

Abstract
The human GABAB receptor—a member of the class C family of G-protein-coupled receptors (GPCRs)—mediates inhibitory neurotransmission and has been implicated in epilepsy, pain and addiction1. A unique GPCR that is known to require heterodimerization for function2,3,4,5,6, the GABAB receptor has two subunits, GABAB1 and GABAB2, that are structurally homologous but perform distinct and complementary functions. GABAB1 recognizes orthosteric ligands7,8, while GABAB2 couples with G proteins9,10,11,12,13,14. Each subunit is characterized by an extracellular Venus flytrap (VFT) module, a descending peptide linker, a seven-helix transmembrane domain and a cytoplasmic tail15. Although the VFT heterodimer structure has been resolved16, the structure of the full-length receptor and its transmembrane signalling mechanism remain unknown. Here we present a near full-length structure of the GABAB receptor, captured in an inactive state by cryo-electron microscopy. Our structure reveals several ligands that preassociate with the receptor, including two large endogenous phospholipids that are embedded within the transmembrane domains to maintain receptor integrity and modulate receptor function. We also identify a previously unknown heterodimer interface between transmembrane helices 3 and 5 of both subunits, which serves as a signature of the inactive conformation. A unique ‘intersubunit latch’ within this transmembrane interface maintains the inactive state, and its disruption leads to constitutive receptor activity.
Similar content being viewed by others


Main
We assembled the heterodimeric human GABAB receptor by using baculovirus-infected mammalian cells. Each subunit was truncated at the carboxy-terminal end according to the domain boundary of the intracellular coiled-coil17, in order to eliminate flexible regions (Supplementary Fig. 1). Upon extraction and purification with detergent (Extended Data Fig. 1a, b and Supplementary Fig. 2), the complex bound to radioactive [3H]GABA with a dissociation constant comparable to the GABA affinity reported for native receptors3,4,7 (Extended Data Fig. 1c). Functional analysis incorporating a chimaeric Gαqi5 protein and inositol phosphate assay18 in mammalian cells showed that the agonist baclofen activated the C-terminally truncated and full-length receptor with similar potency and efficacy, indicating that the deleted regions in the GABAB1b and GABAB2 cytoplasmic tails are not required for ligand-mediated G-protein activation (Extended Data Fig. 1d).
We determined the structure of the heterodimeric GABAB receptor by cryo-electron microscopy (cryo-EM) to an overall resolution of 3.3 Å (Extended Data Fig. 2a–e and Supplementary Table 1). The global density map displayed directional anisotropy owing to linker flexibility (Extended Data Fig. 2f). Performing local refinement separately on the extracellular domains (ECDs) and transmembrane domains yielded reconstructions at resolutions of 3.1 Å and 3.4 Å, respectively (Extended Data Fig. 2g, h). We used a composite map that combined the ECD and transmembrane reconstructions for model building and refinement (Extended Data Fig. 3). By applying three-dimensional variability analysis to the data, we found that the receptor is in dynamic motion, and that its functional state corresponds to a continuum of conformations along multiple dimensions (Supplementary Videos 1–4).
The heterodimeric GABAB receptor is assembled when GABAB1b and GABAB2 subunits interact side-by-side while facing in opposite directions (Fig. 1a, b and Extended Data Fig. 4). Both the VFT and the transmembrane components of the two subunits are related by pseudo-twofold axes. Extracellular and intracellular loops (ECLs and ICLs) that interconnect adjacent helices within each transmembrane domain are visible in the density map, except for ICL2. The cytoplasmic tail including the coiled-coil domain is disordered, possibly because of its flexible attachment to the transmembrane domain.

a, Ribbon representation of the GABAB-receptor structure, composed of GABAB1b (blue) and GABAB2 (green) subunits. Ca2+ is shown as a green sphere; phospholipids (PE 38:5 and PC 38:2) are shown as yellow space-filling models; N-linked glycans (NAGs) and cholesterols (CLRs) are shown, respectively, as grey and pink ball-and-stick models. Transmembrane helices 1–7 are marked for each subunit. b, Cryo-EM density map of the GABAB receptor, composed of local reconstructions for extracellular (3.1 Å) and transmembrane (3.4 Å) domains, in an orthogonal view from a. c, Linker domains of GABAB1b and GABAB2, showing the main-chain and side-chain hydrogen-bonding patterns between the linker region and ECL2.
The elongated peptide linker that joins the VFT to the transmembrane domain is buttressed through its interaction with a β-hairpin formed by ECL2 (Fig. 1c). ECL2 twists across the linker, forming a united mechanical junction that transmits the conformational changes in the VFT to the transmembrane domain and vice versa. Additionally, the ECD and transmembrane domains of the receptor spontaneously flex back and forth about the linker, exhibiting the region’s intrinsic flexibility (Supplementary Video 1).
Given that no ligand was added during protein purification, we expected the receptor to be in an apo form and an inactive conformation. To our surprise, we observed several ligands bound to the receptor. GABAB1b contains a Ca2+ ion at the interdomain cleft of VFT. In addition, an endogenous phospholipid is bound within the transmembrane pocket of each subunit, with a phosphatidylethanolamine (PE 38:5) in GABAB1b, and a phosphatidylcholine (PC 38:2) in GABAB2. Finally, ten cholesterol or cholesteryl hemisuccinate molecules, which we modelled as cholesterol (see Methods), are distributed around the exterior of the transmembrane complex, including two that interface with both subunits.
Inactive conformation of GABAB receptor
The cryo-EM structure of the GABAB receptor appears to occupy an inactive conformation, given its similarity to known crystal structures of the GABAB VFT in its apo and antagonist-bound states16. First, the VFT module, composed of the lobe-shaped LB1 and LB2 domains, adopts an open interdomain conformation in both subunits (Extended Data Fig. 5a, b). Second, although an amino-terminal LB1–LB1 dimer interface is present in all functional states, the distinct lack of a heterodimer interface between the membrane-proximal LB2 domains is shared by the near full-length cryo-EM structure and inactive-state VFT structures (Extended Data Fig. 5c, d). By contrast, a hallmark of the active-state VFT structures is a unique heterodimer interface between LB2 domains, which results from agonist-induced GABAB1b closure16.
Using conformational variability analysis, we observed that the LB2 domains of both subunits fluctuate by approaching and withdrawing from the central vertical axis of the heterodimer, yet never make contact as they do in active-state VFT crystal structures (Supplementary Videos 2, 3). The motion exhibited by the LB2 domains and their associated linkers suggests that the inactive functional state corresponds to an ensemble of conformations, where the separation between the membrane-proximal regions falls within a small range around the coordinates of our structure.
Inactive transmembrane heterodimer interface
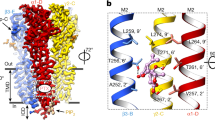
We identified a hitherto unknown heterodimer interface between the transmembrane TM5 and TM3 helices of both subunits that embodies the signature of the GABAB receptor’s inactive conformation (Fig. 2a). A TM5–TM5 contact has previously been detected through crosslinking of the GABAB receptor19, but transmembrane dimer interfaces of any kind are yet to be found in other inactive class C GPCRs, including the recent inactive metabotropic glutamate receptor mGlu5 structure20 (Extended Data Fig. 5e).

a, The transmembrane heterodimer interface formed by TM5 and TM3 of both subunits. Three layers (I, II and III) of interfacial contacts are identified by dotted circles. Direct heterodimer contacts within each layer are shown on the right. b, c, Functional analysis of the ‘intersubunit latch’. Basal activity (b) and dose-dependent baclofen-stimulated receptor response (c) in cells transiently expressing a Gαqi5 chimaera (abbreviated as Gqi) with different combinations of GABAB wild-type subunits (WT) and mutant subunits (GABAB1b E673R, abbreviated as E673R; GABAB2 H579E, abbreviated as H579E). Inositol-1-phosphate (IP1) accumulation was measured in the presence or absence of 20 μM CGP54626 (abbreviated as INV). Cells transfected with empty pcDNA3.1 vector (abbreviated as pcDNA), Gαqi5 alone, or WT GABAB subunits in the absence of Gαqi5 were used as controls. Relative activity is expressed as percentage of maximum wild-type activity induced by baclofen relative to the activity of Gαqi5 alone. Data points represent averages ± s.e.m. of multiple experiments (n values shown), each with quadruplicate measurements. ****P < 0.0001; one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test was used to calculate statistical differences in basal activity (b). The level of cell surface expression was 90% for E673R/WT and 76% for WT/H579E mutants in comparison with the WT/WT heterodimer.
Positioned at 30° from the extracellular dimer interface, the transmembrane dimer interactions bury approximately 740 Å2 of surface area and exhibit high shape complementarity (Extended Data Fig. 5f). The pair of TM5 helices scissor at their central residues before contacting the transverse TM3 of opposing subunits at their intracellular ends (Fig. 2a). TM5 extracellular ends display conformational variance wherein they approach and withdraw but fail to make contact (Supplementary Video 4). All direct heterodimer interactions occur near the cytoplasmic membrane surface and can be divided into three core layers (I–III) along the helical path of TM5 (Fig. 2a).
The surface layer I caps the extracellular end of the transmembrane heterodimer interface. It is composed of hydrophobic contacts between four leucine residues of both subunits. The middle layer II lies directly beneath layer I, and consists of three phenylalanine residues packing against one another, as well as their neighbouring leucine residues. Both layers solely incorporate TM5 residues.
Layer III, consisting of sections IIIa and IIIb, completes the interface at the intracellular end. IIIa possesses a network of salt bridges that tether the cytoplasmic ends of GABAB1b and GABAB2 transmembrane domains. This critical interaction is mediated by a quartet of charged residues from the TM3 and TM5 helices (His 5723.55 and Glu 6735.60 in GABAB1b; His 5793.55 and Glu 6775.60 in GABAB2; Supplementary Table 2), a feature that we refer to as the ‘intersubunit latch’ for securing the transmembrane orientation of the two subunits in the inactive conformation. One of the ‘intersubunit latch’ residues shares nonpolar contacts with a lysine of GABAB2 TM5, establishing the accessory site IIIb.
In addition, the central layers of direct heterodimer contacts are flanked on each side by a cholesterol molecule. One mediates the interaction between TM5 helices of both subunits (CLR3), while the other bridges TM3 of GABAB1b and TM5 of GABAB2 (CLR6) (Extended Data Fig. 5g).
The ‘intersubunit latch’
To determine the importance of the ‘intersubunit latch’ in controlling the inactive state of the GABAB receptor, we examined the effect of single charge-repelling mutations (GABAB1b E673R and GABAB2 H579E) within the motif. Wild-type GABAB receptor exhibited basal activity in the absence of agonist (Fig. 2b), as reported previously14. Both mutants substantially increased basal activity when compared to the wild type, suggesting that each mutation promotes agonist-independent constitutive activity (Fig. 2b). The basal activity displayed by these mutants reached approximately 80–90% of the maximal agonist-dependent wild-type response (Fig. 2b, c). Application of the agonist baclofen raised receptor activity further to the highest wild-type level (Fig. 2c). Neither mutation altered the agonist potency. After each construct was treated with the inverse agonist CGP54626, the basal activity of mutants still remained higher than that of the wild type, providing further evidence that the ‘intersubunit latch’ mutations serve to shift the conformational equilibrium of GABAB towards an active state. Taken together, our mutational data indicate that the ‘intersubunit latch’ is fundamental to maintaining the inactive state of the receptor.
Endogenous ligands bound to GABAB1b VFT
We identified a new potential Ca2+-binding site in the vicinity of the orthosteric ligand-binding cleft on the LB2 surface of GABAB1b (Fig. 3a and Extended Data Fig. 6a, b). The metal ion density has remarkable peak height in the cryo-EM density map (10σ) and is surrounded by residues that are chemically favourable for Ca2+ coordination. The Ca2+ is anchored by the carboxylate groups of three acidic residues (Asp 281, Glu 309 and Glu 423) and by the backbone carbonyl atoms of two additional residues (Gly 277 and Tyr 279) (Fig. 3b). The Ca2+–O bond distances are between 3.0 Å and 4.6 Å, suggesting that the ion is in a hydrated state. The Ca2+ location in GABAB1b is different from any of the four sites found in the calcium-sensing (CaS) receptor21, another class C GPCR (Fig. 3c).

a, Ribbon representation of the GABAB1b subunit, showing the location of the Ca2+-binding site at the interdomain crevice of VFT. b, Specific interactions between GABAB1b and Ca2+. The mesh represents the cryo-EM density map contoured at 7.5σ surrounding Ca2+. c, Crystal structure of the CaS receptor extracellular domain (Protein DataBank (https://www.rcsb.org) code 5K5S), highlighting its bound Ca2+ (black spheres, numbered 1–4 and 1′–4′ in the two protomers) and the corresponding location of the Ca2+-binding site in GABAB1b (green sphere).
Using inductively coupled plasma mass spectroscopy, we detected the presence of Ca and Cu above background level in purified GABAB receptor. The amounts of other metal elements were negligible (Supplementary Table 3). The molar ratios of Ca and Cu relative to the receptor protein (0.43:1 and 0.51:1) suggest partial occupancy of the ion-binding sites. An unmodelled density within the interdomain cleft, coordinated by tryptophan and histidine, may serve as a potential Cu2+-binding site but lacks sufficient signal to be labelled confidently.
In exploring the functional role of the bound ion, we found that the Ca2+ chelator EGTA substantially reduced GABAB basal activity, and that mutating specific coordinating residues (GABAB1b E309K and GABAB1b E423R) had similar, although less drastic, effects (Extended Data Fig. 6c, d). Consistent with previous findings, these data suggest that Ca2+ may act as a positive allosteric modulator of the GABAB receptor22,23. Our structure implies that Ca2+ stabilizes residues adjacent to the critical agonist-binding residue Trp 278, thereby indirectly reinforcing its conformation (Fig. 3b).
We also found density in the orthosteric agonist-binding site of GABAB1b, and its shape suggests a GABA-like endogenous ligand (Extended Data Fig. 6a, b). GABA is a potential candidate because it fits the density and was detected in the lysate of cells used to express the receptor (Extended Data Fig. 6e, f). This GABA-like endogenous ligand bound in an inactive receptor conformation may reflect a preactivation state; however, further investigation is required.
Discovery of endogenous phospholipid ligands
Our structure revealed endogenous phospholipids within the transmembrane domains of both GABAB subunits. Using the CaS receptor as a control in mass spectrometry, we identified two phospholipids specifically bound to the GABAB receptor, PE 38:5 and PC 38:2 (Fig. 4a–d). Both lipids consist of two long-chain fatty acyl moieties of 18 and 20 carbons. We further assigned PE 38:5 to GABAB1b and PC 38:2 to GABAB2 on the basis of the size difference between the phosphoethanolamine and phosphocholine head groups of the two lipids (Extended Data Fig. 7a, b). The lipid density in the GABAB2 transmembrane domain has a bulkier head group that can better accommodate the larger choline moiety of PC 38:2.

a, c, Liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis of phospholipids bound to the GABAB receptor (n = 1). The LC traces show the abundance of phospholipids PC 38:2 (a) and PE 38:5 (c) in GABAB preparation relative to a CaS receptor control. b, d, High-resolution MS spectra of the peaks in a, c (black) matched with standard spectra of PC 38:2 (b) and PE 38:5 (d) in red. NL, neutral loss: a fragment in a neutral charge state during collision-induced dissociation; SN1 and SN2, stereospecific numbering of glycerophospholipids, marking the fatty acyl groups at respectively the first and second carbons of the glycerol. e, f, Molecular surface of the GABAB1b (e) and GABAB2 (f) transmembrane domains, showing the binding pockets for PE 38:5 and PC 38:2, respectively. g–j, Three views of the specific interactions between phospholipids and each GABAB subunit. Contacts are shown between the phospholipid head group and residues from GABAB1b (g) and GABAB2 (h), and between phospholipid fatty acyl chains and GABAB1b (i) and GABAB2 (j).
Mirroring the amphipathicity of phospholipids, the lipid-binding pocket of each GABAB subunit retains a hydrophilic trunk and two lipophilic branches for binding the lipid polar head and nonpolar tails, respectively. The trunk consists of a negatively charged patch covering the amine moiety, and a positively charged area surrounding the phosphate (Extended Data Fig. 7c, d). The lipid-binding pockets are notably deep, extending from the extracellular membrane surface to the centre of the transmembrane domain (Fig. 4e, f). Each lipid occupies nearly the entire range of ligand-binding positions in class A, B, C and F GPCRs (Extended Data Fig. 7e–h).
Each GABAB subunit makes extensive contacts with the bound lipid, using a majority of the transmembrane helices, including TM2, 3, 5, 6 and 7 (Extended Data Fig. 7a, b, i, j). Approximately 2,400 Å2 of surface area is buried by either lipid–subunit pair. In addition, ECL2 directly contacts PE 38:5 in GABAB1b, while the linker and all three ECLs interact with PC 38:2 in GABAB2.
Key elements of the lipid–receptor interactions are conserved in GABAB1b and GABAB2 (Fig. 4g–j, Extended Data Fig. 7i, j and Supplementary Fig. 3). First, the hydrophilic head of each lipid is anchored through interactions with conserved histidine and arginine residues (His 643 of ECL2 and Arg 5493.32 in GABAB1b; His 647 of ECL2 and Arg 5563.32 in GABAB2) (Fig. 4g, h). GABAB2 also incorporates Arg 714 of ECL3, rendering the lipid-binding pocket more electropositive than that of GABAB1b (Fig. 4h).
Second, the 20-carbon fatty acyl chain of both lipids follows a perpendicular turn to pass between two aromatic residues (Phe 5573.40 and Tyr 6575.44 in GABAB1b; Tyr 5643.40 and Tyr 6615.44 in GABAB2) (Fig. 4i, j). A cis double bond in each fatty acyl chain forms π interactions with the aromatic side chains. The bend is further buttressed by extensive nonpolar contacts with the aliphatic part of a conserved lysine (GABAB1b Lys 6605.47; GABAB2 Lys 6645.47) lying parallel to the chain. Finally, the 18-carbon fatty acyl chain of both lipids is relatively straight, extending towards the cytoplasm in a binding pocket lined by small aliphatic residues on TM2, TM3 and TM7 (Fig. 4i, j).
The GABAB receptor’s transmembrane domains are covered by the linker and ECLs, which form a lid over the lipid-binding pocket. Phospholipids may access the lipid-binding pocket of designated subunits laterally through gaps between TM5 and TM6 (Extended Data Fig. 8a–d). One of the fatty acyl tails of each phospholipid even protrudes through this opening. The lipid-binding sphingosine-1-phosphate (S1P1) receptor possesses a similar gap between TM1 and TM7 (ref. 24; Extended Data Fig. 8e, f). The endogenous lipids of other GPCRs can be readily replaced24, but the size and engagement of the endogenous lipids bound to the GABAB receptor suggest that they are essential for maintaining receptor integrity and stability.
To explore the physiological relevance of the endogenous phospholipids, we mutated residues that hydrogen bonded with the phosphate head group. We identified R714A in GABAB2 ECL3, which displayed a small gain of function despite reduced cell surface expression (Extended Data Fig. 8g, h). This mutation is expected to enhance the movement of PC 38:2 within GABAB2 by eliminating a critical interaction with the lid. Our results suggest that PC 38:2 may act as a negative allosteric modulator of GABAB receptor by stabilizing the inactive conformation of GABAB2.
Comparison of subunits with other GPCRs
The GABAB1b and GABAB2 subunits have highly similar VFT and transmembrane components but differ in their relative orientation (Extended Data Fig. 9a, b). Each subunit differs from all other class C GPCRs in possessing an extended peptide linker between the VFT and transmembrane domains instead of a cysteine-rich domain (Fig. 1c).
GABAB subunits also display distinct helix positions in the seven-helix bundles among inactive GPCRs (Extended Data Fig. 9c–f). However, these differences are minor compared with the dramatic movement of TM6 in class A and B GPCRs when activated by G-protein coupling25,26. This corroborates our conclusion that we have captured an inactive conformation of the GABAB receptor.
The GABAB receptor presents unique variations of conserved transmembrane motifs (Extended Data Fig. 10a, b). In most class A GPCRs such as rhodopsin27, the ‘ionic lock’ tethers TM3 and TM6 to stabilize the inactive state of an individual transmembrane domain27 (Extended Data Fig. 10c). The ‘ionic locks’ of both GABAB subunits consist of an aspartate from ICL3 and a lysine from TM3 (Lys3.50) (Extended Data Fig. 10a, b). Although only the Lys/Asp pair in GABAB1b are within hydrogen-bond distance, their backbone Cα–Cα separations (9.3–9.4 Å) are comparable to that of rhodopsin27 (8.7 Å) and inactive mGlu receptors28,29 (10.9–11.2 Å), indicating that the ‘ionic lock’ is intact within both GABAB subunits (Extended Data Fig. 10a–e).
The ‘ionic lock’ in each GABAB subunit resides in close proximity to an FxPKxx sequence in TM7, which is the counterpart of the NPxxY(x)5,6F motif in class A GPCRs25. Specifically, the conserved Lys7.51 participates in a network of hydrophilic contacts with the ‘ionic lock’ through Asn2.39 and a serine in ICL1 (Extended Data Fig. 10a, b and Supplementary Fig. 3). These interactions unite the ‘ionic lock’ and FxPKxx motif into a larger and integral system for maintaining the inactive transmembrane conformation of individual GABAB subunits.
Conclusion
The combination of our previous VFT structures and present cryo-EM data supports the occurrence of three essential events during activation of the GABAB receptor: first, agonist-induced closure of the GABAB1b VFT; second, association of membrane-proximal LB2 domains; and third, dissociation of the ‘intersubunit latch’ and ensuing rearrangement of the transmembrane heterodimer interface. This hypothesis is consistent with our finding that an inverse agonist bound to the extracellular domain can inhibit the constitutive activity stemming from the spontaneous closure of the GABAB1b VFT, but not that resulting from direct ‘downstream’ disruption of the ‘intersubunit latch’.
The GABAB-receptor structure also yields surprising findings regarding its endogenous ligand composition. We suspect that the phospholipids discovered well inside each transmembrane cavity are necessary structural components, as they are prebound within each subunit and their interactions with the receptor are extensive. These endogenous lipids may be unique to the GABAB receptor, as the lipid-binding residues are not conserved among class C GPCRs (Supplementary Fig. 3). Preoccupation of the transmembrane pocket suggests that GABAB allosteric modulators may bind to yet unknown sites, with the heterodimer interface being a potential location. An active structure of the GABAB receptor would confirm whether the phospholipids are integral receptor components or functional modifiers.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Protein expression and purification
Human GABAB1b (UniProt (https://www.uniprot.org) code Q9UBS5-2) and GABAB2 (UniProt code O75899) subunits were each cloned into a modified pEG BacMam vector30 for coexpression in baculovirus-infected mammalian cells. GABAB1a and GABAB1b are two major isoforms of GABAB1, and have identical pharmacological profiles7. Different C-terminal truncations of each receptor subunit were tested for heterodimeric receptor assembly. The optimal GABAB1b expression construct consisted of residues 1–802 (GABAB1b(1–802)), while the GABAB2 construct included residues 1–819 (GABAB2(1–819)). This allowed the heterodimeric construct to be transported to the cell membrane, as it retained the intracellular coiled-coil region present in the intracellular tail of each subunit31,32. Signal peptides for GABAB1b and GABAB2 occupied residues 1–29 and 1–41, respectively. A Flag tag was engineered at the C terminus of each subunit to facilitate affinity purification.
Human embryonic kidney (HEK) 293 GnTI– cells33 were grown in suspension culture at 37 °C in 8% CO2 using 293 freestyle media (Life Technology, Carlsbad, CA). The cells were coinfected with recombinant baculoviruses carrying the GABAB1b(1–802) and GABAB2(1–819) genes at 37 °C. To enhance the expression level, we added 10 mM sodium butyrate 18 h post infection, and incubated the cells for a further 72 h at 30 °C before harvest.
Cell membrane was isolated by differential centrifugation. The cells were lysed using an EmulsiFlex-C3 high pressure homogenizer (Avestin, Ottawa, Canada) in a buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol and a cocktail of protease inhibitors (Roche, Basel, Switzerland). Cell debris was removed by centrifugation of the lysed cell suspension at 10,000 r.p.m. The cell membrane was then pelleted by ultracentrifugation at 45,000 r.p.m.
The GABAB receptor was extracted from the cell membrane with 50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% lauryl maltose neopentyl glycol (LMNG) (Anatrace, Maumee, OH) and 0.2% cholesteryl hemisuccinate (CHS) (MilliporeSigma, St. Louis, MO) at 4 °C overnight. After the insoluble matter was removed by centrifugation, the supernatant was applied to an anti-Flag M2 antibody affinity column (MilliporeSigma, St. Louis, MO). The column was washed stepwise with decreasing concentrations of detergent, from 0.1% to 0.002% LMNG. The heterodimeric GABAB1b(1–802)–GABAB2(1–819) complex was then eluted with 50 mM HEPES, pH 7.5, 50 mM NaCl, 0.002% LMNG, 0.0004% CHS and 0.2 mg ml−1 Flag peptide.
The receptor was further purified by Mono Q (GE Healthcare, Chicago, IL) ion-exchange chromatography using a linear salt gradient from 50 mM to 1 M NaCl in 50 mM HEPES, pH 7.5, 0.002% LMNG and 0.0004% CHS. Finally, the assembled GABAB receptor was subjected to Superose 6 (GE Healthcare, Chicago, IL) size-exclusion chromatography in 50 mM HEPES, pH 7.5, 50 mM NaCl, 0.002% LMNG and 0.0004% CHS.
HEK 293 GnTI− cells were purchased from and authenticated by the American Type Culture Collection (ATCC catalogue number CRL-3022). Cell morphology was examined for each passage of cells. The cells were certified by ATCC to be free of mycoplasma contamination, but they were not tested again during culturing.
Cryo-EM specimen preparation and data acquisition
Specimens were composed of vitrified GABAB protein samples occupying UltraAuFoil R 0.6/1, 300-mesh holey Au/Au grids (Quantifoil Micro Tools, Jena, Germany). The surfaces of the grids were rendered hydrophilic by glow-discharging using H2 and O2 for 25 s at 10 W with a Solarus 950 plasma cleaner system (Gatan, Pleasanton, CA). For vitrification, 3 μl of purified GABAB receptor at a concentration of approximately 0.3 mg ml−1 was applied to each grid, blotted for 4 s at a blot force of 3 inside a Vitrobot Mark IV (Thermo Fisher Scientific, Waltham, MA), and plunge-frozen in a liquid propane:ethane mixture (63:37, v/v) cooled with liquid nitrogen.
Data collection was performed on a Titan Krios transmission electron microscope (Thermo Fisher Scientific, Waltham, MA) equipped with a K2 Summit direct electron detection camera (Gatan, Pleasanton, CA) in counting mode and a post-column GIF Quantum energy filter (Gatan) in zero-energy-loss mode with a slit width of 20 eV. Micrographs were accrued at a calibrated pixel size of 1.06 Å and with nominal defocus range of −0.5 to −2 μm. Each micrograph consisted of 60 frames collected over a 12-s exposure at a dose rate of roughly 8 electrons per pixel per second for a total dose of roughly 85 electrons per Å2. A total of 3,435 micrographs were acquired as dose-fractionated image stacks.
Cryo-EM image processing
Image processing began with frame alignment and dose-weighting of the image stacks using the CPU-based implementation of MotionCor2 (ref. 34) in Relion 3.0 (ref. 35). Estimation of contrast transfer function (CTF) for each non-dose-weighted micrograph was determined by Gctf36 version 1.06. After visual inspection of the micrographs and their power spectra, 3,334 were selected for further processing.
Approximately 3,000 particles were manually picked in Relion 3.0 (ref. 35), and extracted from a 4× binned data set with a pixel size of 4.24 Å. These data produced an initial set of two-dimensional (2D) classes that were used as templates to select 1,048,241 particles automatically, all of which were subsequently imported into cryoSPARC version 2 (ref. 37) for extensive 2D classification. After elimination of unfit classes, a total of 312,840 particles from the high-quality 2D classes were combined to produce an ab initio three-dimensional (3D) reference in cryoSPARC version 2 (ref. 37). On the basis of the ab initio model, particles were re-extracted at full scale from the unbinned data set with a pixel size of 1.06 Å in Relion 3.0 (ref. 35), and re-introduced into cryoSPARC version 2 (ref. 37) for 3D refinement. Homogeneous refinement of the ab initio 3D model against the unbinned set of particles yielded a density map with nominal resolution of 3.6 Å according to the Fourier shell correlation (FSC) = 0.143 gold standard criterion38. Heterogenous refinement of multiple models obtained before and after homogeneous refinement allowed removal of additional poor-quality particles and reduced the particle count to 233,737. Nonuniform refinement then improved the resolution to 3.5 Å. At this point, CTF refinement and Bayesian polishing were conducted in Relion 3.0 (ref. 35), followed by an additional round of nonuniform refinement in cryoSPARC version 2 (ref. 37), further improving the resolution to 3.3 Å.
Although the transmembrane domains of the GABAB receptor exhibit pseudo twofold symmetry, bulky carbohydrate densities that are visible only in the ECD of the GABAB1b subunit resulted in sufficient low-resolution asymmetry to prevent particle misalignment. These densities include partial carbohydrate densities attached to Asn 323 and Asn 365 of GABAB1b that do not have counterparts in GABAB2.
The global density map exhibited directional anisotropy39 that is caused by interdomain movement about a flexible linker. To eliminate the adverse effect of such movement on map quality, we performed local refinement of the ECD and transmembrane domains of the GABAB receptor independently. A mask was created covering each region, and the nonuniform refinement algorithm was used as implemented in cryoSPARC version 2 (ref. 37). The resulting reconstructions for the individual ECD and transmembrane domains reached 3.1 Å and 3.4 Å resolution, respectively. A composite map was generated in UCSF Chimera40 by taking the maximum values pointwise from the two locally refined maps after alignment to the global reconstruction (‘vop maximum’ command in UCSF Chimera40). This composite map was used for subsequent model building and refinement.
Resolutions of cryo-EM reconstructions were determined using a cutoff value of 0.143 in gold standard half-map FSC curves38.
We carried out 3D variability analysis41 in cryoSPARC version 2 (ref. 37) using the 233,737 particles from global nonuniform refinement as input. Calculations were performed for the entire receptor, the ECDs and the transmembrane domains, respectively. In each case, multiple modes of variability were solved, and represented as eigenvectors along which conformational changes occur. To visualize the transformation of density, we calculated five reconstructions along each eigenvector, with a filter resolution of 4.5 Å. A movie that combines these reconstructions as frames was generated in Chimera40 for each dimension of motion.
Model building and refinement
Model building was carried out in COOT42. The crystal structure of a complex of human GABAB1b VFT and GABAB2 VFT in the apo form (Protein DataBank code 4MQE) was used as the initial model for the extracellular domain of the receptor. The VFT modules of GABAB1b and GABAB2 were separately placed into density as rigid bodies. Individual residues were then adjusted to optimize the fit. The linker and transmembrane domain of each subunit were traced de novo based on the density. The final model contained residues 48–368, 377–576 and 588–747 of GABAB1b, and 54–294, 302–376, 385–584 and 595–749 of GABAB2.
In addition to the polypeptide chains, we built models for a Ca2+ ion in the extracellular domain of GABAB1b, and for one phospholipid (PE 38:5 in GABAB1b and PC 38:2 in GABAB2) within the transmembrane domain of each subunit. Density for carbohydrate was observed at three N-linked glycosylation sites on GABAB1b (Asn 365, Asn 385 and Asn 397) and one site on GABAB2 (Asn 404). An N-glucosamine residue was modelled at each of these glycosylation sites. Continuous density was also identified for ten cholesterol or CHS molecules surrounding the transmembrane domains of both GABAB subunits. Cholesterols were modelled to optimize the fit with density; however, these densities may belong to CHS molecules with disordered parts. Density for an endogenous ligand was found at the interdomain cleft of the GABAB1b VFT. Although this density could be fit with GABA, it was not modelled because the origin and identity of the ligand remained ambiguous without confirmation by an independent method.
The entire structure was refined by the real-space refinement algorithm and validated with the comprehensive validation application as implemented in Phenix43. Ramachandran statistics were calculated using MolProbity44. The refined model also has an overall EMRinger45 score of 2.7, while the extracellular and transmembrane domains have scores of 3.4 and 1.8, respectively. The final model revealed that VFT and transmembrane components are related to their counterparts in the other subunit by 177° and 179° rotations about the vertical axis, respectively.
Pairwise structural alignment was performed using LSQMAN46. Figures were generated using Pymol Molecular Graphics System version 2.3 (Schrödinger), UCSF Chimera40 and UCSF ChimeraX47. Software installation support was provided by SBGrid48.
Scintillation proximity assay
Binding of [3H]GABA (60 Ci mmol−1; American Radiolabelled Chemicals, St. Louis, MO) to the GABAB receptor was measured with a scintillation proximity assay (SPA)49,50. Purified GABAB1b(1–802)–GABAB2(1–819) complex (100 ng) was extensively dialysed against the purification buffer to remove any residual endogenous ligand. The dialysed protein was then immobilized to yttrium silicate (YSi) protein A SPA beads (62.5 μg; PerkinElmer, Waltham, MA) using anti-Flag M2 antibody (12.5 pg; Millipore Sigma, St. Louis, MO), and incubated at 4 °C for 30 min in the same buffer as used for the final step of protein purification (50 mM Hepes, pH 7.5, 50 mM NaCl, 0.002% LMNG, 0.004% CHS). Increasing concentrations (ranging from 0.2 μM to 25 μM) of [3H]GABA (2.5 Ci mmol−1 final specific radioactivity) were added to the protein/antibody/SPA-bead mixture, and the samples were allowed to reach equilibrium at 4 °C for 16 h. A reaction performed with an antibody/SPA-bead mixture in the absence of GABAB receptor was used to determine the nonproximity signal originating from nonspecific interaction between the radioligand and SPA beads.
All samples were counted in a Microbeta counter (PerkinElmer, Waltham, MA) in counts per minute (c.p.m.) in the SPA mode. The efficiency of detection was calculated with a standard curve of known [3H]GABA concentrations, and this was used to transform c.p.m. into pmol. Specific binding was determined by subtracting the nonproximity signal (nonspecific binding) from the total binding signal and was plotted as a function of free radioligand concentration. Nonlinear regression fitting of the data was performed in SigmaPlot 13.0 to obtain the dissociation constant (Kd) and the molar ratio of GABA-to-receptor binding.
Identification of phospholipid ligands and GABA
Identification of bound endogenous lipids was conducted as described51 with modifications. Briefly, intact GABAB and control CaS receptor were digested with trypsin overnight at 37 °C. Digested proteins were dried and extracted with 1 ml of ice-cold methanol:water (9:1, v/v). The supernatants were dried and resuspended with methanol:toluene (9:1, v/v) to an equivalent concentration of 2 μM. For LC-MS/MS analysis52, the lipid extracts were separated on a Waters Acquity UPLC CSH C18 column (100 × 2.1 mm; 1.7 μm) (Waters, Milford, MA) coupled to an Acquity UPLC CSH C18 VanGuard precolumn (5 × 2.1 mm; 1.7 μm) (Waters). The column was maintained at 65 °C at a flow rate of 0.6 ml min−1. The mobile phases consisted of A, acetonitrile:water (60:40, v/v) with ammonium formate (10 mM) and formic acid (0.1%), and B, 2-propanol:acetonitrile (90:10, v/v) with ammonium formate (10 mM) and formic acid (0.1%). The 15-min separation was conducted under the following gradient: 0 min 15% B; 0−2 min 30% B; 2−2.5 min 48% B; 2.5−11 min 82% B; 11−12 min 99% B; 12−15 min 15% B. A Q Exactive HF mass spectrometer (Thermo Fisher Scientific, Waltham, MA) was operated in electrospray ionization (ESI) in positive mode with the following parameters: mass range 100−1,500 m/z; spray voltage +3.6 kV; sheath gas (nitrogen) flow rate 60 units; auxiliary gas (nitrogen) flow rate 25 units; capillary temperature 320 °C; full scan MS1 mass resolving power 60,000; data-dependent MS/MS acquisition (dd-MS/MS) four scans per cycle; dd-MS/MS mass resolving power 15,000. The mass features that were differentially higher in the GABAB receptor were subjected to targeted MS/MS in reinjections to acquire tandem mass spectra. Thermo Xcalibur version 4.0.27.19 was used for data acquisition. Data processing and identification were performed in MS-DIAL version 3.40. Identification was conducted by matching accurate mass, tandem mass spectra and chromatographic retention time with built-in lipid library LipidBlast53. The identified endogenous lipids bound to GABAB receptor include phosphatidylcholine (PC) 38:2 (PC(18:1_20:1); International Chemical Identifier (InChI) key: QLEJPADMSQQACL-WWUFLCHTSA-N), and phosphatidylethanolamine (PE) 38:5 (PE(18:1_20:4); InChIKey VFUVYNGTMNUBMF-ZRVIQYDLSA-N). Although many isoforms exist for these lipids, both phospholipids share two long-chain fatty acyl moieties of 18 and 20 carbons based on mass spectrometry fragmentation pattern and biological relevance.
Identification of GABA was conducted with targeted LC-MS/MS. Briefly, cell supernatant, cell lysate, culture media and lysis buffer controls were dried from 1 ml and extracted with 1 ml of ice-cold methanol:water (9:1, v/v). The supernatants were dried and resuspended with 200 μl of acetonitrile:water (8:2, v/v). The extracts were separated on a Waters Acquity UPLC BEH Amide column (150 × 2.1 mm; 1.7 μm) (Waters, Milford, MA) coupled with an Acquity UPLC BEH Amide VanGuard precolumn (5 × 2.1 mm; 1.7 μm) (Waters). The column was maintained at 45 °C at a flow rate of 0.4 ml min−1. Mobile phase A was water with ammonium formate (10 mM) and formic acid (0.125%) while B was acetonitrile:water (95:5, v/v) with ammonium formate (10 mM) and formic acid (0.125%). Separation was conducted using the gradient: 0−2 min 100% B; 2−7.7 min 70% B; 7.7−9.5 min 40% B; 9.5−12.5 min 30% B; 10.25−12.75 100% B, 12.75−16.75 100% B. A Q Exactive HF mass spectrometer was operated with the same parameters as above. A GABA standard was injected along with the samples to confirm its spectrum and retention time. The responses of GABA in samples were normalized to that of GABA standard with known concentration.
Cell surface expression
Full-length human GABAB1b and GABAB2 were each cloned into a pcDNA3.1(+) vector (Life Technologies, Carlsbad, CA), with a Flag tag inserted after the signal peptide of GABAB1b, and a haemagglutinin (HA) tag after the signal peptide of GABAB2. Similar constructs were also generated for the C-terminally truncated GABAB1b(1–802) and GABAB2(1–819). Single mutants of full-length GABAB1b (E673R, E309K and E423R) and GABAB2 (H579E and R714A) were constructed using the QuikChange mutagenesis system (Agilent Technologies, Santa Clara, CA).
The cell surface expression levels of wild-type and mutant GABAB receptor were measured as described17. Briefly, HEK293 T/17 cells (ATCC) were cultured in monolayer in DMEM/F12 media (Life Technology, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) at 37 °C in the presence of 5% CO2. The cells were co-transfected with GABAB1b and GABAB2 plasmids using Lipofectamine 3000 (Life Technologies, Carlsbad, CA). Each GABAB1b and GABAB2 mutant was paired with its wild-type partner. Given that GABAB1b is retained inside the cells unless it is chaperoned by GABAB2, we used the surface expression level of GABAB1b on intact cells to measure the amount of assembled heterodimeric GABAB receptor on the cell surface. The amount of surface GABAB1b protein detected for each pair of constructs was normalized with the cell count in each experiment. The cell surface expression level of each mutant is calculated as a percentage of the wild-type receptor.
After blocking with 1% BSA, the cells were incubated with mouse anti-Flag M1 antibody (MilliporeSigma, St. Louis, MO) as the primary antibody to measure GABAB1b expression, followed by donkey anti-mouse IRDye 800-labelled antibody (Li-Cor Biosciences, Lincoln, NE) as the secondary antibody. Fluorescent signals were measured with an Odyssey Infrared Imager (Li-Cor Biosciences). Each experiment was performed in triplicate.
HEK 293 T/17 cells were purchased from and authenticated by ATCC (catalogue number CRL-11268). Cell morphology was examined for each passage of cells. The cells were certified by ATCC to be free of mycoplasma contamination, but they were not tested again during culturing.
Inositol phosphate measurement
HEK293 T/17 cells were cotransfected with plasmids encoding GABAB1b, GABAB2, and Gαqi5. The Gαqi5 chimaera was constructed by replacing the five C-terminal amino acids of murine Gαq with those of murine Gαi (ref. 18). Exchanging the C-terminal end of Gαq with that of Gαi/o permits it to couple with the GABAB receptor and allows the functional activity of the receptor to be tracked through the activity of phospholipase C (PLC). Control experiments were conducted using cells transfected with an empty pcDNA3.1(+) vector, Gαqi5 alone, or wild-type GABAB1b and GABAB2 in the absence of Gαqi5.
Inositol phosphate accumulation was quantified with the homogenous time-resolved fluorescence (HTRF) IP-one Tb kit (Cisbio Bioassays, Codolet, France), which measures the accumulation of inositol-1-monophosphate (IP1), a metabolite of inositol-1,4,5-triphosphate (IP3). One day after transfection, the cells were stimulated with increasing concentrations of baclofen for one hour at 37 °C. The stimulated cells were lysed, and the native IP1 that had been produced was incubated with a d2 fluorophore-labelled IP1 analogue (the acceptor) to compete for binding to an Eu Cryptate-coupled anti-IP1 monoclonal antibody (the donor). Fluorescence data were collected at 620 nm and 665 nm with a PHERAstar FS plate reader (BMG Labtech, Cary, NC) after laser excitation at 320 nm. The fluorescence resonance energy transfer (FRET) signal was calculated as the fluorescence ratio (665 nm/620 nm) and is inversely proportional to the concentration of native IP1 produced following GABAB activation through a chimaeric Gαqi5 G protein. The agonist-induced receptor response of each mutant was calculated as a percentage of the maximum activity of wild-type receptor relative to the activity observed for Gαqi5 alone. Basal activity was determined in the absence of baclofen stimulation and calculated similarly to the agonist-dependent receptor response. Data analysis was performed using the nonlinear regression algorithms in Prism (GraphPad Software, San Diego, CA). Data points represent averages ± s.e.m. of multiple experiments, each consisting of quadruplicate measurements.
Application of a known antagonist, CGP54626, reduced agonist potency as expected. In addition, the compound lowered the basal activity of the GABAB receptor, indicating that it has inverse agonist activity, as previously reported54. Therefore, we refer to the compound as an inverse agonist.
Inductively coupled plasma mass spectrometry
Purified GABAB receptor (200 μl, 11.1 μg μl−1 or 57.7 μM) was collected in metal-free tubes and digested overnight by adding 1 ml of concentrated nitric acid (HNO3) (Fisher Scientific, Hampton, NH; Optima grade). The digested protein was then diluted to a total volume of 10 ml with 8.7 ml of deionized water supplemented with 500 μg l−1 of gold (Au) and 100 μl of an internal standard solution containing 500 μg l−1 each of gallium (Ga) and rhodium (Rh) in 2% HNO3. The protein purification buffer (200 μl), containing 10 mM HEPES, pH 7.5, 50 mM NaCl, 0.002% LMNG and 0.0004% CHS, was similarly mock digested and diluted for analysis.
Inductively coupled plasma mass spectrometry (ICP-MS) was conducted using a NexION 350S ICP-MS instrument (Perkin Elmer, Waltham, MA) equipped with dynamic reaction cell (DRC) feature and a SC-4 DX FAST Autosampler (Elemental Scientific, Omaha, NE). The DRC-ICP-MS experimental method was developed from published procedures55,56 and a laboratory protocol for multi-element DCR-ICP-MS from the Centers for Disease Control (CDC) (https://www.cdc.gov/nchs/data/nhanes/nhanes_13_14/UM_UMS_UTAS_UTASS_H_MET.pdf). The concentrations of magnesium (Mg), calcium (Ca), manganese (Mn), iron (Fe), cobalt (Co), nickel (Ni), copper (Cu), zinc (Zn) and strontium (Sr) in the digested protein and buffer samples were measured. Data points represent averages ± coefficient of variance from eight measurements within two experiments.
One multi-element calibration standard was prepared from concentrated single-element stocks, and used for instrument calibration. The calibration standard was diluted to various concentrations using a solution containing 10% HNO3 and 500 μg l−1 of Au to cover the expected concentration range of each analyte in the protein sample: 0.01, 0.02, 0.05, 0.1, 0.2 μg l−1 for Co and Sr; 0.05, 0.1, 0.25, 0.5, 1.0 μg l−1 for Mn and Ni; 0.25, 0.5, 1.25, 2.5, 5.0 μg l−1 for Mg and Cu; 0.5, 1.0, 2.5, 5.0, 10.0 μg l−1 for Fe and Zn; and 2.5, 5.0, 12.5, 25, 50 μg l−1 for Ca.
The instrument was also calibrated against a set of blank solutions, including commercially available quality controls containing digested hair samples from Public Health Expertise and Reference Center, Quebec (INSPQ, Quebec, Canada), and a water sample containing a broad range of metals from the National Institute of Technology (NIST, Gaithersburg, MD).
Special attention was paid to correction for matrix-induced interferences. Matrix suppression was compensated by the selection of suitable internal standards, which were matched to masses and, if possible, to ionization properties of the analytes. The internal standards were added to each calibration standard and quality control sample to the same final concentration as that in the protein sample and buffer (5 μg l−1 each of Ga and Rh). The elements Mg, Ca, Mn, Fe, Co, Ni, Cu, and Zn were corrected with Ga, while Sr was corrected with Rh. Polyatomic interferences were suppressed with the instrument’s DRC technology feature, using ammonia as a second gas for Mn and Fe, while Mg, Ca, Sr, Co, Ni, Cu, and Zn were measured in standard mode without a second gas.
Matrix-assisted laser desorption/ionization
Purified GABAB1b(1–802)–GABAB2(1–819) complex (0.3 mg ml−1, 1 μl) was mixed with 1 μl sinapinic acid matrix solution (Bruker Daltonics, Billerica, MA) containing 10 mg of sinapinic acid in 1 ml of 2.5% trifluoroacetic acid (MilliporeSigma, St. Louis, MO) and 50% acetonitrile (MilliporeSigma). The protein–matrix suspension (2 μl) was added to the ground-steel matrix-assisted laser desorption/ionization (MALDI) target plate and dried at room temperature. Mass spectra were collected using a UltrafleXtreme MALDI time-of-flight (TOF)/TOF mass spectrometer (Bruker Daltonics) operated with FlexControl software in linear positive mode—that is, using a mass range of 30,000–120,000 daltons. The instrument was externally calibrated with a Proteins MALDI-MS calibration kit (MilliporeSigma, St. Louis, MO). Each individual mass spectrum was analysed and adjusted for smoothness and baseline using FlexAnalysis software 3.0 (Bruker Daltonics). We determined the molecular mass of the heterodimeric GABAB1b(1–802)–GABAB2(1–819) complex to be 192,647.967 daltons.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data are included in this paper and its Supplementary Information. Cryo-EM density maps of the GABAB receptor have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/) under accession code EMD-21685. Atomic coordinates for the GABAB receptor structure have been deposited in the RCSB Protein Data Bank under accession code 6WIV. Raw cryo-EM images have been deposited in the Electron Microscopy Public Image Archive (https://www.ebi.ac.uk/pdbe/emdb/empiar/) under accession code EMPIAR-10410.
Change history
15 July 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41586-020-2543-y
References
Bettler, B., Kaupmann, K., Mosbacher, J. & Gassmann, M. Molecular structure and physiological functions of GABAB receptors. Physiol. Rev. 84, 835–867 (2004).
Jones, K. A. et al. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature 396, 674–679 (1998).
Kaupmann, K. et al. GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature 396, 683–687 (1998).
White, J. H. et al. Heterodimerization is required for the formation of a functional GABAB receptor. Nature 396, 679–682 (1998).
Kuner, R. et al. Role of heteromer formation in GABAB receptor function. Science 283, 74–77 (1999).
Ng, G. Y. et al. Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J. Biol. Chem. 274, 7607–7610 (1999).
Kaupmann, K. et al. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature 386, 239–246 (1997).
Malitschek, B. et al. The N-terminal domain of γ-aminobutyric acidB receptors is sufficient to specify agonist and antagonist binding. Mol. Pharmacol. 56, 448–454 (1999).
Galvez, T. et al. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J. 20, 2152–2159 (2001).
Margeta-Mitrovic, M., Jan, Y. N. & Jan, L. Y. Function of GB1 and GB2 subunits in G protein coupling of GABAB receptors. Proc. Natl Acad. Sci. USA 98, 14649–14654 (2001).
Robbins, M. J. et al. GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J. Neurosci. 21, 8043–8052 (2001).
Duthey, B. et al. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABAB receptor. J. Biol. Chem. 277, 3236–3241 (2002).
Havlickova, M. et al. The intracellular loops of the GB2 subunit are crucial for G-protein coupling of the heteromeric γ-aminobutyrate B receptor. Mol. Pharmacol. 62, 343–350 (2002).
Monnier, C. et al. Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J. 30, 32–42 (2011).
Pin, J. P. & Bettler, B. Organization and functions of mGlu and GABAB receptor complexes. Nature 540, 60–68 (2016).
Geng, Y., Bush, M., Mosyak, L., Wang, F. & Fan, Q. R. Structural mechanism of ligand activation in human GABAB receptor. Nature 504, 254–259 (2013).
Burmakina, S., Geng, Y., Chen, Y. & Fan, Q. R. Heterodimeric coiled-coil interactions of human GABAB receptor. Proc. Natl Acad. Sci. USA 111, 6958–6963 (2014).
Conklin, B. R., Farfel, Z., Lustig, K. D., Julius, D. & Bourne, H. R. Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature 363, 274–276 (1993).
Xue, L. et al. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 10, 2765 (2019).
Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019); correction 567, E10 (2019).
Geng, Y. et al. Structural mechanism of ligand activation in human calcium-sensing receptor. eLife 5, e13662 (2016).
Wise, A. et al. Calcium sensing properties of the GABAB receptor. Neuropharmacology 38, 1647–1656 (1999).
Galvez, T. et al. Ca2+ requirement for high-affinity gamma-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol. Pharmacol. 57, 419–426 (2000).
Hanson, M. A. et al. Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 (2012).
Rasmussen, S. G. et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Thal, D. M., Glukhova, A., Sexton, P. M. & Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018).
Palczewski, K. et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 (2000).
Doré, A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014).
Wu, H. et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64 (2014).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protocols 9, 2574–2585 (2014).
Margeta-Mitrovic, M., Jan, Y. N. & Jan, L. Y. A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27, 97–106 (2000).
Pagano, A. et al. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J. Neurosci. 21, 1189–1202 (2001).
Reeves, P. J., Callewaert, N., Contreras, R. & Khorana, H. G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl Acad. Sci. USA 99, 13419–13424 (2002).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Scheres, S. H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Punjani, A. & Fleet, D. J. 3D variability analysis: directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images. bioRxiv https://doi.org/10.1101/2020.04.08.032466 (2020).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Novotny, M., Madsen, D. & Kleywegt, G. J. Evaluation of protein fold comparison servers. Proteins 54, 260–270 (2004).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013).
Geng, Y. et al. Structure and functional interaction of the extracellular domain of human GABAB receptor GBR2. Nat. Neurosci. 15, 970–978 (2012).
Quick, M. & Javitch, J. A. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc. Natl Acad. Sci. USA 104, 3603–3608 (2007).
Gupta, K. et al. Identifying key membrane protein lipid interactions using mass spectrometry. Nat. Protocols 13, 1106–1120 (2018).
Mafu, S. et al. Biosynthesis of the microtubule-destabilizing diterpene pseudolaric acid B from golden larch involves an unusual diterpene synthase. Proc. Natl Acad. Sci. USA 114, 974–979 (2017).
Kind, T. et al. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 10, 755–758 (2013).
Mukherjee, R. S., McBride, E. W., Beinborn, M., Dunlap, K. & Kopin, A. S. Point mutations in either subunit of the GABAB receptor confer constitutive activity to the heterodimer. Mol. Pharmacol. 70, 1406–1413 (2006).
Chen, K. L. B., Amarasiriwardena, C. J. & Christiani, D. C. Determination of total arsenic concentrations in nails by inductively coupled plasma mass spectrometry. Biol. Trace Elem. Res. 67, 109–125 (1999).
Pruszkowski, E., Neubauer, K. & Thomas, R. An overview of clinical applications by inductively coupled plasma mass spectrometry. Atomic Spectroscopy 19, 111–115 (1998).
Hollenstein, K. et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013).
Wang, C. et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343 (2013).
Acknowledgements
We thank R. Henderson for early-stage cryo-EM investigation and critical reading of the manuscript; C. S. Zuker for advice and financial support; C. Karan and R. Realubit for assistance with the EnVision plate reader at Columbia Genome Center; A. Sobolevsky, K. Saotome and E. Cao for BacMam vectors; Y. H. Wong for a Gαqi5 chimera plasmid; and B. K. Kobilka and B. Skiniotis for advice. Titan Krios data collection was performed at the Simons Electron Microscopy Center, directed by B. O. Carragher and C. Potter, supported by grants from the Simons Foundation (SF349247), NYSTAR and the National Institutes of Health (NIH; GM103310). This work was supported by NIH grants R01GM088454 (to Q.R.F.), R01GM125801 (to Q.R.F., P.A.S. and M.Q.), R01GM107462 (to W.A.H.), P41GM116799 (to W.A.H.), and U2C ES030158 for lipid identification (to O.F.). Q.R.F. was an Irma Hirschl Career Scientist, Pew Scholar, McKnight Scholar and Schaefer Scholar.
Author information
Authors and Affiliations
Contributions
J.P., J.L., Q.R.F. and K.M.R. cultured cells and purified protein; Z.F. and A.F. prepared cryo-EM grids; Z.F., A.F., Q.R.F. and O.B.C. collected cryo-EM data; Z.F. and A.F. performed initial image processing; O.B.C. processed cryo-EM data to high resolution; L.M. and Q.R.F. built and refined models; Q.R.F., L.M., A.F., J.P. and Z.F. analysed structures; T.S. and O.F. identified phospholipids and GABA by mass spectrometry; V.N.S. and J.G. conducted elemental analysis; Q.R.F, J.L., J.P., A.F., J.T., X.L. and J.P.W. performed mutagenesis and cell-based functional assays; M.Q. performed and analysed radioligand binding studies; B.C, Y.G., H.Z. and Y.K. generated expression plasmids and carried out early protein purification trials; R.G., W.J.R., E.T.E., R.K.H. and Z.Y. assisted with cryo-EM data collection; S.C., Z.L., W.J.R. and E.T.E. performed initial cryo-EM characterization; R.K.S. measured molecular mass; B.K. screened detergents; W.A.H. contributed to structural analysis; Q.R.F., P.A.S. and J.A.J. supervised functional analyses; J.F., O.B.C. and H.Y. supervised cryo-EM studies; Q.R.F. and A.F. wrote the paper; T.S., V.N.S., M.Q., Z.F. and R.K.S. contributed Methods sections; all authors contributed to revision of the manuscript; Q.R.F., A.F., J.P., O.B.C., T.S., Z.F., J.L. and M.Q. prepared figures; Q.R.F. conceived and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Ryan Hibbs, Bernhard Bettler and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Purification and functional analysis of the human GABAB receptor.
a, Superose 6 size-exclusion chromatography profile of detergent-purified GABAB1b(1–802)–GABAB2(1–819) complex. b, SDS PAGE gel of the size-exclusion peak fraction from a under reducing conditions. For gel source data, see Supplementary Fig. 2. c, Dose-dependent [3H]GABA binding to purified GABAB1b(1–802)–GABAB2(1–819) complex, reaching maximum molar ratios of GABA-to-receptor binding at 0.98 ± 0.03 mol mol−1. Each data point represents the mean of triplicate measurements from a typical experiment. The experiment was repeated four times with similar results. Data were subjected to nonlinear regression fitting, and the dissociation constant (Kd = 1.3 ± 0.16 µM) is reported as means ± s.e.m. of the fit. d, Functional analysis comparison of full-length and truncated WT GABAB receptor. Shown is the dose-dependent baclofen-stimulated receptor response in cells transiently expressing Gαqi5 (abbreviated as Gqi) with full-length GABAB heterodimer or the C-terminally truncated GABAB1b(1–802)–GABAB2(1–819) complex. Cells transfected with Gαqi5 alone were used as a negative control. Relative agonist-stimulated activity was measured by IP1 accumulation, and is expressed as a percentage of maximum wild-type activity induced by baclofen relative to the activity of Gαqi5 alone. Data points represent averages ± s.e.m. of multiple experiments (n), each with quadruplicate measurements. Cell surface expression level was 106% for the GABAB1b(1–802)–GABAB2(1–819) complex in comparison with the full-length WT/WT heterodimer.
Extended Data Fig. 2 Cryo-EM imaging of human GABAB receptor.
a, Workflow of cryo-EM data processing. b, A representative motion-corrected cryo-electron micrograph of the GABAB receptor. c, Reference-free 2D class averages, highlighting clear density for transmembrane helices. d, Global density map, coloured according to local resolution, in full and clipped views perpendicular to the plane of the membrane. e, Global FSC curve (purple) corrected by high-resolution noise substitution. The overall resolution as determined by an FSC cut-off value of 0.143 (blue line) is 3.3 Å. f, 3D FSC curves, measuring directional resolution anisotropy. Plots show the global half-map FSC (red solid line; left-hand y-axis), together with the spread of directional resolution values within ± 1 standard deviation of the mean (area encompassed by green dashed lines), and a histogram (turquoise; right-hand y-axis) of 100 such directional resolution values sampled evenly over the 3D FSC threshold value of 0.143 (blue line). The sphericity value reported by 3D FSC is 0.958 out of 1. g, h, Separate FSC curves for the locally refined reconstructions of ECDs g) and transmembrane domains (h). The blue lines mark the resolution corresponding to an FSC value of 0.143 (ECD, 3.1 Å; transmembrane region, 3.4 Å). The adjacent diagrams show the masks (translucent surface) used for each local refinement.
Extended Data Fig. 3 Structural model of the GABAB receptor fit within the cryo-EM map.
a–d, Cryo-EM density maps and refined models for the LB1 interface helices (H–B and H–C) in the extracellular VFT (a), the linker between the VFT and transmembrane domains (b), all seven transmembrane helices of the GABAB1b subunit (c), the ten modelled transmembrane cholesterols (d) and all seven transmembrane helices of the GABAB2 subunit (e). The density map is a composite of the locally refined reconstructions for the ECD and transmembrane domains. The N- and C-terminal residues of each helix are labelled.
Extended Data Fig. 4 Architecture of the GABAB receptor.
a, Structure of the GABAB receptor in four views related by 90° rotations about an axis perpendicular to the membrane. GABAB1b (blue) and GABAB2 (green) are rendered as ribbons; Ca2+ is shown as a green sphere; phospholipids (PE 38:5 and PC 38:2) are presented as yellow space-filling models; the observed N-linked glycans (NAGs) and cholesterols (CLRs) are shown respectively as grey and pink ball-and-stick models. Transmembrane helices 1–7, along with N and C termini, are marked for each subunit. b, Cryo-EM density map of the GABAB receptor, in the same orientations and colour scheme as in a. The map is composed of local reconstructions for the ECD and transmembrane domain, which were independently refined to 3.1 Å and 3.4 Å, respectively. c, Linker and transmembrane domain of the GABAB receptor viewed from the extracellular and intracellular sides.
Extended Data Fig. 5 Heterodimer conformation and interface features of the GABAB receptor.
a, b, Cryo-EM structure of near full-length GABAB receptor (cyan) superimposed with the crystal structure of its extracellular VFT module in the inactive state (PDB code 4MQE; purple) (a) or active state (PDB code 4MS3; red) (b). The middle panel shows the heterodimeric receptor structures superimposed based on the LB1 domain of the GABAB1b subunit. The two side panels show superpositions of individual GABAB1b and GABAB2 subunits on the basis of their respective LB1 domains. In b, the green line denotes the axis of rotation that relates the LB2 domains of near full-length and VFT structures of GABAB1b (rotation χ = 28°; screw translation τχ = 0.6 Å), or near full-length and VFT structures of GABAB2 (rotation χ = 7°; screw translation τχ = 0.3 Å). c, d, Extracellular LB2 domains viewed from the C-terminal end. Superposition of near full-length (cyan) and VFT structures (c, inactive state, purple; d, active state, red) based on the LB1 domain of the GABAB1b subunit. Within each heterodimeric complex, the C termini of the LB2 domains in the GABAB1b and GABAB2 subunits are shown as spheres, and the distance between the two C termini is marked by dotted lines. e, Cryo-EM structure of full-length mGlu5 in the inactive (PDB code 6N52) and active (PDB code 6N51) conformations20. f, Molecular surface of the GABAB1b–GABAB2 complex, showing the plane of the heterodimer interface for the extracellular and transmembrane domains. Structural elements involved in heterodimer formation are highlighted in cartoon format (ectodomain, H–B and H–C helices; transmembrane domain, TM5 and TM3 helices). g, The GABAB transmembrane domain viewed from the extracellular side, comparing the locations of core (I, II, IIIa, IIIb) versus peripheral cholesterol-mediated heterodimer contacts from different layers. Heterodimer contacts mediated by two cholesterols (CLR6 and CLR3) are shown at the bottom.
Extended Data Fig. 6 Extracellular ligand binding in GABAB1b.
a, b, Molecular surface (a) and ribbon representation (b) of the GABAB1b subunit, showing the location of the Ca2+-binding site and an unmodelled density at the interdomain crevice of VFT. c, d, Functional analysis of the impact of endogenous Ca2+. Basal activity (c) and dose-dependent baclofen-stimulated receptor response (d) in cells transiently expressing the Gαqi5 subunit (abbreviated as Gqi) with different combinations of WT and mutant GABAB-receptor subunits (GABAB1b E309K, abbreviated as E309K; GABAB1b E423R, abbreviated as E423R). IP1 accumulation by the WT/WT heterodimer was measured in the presence and absence of 2.5 mM EGTA. Cells transfected with Gαqi5 alone were used as negative controls. Relative activity in both graphs is expressed as a percentage of the maximum wild-type activity induced by baclofen relative to the activity of Gαqi5 alone. Data points represent averages ± s.e.m. of multiple experiments (n), each with quadruplicate measurements. **P = 0.0016, ***P = 0.0002, ****P < 0.0001; one-way ANOVA with Bonferroni’s post hoc test was used to calculate statistical differences in basal activity (c). Cell surface expression level was 107% for E309K/WT and 87% for E423R/WT mutants in comparison with the WT/WT heterodimer. e, Fitting of GABA into the extra density (contoured at 7.0σ) at the orthosteric ligand-binding site and its potential interaction with GABAB1b. f, Concentration of endogenous GABA in the supernatant and lysate of HEK 293 GnTI– cells after recombinant expression of the GABAB receptor, as well as cell culture medium and lysis buffer controls, as detected by mass spectrometry.
Extended Data Fig. 7 Endogenous phospholipid-binding sites of the GABAB receptor.
a, b, Ribbon representation of the GABAB1b (a) and GABAB2 (b) transmembrane domains, highlighting the cryo-EM density for phospholipids contoured at 4.0σ. Phospholipids are rendered in ball-and-stick representation. c, d, Electrostatic potential surface of the lipid-binding pocket in GABAB1b (c) and GABAB2 (d). The phospholipids are shown as spheres. Charged residues that directly contact the phosphate group of each lipid are marked. e–h, Comparison of phospholipids bound to GABAB subunits with ligands bound to the class A GPCRs rhodopsin27 (PDB code 1F88) and S1P1 receptor24 (PDB code 3V2Y) (e), the class B GPCR corticotropin-releasing factor receptor 1 (CRF1; ref. 57; PDB code 4K5Y) (f), the class C GPCRs mGlu1 (ref. 29; PDB code 4OR2) and mGlu5 (ref. 28; PDB code 4OO9) (g), and the class F GPCR Smoothened58 (PDB code 4JKV) (h). In each panel, the Cα trace of the GABAB1b linker and transmembrane domain is shown in two orthogonal views in grey, and the superimposed GABAB ligands PE 38:5 and PC 38:2 are shown as blue and green stick models, respectively. Various GPCRs were overlapped onto the transmembrane domain of GABAB1b to bring their bound ligands into superposition. i, j, Schematic diagrams of the specific contacts between GABAB1b and PE 38:5 (i), and between GABAB2 and PC 38:2 (j). Selected contacts between residues and phospholipids are highlighted; hydrogen bonds, red dotted lines; hydrophobic contacts, black wiggled lines; polar interactions, green curved lines; pi-stacking interactions, orange box wave. Red zigzags indicate contacts that are mediated by main-chain atoms. Lipid-interacting residues that are conserved in the two subunits are highlighted in bold and include: (1) head group (GABAB1b, His 643 and Arg 5493.32; GABAB2, His 647 and Arg 5563.32); (2) 20-carbon fatty acyl chain (GABAB1b, Phe 5573.40, Tyr 6575.44 and Ala 7036.54; GABAB2, Tyr 5643.40, Tyr 6615.44 and Ala 7076.54); (3) 18-carbon fatty acyl chain (GABAB1b, Ile 7247.36; GABAB2 Ile7287.36).
Extended Data Fig. 8 Endogenous phospholipid interactions with GABAB receptor.
a, c, e, Orthogonal views of a potential access channel in GABAB1b, GABAB2 and S1P1 in molecular surface representation, along with phospholipids PE 38:5, PC 38:2 or the sphingolipid mimic ML056, in space-filling representation. Side views (left) show an opening between helices TM5 and TM6 in GABAB1b (a) and GABAB2 (c), and between TM1 and TM7 in S1P1 (e), while top views (right) highlight the blocked entrance to the lipid-binding pocket from the extracellular side. In all cases, ECL1 and ECL3 (orange), ECL2 (pale brown), the linker of GABAB-receptor subunits (pink) and the N-terminal helix of S1P1 (pink) are distinguished by colour. b, d, f, The same information as in a, c, e but with ribbon models for GABAB1b (b), GABAB2 (d), and S1P1 (ref. 24) (PDB code 3V2Y) (f). Lipids are in stick format. g, h, Functional effect of a GABAB2 lipid-binding site mutation. Basal activity (g) and dose-dependent baclofen-stimulated receptor response (h) in cells transiently expressing Gαqi5 (Gqi) with WT GABAB receptor or WT GABAB1b and mutant GABAB2 R714A (R714A) heterodimer. Cells transfected with Gαqi5 alone were used as negative controls. Relative activity in both graphs was measured by IP1 accumulation, and expressed as a percentage of the maximum wild-type activity induced by baclofen relative to the activity of Gαqi5 alone. Data points represent averages ± s.e.m. of multiple experiments (n), each with quadruplicate measurements. **P = 0.0016; one-way ANOVA with Bonferroni’s post hoc test was used to calculate statistical differences in basal activity (g). Cell surface expression level was 77% for the WT/R714A mutant in comparison with the WT/WT heterodimer.
Extended Data Fig. 9 Comparison of the GABAB transmembrane domain with other GPCRs.
a, Superposition of GABAB1b and GABAB2 subunits based on their VFT modules. The short purple line denotes the axis of rotation that relates the linker and transmembrane domains of GABAB1b and GABAB2 (rotation χ = 23°; screw translation τχ = 0.01 Å). b, Superposition of the linker and transmembrane domains of the GABAB1b and GABAB2 subunits. c, Superposition of the linker and transmembrane domains of each GABAB subunit with the class C GPCR mGlu5 (ref. 20; PDB code 6N52) in three different views, with arrows revealing inward extracellular shifts in TM5 and TM7, as well as an inward intracellular shift in TM3, in mGlu5 compared with either GABAB subunit. d–f, Superposition of the transmembrane helices of each GABAB subunit with the class A GPCR rhodopsin27 (PDB code 1F88) (d), the class B GPCR CRF1 (ref. 57; PDB code 4K5Y) (e), and the class F GPCR Smoothened58 (PDB code 4JKV) (f). Arrows indicate large shifts in transmembrane helix positions between GABAB subunits and other GPCRs, such as outward extracellular movements in class A (TM2 and TM6), class B (TM1, TM2 and TM7), and class F (TM2, TM4 and TM5). There are also inward intracellular shifts in TM7 in all comparisons, and outward intracellular shifts in TM5 in class A, B and F.
Extended Data Fig. 10 Conserved motifs in GABAB, rhodopsin and mGlu receptors.
a, b, The ‘ionic lock’ and FxPKxx motifs in the GABAB1b (a) and GABAB2 (b) subunits. The ‘ionic locks’ consist of Asp 684 of ICL3 and Lys 5673.50 in GABAB1b, and Asp 688 of ICL3 and Lys 5743.50 in GABAB2. The FxPKxx motifs include the conserved Lys 7397.51 of GABAB1b and Lys 7437.51 of GABAB2, which interact with the ‘ionic locks’ through Asn2.39 (GABAB1b N5132.39; GABAB2 N5202.39) and a serine (GABAB1b S508; GABAB2 S515) in ICL1. c, The ‘ionic lock’ and NPxxY motifs in class A rhodopsin27. d, e, The ‘ionic lock’ and FxPKxY motifs in the class C receptors mGlu1 (ref. 29; d) and mGlu5 (ref. 28; e), which are in close proximity as in GABAB subunits. Key residues of the motifs are displayed as stick models. Hydrogen bonds are indicated by black dotted lines. Interactions between participating residues of the ‘ionic lock’ are denoted by red dotted lines with distances labelled. Distances between the Cα atoms of the ‘ionic lock’ residues are marked by brown dotted lines.
Supplementary information
Supplementary Information
This file contains Supplementary Figs. 1-3, Supplementary Tables 1-3, and Supplementary references.
Supplementary Video 1|
Global conformational variability analysis of GABAB receptor linkers. Motion between the ECD and transmembrane domains as revealed by 3D variability analysis of the entire receptor. The ECD and transmembrane domains flex back and forth about the linker region.
Supplementary Video 2|
Global conformational variability analysis of GABAB receptor membrane-proximal domains. Motion between the membrane-proximal LB2 domains of VFT as revealed by 3D variability analysis of the entire receptor. The LB2 domains of VFT approach and withdraw, as do their associated linkers.
Supplementary Video 3|
Local conformational variability analysis of GABAB receptor membrane-proximal domains. Motion between the membrane-proximal LB2 domains of VFT as revealed by 3D variability analysis of the ECD. The LB2 domains of VFT approach and withdraw, as do their associated linkers.
Supplementary Video 4|
Local conformational variability analysis of GABAB receptor transmembrane domains. Motion between the transmembrane domains as revealed by 3D variability analysis of the transmembrane region. The extracellular ends of the transmembrane domains approach and withdraw.
Rights and permissions
About this article

Cite this article
Park, J., Fu, Z., Frangaj, A. et al. Structure of human GABAB receptor in an inactive state. Nature 584, 304–309 (2020). https://doi.org/10.1038/s41586-020-2452-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2452-0
- Springer Nature Limited
This article is cited by
-
Specific pharmacological and Gi/o protein responses of some native GPCRs in neurons
Nature Communications (2024)
-
Pathology of pain and its implications for therapeutic interventions
Signal Transduction and Targeted Therapy (2024)
-
Structural basis of positive allosteric modulation of metabotropic glutamate receptor activation and internalization
Nature Communications (2024)
-
Structure, function and drug discovery of GPCR signaling
Molecular Biomedicine (2023)
-
Filamin A organizes γ‑aminobutyric acid type B receptors at the plasma membrane
Nature Communications (2023)