, M. et al. A series of Fas receptor agonist antibodies that demonstrate an inverse correlation between affinity and potency. Cell Death Differ. 19, 1187–1195 (2012).
Segal, N. H. et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin. Cancer Res. 24, 1816–1823 (2018).
Rudnick, S. I. et al. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 71, 2250–2259 (2011).
Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 22, 1727–1741 (2015).
Roghanian, A. et al. Antagonistic human FcγRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 27, 473–488 (2015).
Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 43, W314–W319 (2015).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Davis, C. B. et al. Combination of a PD-1 antagonist and a 4-1BB agonist for treating cancer. International patent publication number WO 2015/119923 A1 (2015).
Korman, A. J. et al. Human monoclonal antibodies to programmed death 1 (PD-1) and methods of treating cancer using anti-PD01 antibodies alone or in combination with other immunotherapeutics. International patent publication number WO 2006/121168 A1 (2006).
Keler, T et al. Antibodies that bind human CD27 and uses thereof. US patent 9,169,325 (2015).
Hanke, T et al. Nucleic acids encoding superagonistic anti-CD28 antibodies. US patent 7,585,960 (2009).
Arakawa, F. et al. Cloning and sequencing of the VH and Vκ genes of an anti-CD3 monoclonal antibody, and construction of a mouse/human chimeric antibody. J. Biochem. 120, 657–662 (1996).
Meyer, L. et al. A simplified workflow for monoclonal antibody sequencing. PLoS ONE 14, e0218717 (2019).
Sallusto, F. & Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179, 1109–1118 (1994).
Gate, D. et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020).
Fernandes, R. A. et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature 586, 779–784 (2020).
Austin, C. D. et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 15, 5268–5282 (2004).
Sopp, J. M. et al. On-target IgG hexamerisation driven by a C-terminal IgM tail-piece fusion variant confers augmented complement activation. Commun. Biol. 4, 1031 (2021).
Acknowledgements
We would like to thank the preclinical unit staff for animal husbandry; D. Johnston from the Biomedical Imaging Unit, Southampton General Hospital, Southampton, UK, for assistance with confocal microscopy; D. Kavanagh from the University of Oxford for sharing the dSTORM buffer recipe; and J. Felce from ONI, Oxford, UK, for advice on dSTORM data analysis. The dSTORM microscopy experiments were made possible through the funding of an ONI Nanoimager by the Mark Benevolent Fund. Funding was provided by CRUK grants A20537, A25139, A25169 and DRCDDRPGM-Apr2020\100005. X.Y. is funded by a Careertrack Fellowship provided by the Faculty of Medicine in conjunction with the Cancer Immunology Talent fund.
Author information
Authors and Affiliations
Contributions
X.Y. designed and performed the experiments, analysed and interpreted data and wrote the manuscript. C.M.O., H.T.C.C., S.J., C.A.P., J.K., K.L.C., T.I. and C.I.M. generated or provided key reagents or performed and analysed the experiments. J.W.E. and I.T. supported acquisition of funding and contributed to the conception of the work and approach. M.J.G. designed the study, discussed and interpreted data. M.S.C. designed the study, supervised data collection, discussed and interpreted data and wrote the manuscript with X.Y. All authors commented on and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
M.S.C. acts as a consultant for a number of biotechnology companies, being retained as a consultant for BioInvent and has received research funding from BioInvent, GSK, UCB, iTeos and Roche, and receives institutional payments and royalties from patents and licences relating to antibody immunotherapy. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Martin Dahl, Christoph Wuelfing and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Characterization of anti-CD40 mIgG1 mAb ChiLob 7/4 affinity mutants.
a, SPR of various ChiLob 7/4 m1 affinity mutants injected at 250, 50, 10, 2, 0.4, and 0 nM binding to CD40ECD. Data representative of 3 independent experiments. b, ChiLob 7/4 m1 affinity mutants were evaluated for their binding affinity for CD40ECD by SPR as indicated in a, with affinity constants (ka, kd and KD) calculated. Fold change indicates affinity change compared with WT ChiLob 7/4 m1. c, Ramos cells were incubated with 0.5 μg/mL of AF647-labelled ChiLob 7/4 h1 and various concentrations of competing ChiLob 7/4 m1 affinity mutants as indicated and then washed and bound AF647-labelled ChiLob 7/4 h1 detected. Means ± SEM, n = 3, data representative of 3 independent experiments.
Extended Data Fig. 2 Low affinity anti-CD40 mIgG1 mAb exhibit potent agonism.
a, Purified hCD40Tg mouse B cells were incubated with ChiLob 7/4 m1 mutants for 2 days and then stained for surface expression of CD23. Left plot, exemplar raw data. b, Same experiment as (a). Surface expression of CD86. Left plot, exemplar raw data. c, Purified hCD40Tg mouse B cells were incubated with ChiLob 7/4 m1 mutants for 3 days and then 3H-thymidine was added for 18 h to measure proliferation. Inset cell culture images were taken on day 2. Scale bar, 0.5 mm. For (a—c), Means ± SEM, n = 3, data representative of 3 independent experiments. Rightmost plots illustrate the CD23 expression, CD86 expression or proliferation as a function of the on-rate (ka) or off-rate (kd). d, hCD40Tg mice were inoculated with EG7 cells. 7 days later, mice received OTI cells and the next day were treated with ChiLob 7/4 m1 mutants as indicated. Tumor growth curves are shown with numbers as proportion of tumour free mice at experiment end inset. n = 13–17, data pooled from two to three independent experiments.
Extended Data Fig. 3 Characterization of anti-CD40 hIgG2 mAb ChiLob 7/4 affinity mutants.
a, SPR of various ChiLob 7/4 h2 affinity mutants injected at 250, 50, 10, 2, 0.4, and 0 nM binding to CD40ECD. Data representative of 3 independent experiments. b, ChiLob 7/4 h2 affinity mutants were evaluated for their binding affinity for CD40ECD by SPR as indicated in a, with affinity constants (ka, kd and KD) calculated. Fold change indicates affinity change compared with WT ChiLob 7/4 h2. c, Ramos cells were incubated with 0.5 μg/mL of AF647-labelled ChiLob 7/4 h1 and various concentrations of competing ChiLob 7/4 h2 affinity mutants as indicated and then washed and bound AF647-labelled ChiLob 7/4 h1 detected. Means ± SEM, n = 3, data representative of 3 independent experiments. d, e, Purified hCD40Tg mouse B cells were incubated with ChiLob 7/4 h2 affinity mutants as indicated for 2 days and then stained for surface expression of CD23 (d, left plot, exemplar raw data) and CD86 (d, right plot, exemplar raw data). B cell proliferation was assessed by 3H-thymidine incorporation (e). Plots show affinity (KD) vs maximum CD23 MFI, maximum CD86 MFI or maximum proliferation. Means ± SEM, n = 3, data representative of 3 independent experiments. f, OTI cells were adoptively transferred into hCD40Tg mice 1 day before treatment with ChiLob 7/4 h2 mutants with peripheral SIINFEKL+ CD8 cells identified by flow cytometry on day 4. Mean ± SEM, n = 7, data pooled from two independent experiments. Each dot represents one mouse. Two-tailed, non-paired Student’s t test, the p values for WT h2 vs FW-12 h2, vs FW-22 h2, vs FW-16 h2 are (from left to right) 0.0023, 0.0023 and 0.0023. g, Purified human B cells were incubated with ChiLob 7/4 h2 affinity mutants for 2 days and then stained for surface expression of CD23 (left plot, exemplar raw data) and CD86 (right plot, exemplar raw data). h, Purified human B cells were incubated with ChiLob 7/4 h2 mutants as indicated for 3 days and then 3H-thymidine was added for 18 h to measure proliferation. Inset cell culture images were taken on day 2. For g, h, Means ± SEM, n = 3, data representative of 3 independent experiments. i, Purified human B cells were incubated with ChiLob 7/4 h2 affinity mutants as indicated for 2 days. B cell proliferation was assessed by 3H-thymidine incorporation. Plots show affinity (KD) vs maximum CD23 MFI, maximum CD86 MFI or maximum proliferation. Means ± SEM, n = 3, data representative of 3 independent experiments.
Extended Data Fig. 4 Low affinity anti-CD40 mAb exhibit potent agonism in human systems.
a, Human DCs were stimulated with various ChiLob 7/4 h2 affinity mutants for 2 days and then evaluated for CD86 expression. The ranking of CD86 MFI was plotted against KD. Means ± SEM, each dot represents an average value from 9 donors. b, Gating strategy and representative histograms for a. c, Human DCs were pre-treated with ChiLob 7/4 h2 affinity mutants for 2 days and co-cultured with allogeneic CD4+ T cells for 5 days. CD4+ T cell proliferation was measured by 3H-thymidine incorporation. Ranking of CD4+ T cell proliferation was plotted against affinity (KD). Means ± SEM, each dot represents the average from 7 donors. d, Human monocyte-derived DCs were pre-treated with ChiLob 7/4 h2 affinity mutants for 2 days and then co-cultured with allogeneic CD4+ T cells at different ratios for 5 days. CD4+ T cell proliferation was measured by 3H-thymidine incorporation. The ranking of CD4+ T cell proliferation was plotted against affinity (KD). Means ± SEM, each dot represents the average value from 7 healthy donors. e, CFSE-labelled PBMCs were stimulated with antigenic peptides and ChiLob 7/4 h2 affinity mutants for 5 days and proliferating CD8+ T cells evaluated for surface expression of CD25. Ranking of CD25 MFI plotted against KD. Means ± SEM, each dot represents the average from 8 donors. f, Gating strategy and representative histograms for CD25 expression of proliferating CD8+ T cells in PBMCs stimulated with CEFX Ultra SuperStim Pool and ChiLob 7/4 h2 affinity mutants. Data representative of 8 donors.
Extended Data Fig. 5 Low affinity anti-CD40 mIgG1 mAb induce agonism independent of FcγR.
a, Jurkat-NFκB-GFP-CD40 reporter cells were incubated with various ChiLob 7/4 m1 affinity mutants for 6 h and the level of NFκB activation (GFP) assessed. Means ± SEM, n = 3, data representative of 3 independent experiments. Representative flow data shown in left panels. Subsequent plots reflect the dose-response for the different mAb followed by cumulative plots of the data as a function of affinity parameters; ka (top) and kd (bottom), respectively. b—d, Purified hCD40Tg/FcγRnull mouse B cells were incubated with ChiLob 7/4 m1 mutants for 2 days and then stained for surface expression of (b) CD23 and (c) CD86. Representative flow data shown in left panels. Subsequent plots reflect the dose-response for the different mAb followed by cumulative plots of the data as a function of affinity parameters; ka (top) and kd (bottom), respectively.(d) On day 3 3H-thymidine was added for 18 h to assess B cell proliferation. Inset cell culture images were taken on day 2. Means ± SEM, n = 3, data representative of 3 independent experiments. Scale bar, 0.5 mm. Plots reflect the dose-response for the different mAb followed by cumulative plots of the data as a function of affinity parameters; ka (top) and kd (bottom), respectively.
Extended Data Fig. 6 Low affinity anti-CD40 hIgG2 mAb induce agonism independent of FcγR.
a—c, Purified hCD40Tg/FcγRnull mouse B cells were incubated with ChiLob 7/4 h2 mutants for 2 days and then stained for surface expression of (a) CD23 and (b) CD86. Representative flow data shown in left panels. Subsequent plots reflect the dose-response for the different mAb followed by cumulative plots of the data as a function of affinity (KD). (c) On day 3 3H-thymidine was added for 18 h to assess B cell proliferation. Inset cell culture images were taken on day 2. Means ± SEM, n = 3, data representative of 3 independent experiments. Scale bar, 0.5 mm. Plots reflect the dose-response for the different mAb followed by cumulative plots of the data as a function of affinity (KD).
Extended Data Fig. 7 Low affinity anti-CD40 mAb induce agonism through receptor clustering with minimal receptor internalization.
a, Schematic of the method for calculating clustering index. Confocal images through the centre of cells were opened in LAS X software and fluorescence intensity measurements taken for regions of interest at the cell:cell junctions (red) or at the periphery of the cells (blue). b, Jurkat-CD40-GFP cells were incubated with ChiLob 7/4 m1 affinity mutants as indicated for 3 h at 37 °C and then imaged by confocal. Green: CD40-GFP. Scale bar: 4 μm. Image representative of at least fifteen images taken from 3 independent experiments. c, Same experiment as (b) Left panel: cell circularity was measured by ImageJ for five confocal images per treatment and the results of three independent experiments were pooled. Each dot represents one cell. Right panel: plot showing cumulative circularity data as a function of B cell proliferation. Means ± SEM. d, Binding of Fab fragments of ChiLob 7/4 m1 affinity mutants to Ramos cells. Data representative of 3 independent experiments. e, Jurkat-NFκB-GFP-CD40 reporter cells were incubated with various ChiLob 7/4 m1 affinity mutant IgG versus Fab pairs for 6 h and the level of NFκB activation (GFP) assessed. Means ± SEM, n = 3, data representative of 3 independent experiments. f, Same experiment as (b) evaluating the clustering potential of 4 different ChiLob 7/4 m1affinity mutant Fab versus an IgG positive control. Clustering index calculated as indicated in (a). Means ± SEM, n = 5, data representative of 3 independent experiments. g, Ramos cells were treated with AF488-labelled ChiLob 7/4 m1 affinity mutants for 10, 30, 60, 120 or 180 min (left to right) at 37 °C or 4 °C as indicated. Cells were then washed and half the cells treated with anti-AF488 mAb at 4 °C to quench cell surface associated AF488 fluorescence. Remaining cell surface-bound CD40 was expressed as % Total expression. Data representative of 3 independent experiments. h, Jurkat-CD40-GFP cells were incubated with ChiLob 7/4 m1 affinity mutants as indicated for 3 h at 37 °C and then fixed with PFA, counterstained with DAPI and imaged by confocal. Orthogonal images shown. Blue: nucleus; Green: CD40-GFP. Image representative of at least ten images taken from 3 independent experiments.
Extended Data Fig. 8 Super-resolution dSTORM analysis of receptor clusters induced by low affinity anti-CD40 mAb or CD40L.
a, Jurkat-CD40-GFP cells were incubated with various CD40 agonists as indicated for 1 h, and then CD40-GFP detected with AF647-conjugated anti-GFP nanobody and visualised by wide field fluorescence microscopy and dSTORM. A region of interest was drawn around the cell:cell junctions and clustering analysis was performed using HDBSCAN, example results of subclusters are shown. Scale bars; 10 μm (wide field), 5 μm (dSTORM), 1 μm (HDBSCAN) b, Subcluster density (number of localisation per unit area). One-way ANOVA followed by Kruskal-Wallis test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n= number of subclusters, y=number of cell:cell junctions examined: Untreated, n = 281, y = 28; ChiLob 7/4 h2, n = 664, y = 26; CD40L, n = 427, y = 25; WT m1, n = 138, y = 23; FW-16 m1, n = 642, y = 29; FW-32 m1, n = 151, y = 30. c, Plot of subcluster area versus density. Results shown are representative of 3 independent experiments.
Extended Data Fig. 9 Characterization of low affinity utomilumab and nivolumab variants.
a, Utomilumab affinity mutants were evaluated for their binding affinity for 4-1BBECD by SPR, with affinity constants (ka, kd and KD) calculated as well as fold change which indicates affinity change compared with WT utomilumab. b, Jurkat-4-1BB-GFP cells were incubated with utomilumab affinity mutants and then imaged by confocal. Green: 4-1BB-GFP. Scale bar: 4 μm. Images representative of at least fifteen images taken from 3 independent experiments. c, Same experiment as (b) Left panel: cell circularity was measured by ImageJ for five confocal images per treatment and the results of three independent experiments were pooled. Each dot represents one cell. Right panel: plot showing cumulative circularity data as a function of NFκB activation. Means ± SEM. d, The expression level of 4-1BB on Ramos-4-1BB cells was analysed by flow cytometry. e, The expression level of 4-1BB on IIA1.6-4-1BB cells was analysed by flow cytometry. f, SPR of various nivolumab affinity mutants injected at 250, 50, 10, 2, 0.4, and 0 nM binding to PD-1ECD. Plots display sensorgram data representative of 3 independent experiments. g, Nivolumab affinity mutants were evaluated for their binding affinity for PD-1ECD by SPR as indicated in f, with affinity constants (ka, kd and KD) calculated. Fold change indicates affinity change compared with WT nivolumab. h, PD-1-transfected Jurkat cells were incubated with various nivolumab affinity mutants as indicated and then washed and bound hIgG detected. Means ± SEM, n = 3, data representative of 3 independent experiments.
Extended Data Fig. 10 Low affinity nivolumab variants induce PD-1 agonism through receptor clustering.
a, Schematic of assays investigating the antagonistic and agonistic properties of anti-PD-1 affinity mutants. Left panel: Assay to evaluate the ability of mAb to block PD-L1-mediated T cell suppression. Middle panel: Assay to evaluate the ability of mAb to induce PD-1 signalling. Right panel: Assay to evaluate the ability of mAb to suppress anti-CD3 mAb-mediated T cell activation. b, Histogram showing CHO-SB2H2-scFv-CD8α cells binding to various nivolumab affinity mutants, data representative of 3 independent experiments. c, CHO-SB2H2-scFv-CD8α cells were opsonized with OKT3 and nivolumab affinity mutants as indicated and then co-cultured with Jurkat-NFAT-Luc-PD-1 for 6 hours. NFAT signalling activity was then assessed. Plots showing relative luciferase units (RLU) vs ka and RLU vs kd. Means ± SEM, n = 3, data representative of 3 independent experiments. d, Same experiment as (c). Plots showing CD69 MFI vs ka and RLU vs CD69 MFI. e, IIA1.6-PD-1-GFP cells were incubated with nivolumab affinity mutants for 3 h and then imaged using confocal. Green: PD-1-GFP. Scale bar: 4 μm. Image representative of at least 15 images taken from 3 independent experiments. f, IIA1.6-PD-1-GFP cells were incubated with nivolumab affinity mutants as indicated for 3 h and then fixed, counterstained with DAPI and imaged using confocal. Z-stack projections shown. Blue: nucleus; Green: PD-1-GFP. Scale bar: 4 μm. Image representative of at least ten images taken from two independent experiments. g, Jurkat-NFκB-GFP-PD-1 reporter cells were incubated with various nivolumab affinity mutants as indicated for 30 min then, Left panel: washed with the level of mAb remaining bound after various periods quantified or Right panel: the level of NFκB activation (GFP) assessed after various periods. Means ± SEM, n = 3, data representative of 3 independent experiments. h, Same experiment as (e) Left panel: cell circularity was measured by ImageJ for five confocal images per treatment and the results of three independent experiments were pooled. Each dot represents one cell. Right panel: plot showing cumulative circularity data as a function of CD69 MFI, Means ± SEM, n = 34, 32, 39, 39, 36, 38, 33, 36, 33, 37 (from left to right).
Extended Data Fig. 11 Low affinity mutants retain target-specific binding and exhibit agonism at multiple receptor densities.
a, Heat map showing MFI values of ChiLob 7/4 m1 affinity mutants binding to Jurkat-NFκB-GFP-TNFRII cells versus a positive control anti-TNFRII mAb. b, Heat map showing SPR response of ChiLob 7/4 m1 affinity mutants binding to soluble TNFRII immobilized onto a CM5 chip versus a positive control anti-TNFRII mAb. c, Heat map showing MFI values of utomilumab affinity mutants binding to Jurkat-NFκB-GFP-CD27 cells versus a positive control anti-CD27 mAb. d, Heat map showing SPR response of utomilumab affinity mutants binding to soluble CD27 immobilized onto a CM5 chip versus a positive control anti-CD27 mAb. e, Heat map showing MFI values of nivolumab affinity mutants binding to Jurkat-CD28-GFP cells versus a positive control anti-CD28 mAb. f, Heat map showing SPR response of nivolumab affinity mutants binding to soluble CD28 immobilized onto a CM5 chip versus a positive control anti-CD28 mAb. g, Representative z plane images covering the entire z-axis of Jurkat-CD40-GFP cells treated with anti-CD40 mAb. Blue: nucleus; Green: PD-1-GFP. Scale bar: 4 μm. Data representative of 3 independent experiments. h, Representative z plane images covering the entire z-axis of Jurkat-4-1BB-GFP cells treated with anti-4-1BB mAb. Blue: nucleus; Green: PD-1-GFP. Scale bar: 4 μm. Data representative of 3 independent experiments. i, Representative z plane images covering the entire z-axis of IIA1.6-PD-1-GFP cells treated with anti-PD-1 mAb. Blue: nucleus; Green: PD-1-GFP. Scale bar: 4 μm. Data representative of 3 independent experiments. j, Jurkat-NFκB-GFP reporter cells expressing low, medium or high levels of CD40 were incubated with various ChiLob 7/4 m1 affinity mutants for 6 h and the level of NFκB activation (GFP) assessed. k, Quantification of CD40 receptor number (as expressed by Molecules of Equivalent Soluble Fluorochrome, MESF) on various cell lines and primary cells as indicated. l, Jurkat-NFκB-GFP reporter cells expressing low, medium or high levels of PD-1 were incubated with various nivolumab affinity mutants for 6 h and the level of NFκB activation (GFP) assessed. m, Quantification of PD-1 receptor number (MESF) on various cell lines and primary cells as indicated. n, Jurkat-NFκB-GFP reporter cells expressing low, medium or high levels of 4-1BB were incubated with various utomilumab affinity mutants for 6 h and the level of NFκB activation (GFP) assessed. o, Quantification of 4-1BB receptor number (MESF) on various cell lines and primary cells as indicated. p, mAb (black) or soluble ligands (red) were injected at 250, 50, 10, 2, 0.4, and 0 nM to evaluate binding by SPR to their cognate soluble receptor CD40, 4-1BB or PD-1, as indicated. Values indicate the equilibrium affinity KD of the ligands for their cognate receptor. Means ± SEM, n = 3, data representative of 3 independent experiments.
Supplementary information
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yu, X., Orr, C.M., Chan, H.T.C. et al. Reducing affinity as a strategy to boost immunomodulatory antibody agonism. Nature 614, 539–547 (2023). https://doi.org/10.1038/s41586-022-05673-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05673-2
- Springer Nature Limited
This article is cited by
-
Development of a new affinity maturation protocol for the construction of an internalizing anti-nucleolin antibody library
Scientific Reports (2024)
-
i-shaped antibody engineering enables conformational tuning of biotherapeutic receptor agonists
Nature Communications (2024)
-
Fcγ receptors and immunomodulatory antibodies in cancer
Nature Reviews Cancer (2024)
-
4-1BB immunotherapy: advances and hurdles
Experimental & Molecular Medicine (2024)
-
Cancer organoids 2.0: modelling the complexity of the tumour immune microenvironment
Nature Reviews Cancer (2024)