

Abstract
‘Pioneer’ transcription factors are required for stem-cell pluripotency, cell differentiation and cell reprogramming1,2. Pioneer factors can bind nucleosomal DNA to enable gene expression from regions of the genome with closed chromatin. SOX2 is a prominent pioneer factor that is essential for pluripotency and self-renewal of embryonic stem cells3. Here we report cryo-electron microscopy structures of the DNA-binding domains of SOX2 and its close homologue SOX11 bound to nucleosomes. The structures show that SOX factors can bind and locally distort DNA at superhelical location 2. The factors also facilitate detachment of terminal nucleosomal DNA from the histone octamer, which increases DNA accessibility. SOX-factor binding to the nucleosome can also lead to a repositioning of the N-terminal tail of histone H4 that includes residue lysine 16. We speculate that this repositioning is incompatible with higher-order nucleosome stacking, which involves contacts of the H4 tail with a neighbouring nucleosome. Our results indicate that pioneer transcription factors can use binding energy to initiate chromatin opening, and thereby facilitate nucleosome remodelling and subsequent transcription.
Similar content being viewed by others

Main
Transcription of the human genome is controlled by about 1,600 transcription factors4. Transcription factors recognize DNA motifs and recruit protein complexes that enable transcription initiation5. The binding of most transcription factors is restricted to regions of the genome that are not packaged into chromatin6. Some transcription factors can, however, bind to chromatin via contacts to its fundamental unit, the nucleosome7. These pioneer transcription factors can initiate transcription in silent chromatin regions8, and are required for embryo development, cell differentiation and cell reprogramming9.
SOX2 and OCT4 (also known as POU5F1) are pioneer factors that are widely used for the reprogramming of adult cells to induced pluripotent stem cells2,10,11. SOX2 and OCT4 can interact with nucleosomes in vitro and in vivo12,13. SOX2 alone can direct chromatin opening14 and bind target DNA sites before OCT4 in vivo10, and SOX2 binding to DNA can also follow OCT4 binding in vitro15. Most factors in the SOX family (hereafter, SOX factors) show pioneer factor function7 and are essential for developmental processes1,9. The mutation of SOX factors can lead to severe developmental defects and cancer16. How pioneer transcription factors such as SOX factors bind to the nucleosome, and how they make DNA accessible for their non-pioneer partner proteins, is unknown.
To investigate this, we determined the cryo-electron microscopy (cryo-EM) structure of human SOX2 in complex with a nucleosome (Methods). We used a 147-bp nucleosomal DNA sequence (hereafter referred to as DNA-1) (Extended Data Fig. 1) that was previously selected for binding the closely related factor SOX117. The DNA-binding domains (DBDs) of SOX2 and SOX11 share 83% sequence similarity (Extended Data Fig. 2), and bind the same DNA motif (TTGT)17. Base pairs of the TTGT core motif are specifically contacted by amino acid residues in the RPMNAFMVW signature motif of the SOX-factor HMG box18. SOX2 and SOX11 bind the same target sites in cells, substantially differ only in regions that flank their DBDs, and recruit different factors19,20.
Nucleosomes containing DNA-1 bound recombinant SOX2 or SOX11 DBDs (Extended Data Fig. 3). We added the purified SOX2 DBD in excess to reconstituted nucleosomes, plunge-froze cryo-EM grids and collected cryo-EM data (Methods). A subset of 32,301 particles resulted in an approximately 5.5 Å resolution map that showed extra density on the nucleosome surface (Fig. 1, Extended Data Fig. 4, Extended Data Table 1). We fitted the map with structures of the nucleosome21 and the SOX2 DBD22 (Extended Data Figs. 4, 5).

Structure of the nucleosome–SOX2 complex reveals SOX2 binding at SHL +2. Top and side views are related by a 90° rotation around the dyad (dashed line). Superhelical locations –3 to +3 are labelled. SOX2 DBD is shown in pink; histones H2A, H2B, H3 and H4 are shown in yellow, red, blue and green, respectively; DNA is shown in dark and light blue. h1–h3, helices 1–3.
The nucleosome–SOX2 structure that we obtained revealed a single copy of the SOX2 DBD bound to DNA at superhelical location (SHL) +2 (Fig. 1). The observed SOX binding involves specific interactions with the DNA motif, as confirmed by site-directed mutagenesis of involved residues in SOX11 and by mutagenesis of the DNA-1 sequence (Extended Data Figs. 3, 6). In further agreement with our structure, in vivo23 SOX factors preferentially occupy target sites that are located near the centre of the nucleosome24. Although DNA-1 contains multiple SOX-binding motifs and can bind multiple copies of SOX2, nucleosomes containing DNA-1 bind only one copy of SOX2 (Extended Data Figs. 1, 3). In the context of the DNA-1 sequence, binding of the SOX DBD to other sites on the nucleosome would result in clashes with DNA and histones (Extended Data Fig. 1d).
Despite extensive efforts, the resolution of our nucleosome–SOX2 structure remained limited. We therefore determined the structure of a nucleosome bound to the DBD of SOX11 (Methods). The cryo-EM dataset contained 222,731 particles and resulted in a detailed reconstruction at 3.7 Å resolution (Extended Data Figs. 4, 5). To aid model building, we additionally determined the crystal structure of the SOX11 DBD in complex with a DNA fragment at 2.5 Å resolution (Extended Data Fig. 2c–f, Extended Data Table 2).
The structure of the nucleosome–SOX11 complex was virtually identical to that of the nucleosome–SOX2 complex (Extended Data Figs. 4g, 5). It is also a good model for nucleosome complexes with the DBDs of other members of the SOX family, which are highly conserved (Extended Data Fig. 2). For comparisons, we further determined the structure of the free nucleosome containing DNA-1 from 368,270 particles at 3.2 Å resolution (Extended Data Figs. 4, 5). This structure was highly similar to the canonical nucleosome structure (Protein Data Bank (PDB) code 6FQ5), root mean square deviation (r.m.s.d.) (P) = 1.0 Å).
Comparison of the nucleosome–SOX11 structure with the free nucleosome structure shows that SOX11 binding leads to strong local DNA distortions at SHL +2 (Fig. 2) (local r.m.s.d. (P) = 3.9 Å; calculated for 12 bp of DNA). SOX11 widens the DNA minor groove by 7 Å and pulls the DNA away from the histone octamer by 3–4 Å (coordinate error of approximately 1 Å), which increases DNA bending (Fig. 2). These DNA distortions are induced by SOX11 binding, and are also observed in our SOX11–DNA crystal structure (r.m.s.d. (P) = 1.4 Å, for 12 bp of DNA). Thus, the SOX factor uses binding energy to distort DNA locally, despite competing histone–DNA interactions.

SOX11 is shown in pink, and DNA bound by SOX is shown in dark and light blue. DNA in the free nucleosome structure is shown in dark and light green. The cryo-EM maps shown here were Gaussian-smoothened. For clarity, SOX density was segmented out on the right (blue models). TF, transcription factor.
In both nucleosome–SOX-factor structures, approximately 2.5 turns of both DNA termini are detached from the histone octamer and not visible in the cryo-EM densities (Fig. 3, Extended Data Fig. 5). This is consistent with the observation that several SOX factors facilitate DNA unwrapping from the nucleosome7, and with the known high dynamics of the terminal DNA25,26. A DNA cleavage assay supports the increase in accessibility of the terminal nucleosomal DNA in the presence of SOX11 (Extended Data Fig. 7). Comparison with the free nucleosome structure indicates that terminal DNA at SHL −7, SHL −6 and SHL −5 is detached from the octamer because of a clash with helix 2 of the SOX factor (Fig. 3c, Supplementary Video 1). Thus SOX factor binding to the nucleosome facilitates DNA detachment and increases accessibility of terminal DNA.

a, Nucleosome invasion by SOX factors and terminal DNA detachment. Schematic of the structures reported here. From left to right, free nucleosome, nucleosome–SOX11*, nucleosome–SOX11 and nucleosome–SOX112 are shown. The histone octamer is shown in orange, SOX in pink and DNA in blue. Detached DNA is shown in grey. The dyad is shown as a dashed line or as a dot. b, Four structures, coloured as in Fig. 1. Detached DNA was modelled as ideal B-DNA (grey). The black box shows a comparison of the nucleosome–SOX* (dark blue) and nucleosome–SOX (pink) structures. c, DNA superposition in the free nucleosome and the SOX factor (surface view) from the nucleosome–SOX structure illustrates the clash between SOX and the second DNA gyre.
Our cryo-EM data also suggest the dynamics that underlie nucleosome invasion by SOX factors. A set of particles from a separate dataset (151-bp DNA-1) (Methods) resulted in an alternative nucleosome–SOX11 structure in which the terminal DNA near SOX11 remained associated with the histone octamer (hereafter, nucleosome–SOX11*) (Fig. 3, Extended Data Figs. 4, 5). Thus, SOX factors may initially bind to their target site without detaching the second DNA gyre. Movement of the DNA-bound SOX factor to the position observed in the nucleosome–SOX11 structure would then lead to a clash that is resolved by terminal DNA detachment. This resulted in a model of nucleosome invasion and DNA unwrapping by SOX-factor binding (Fig. 3, Supplementary Video 1). The proposed mechanism differs from that used by the yeast pioneer factor Reb1, which binds and traps terminal DNA27.
In our nucleosome–SOX factor structures, terminal DNA is detached on both sides of the nucleosome, which suggests an additional allosteric effect of the SOX factor on nucleosome stability. Detachment of terminal DNA on the other side of the nucleosome may be stabilized by binding of a second copy of the SOX factor at SHL –2, which we observed in a subpopulation of our cryo-EM particles (Extended Data Figs. 4, 5e). In this nucleosome–SOX112 structure, the orientation of SOX-factor binding is determined by the asymmetric DNA motifs at both SHL +2 and SHL –2, with the latter apparently having lower affinity (Extended Data Figs. 3, 6). We speculate that SOX factors may also bind to multiple sites of a nucleosome in vivo. For example, a well-studied SOX2-binding genomic location (LIN28) contains two canonical SOX2 DNA motifs within a nucleosome and shows a broad peak of SOX2 occupancy12.
The nucleosome–SOX112 structure shows that SOX11 binding at SHL –2 is incompatible with binding of terminal DNA at SHL +7, SHL +6 and SHL +5, although the predicted clash at this location is with helix 3 and both termini of the SOX-factor DBD. DNA detachment is also observed with the use of Förster resonance energy transfer experiments when SOX11 is present at high concentrations (Extended Data Fig. 7). Thus, SOX factors can induce detachment of both DNA ends and can bind to both sides of the nucleosome (Fig. 3, Supplementary Video 1). These observations agree with the recently described strong preference for SOX2 binding at approximately ±25 bp around the nucleosome dyad in vivo24. However, the possibility that SOX factors may also bind additional nucleosomal positions in other contexts is not excluded.
The nucleosome–SOX11 structure further shows that binding of SOX11 repositions the N-terminal tail of H4 (Fig. 4, Extended Data Fig. 8). In the free nucleosome structure, the H4 tail binds to its canonical site and follows a trajectory towards DNA at SHL +2. However, in the nucleosome–SOX11 structure, the binding site of the H4 tail at SHL +2 is occupied by the SOX11 C-terminal tail (Fig. 4, Extended Data Fig. 8). The H4 tail is displaced, rotated by about 90° and extends towards SHL +1. The functionally important residue lysine 16 (K16) moves by around 33 Å. However, at SHL −2 SOX11 is oriented differently and does not displace the H4 tail (Extended Data Fig. 8c).

Top and side views of SHL +2 with SOX transcription factor (pink). Histones are grey, except for H4. H4 from the free nucleosome is shown in green where the H4 N-terminal (N) tail would clash with the C-terminal (C) SOX tail. In the nucleosome–SOX structure, the H4 tail is repositioned (orange). Residue K16 is marked with a coloured circle.
We speculate that SOX-factor binding may be incompatible with the formation of canonical nucleosome–nucleosome contacts28 (Extended Data Fig. 8). Formation of nucleosome arrays depends on the H4 tail and is impaired by K16 acetylation or tail truncation29,30,31. Nucleosome stacking is mediated by H4 tail residues K16–R19 that interact with the acidic patch of the H2A–H2B histone dimer of the neighbouring nucleosome21,32. Modelling the SOX DBD onto a nucleosome array32 suggests that the pioneer factor could be accommodated. SOX binding at SHL +2 and SHL −2 might be preferred over binding at the nucleosome dyad, which would be occluded by the H1 linker histone. However, for efficient chromatin opening, SOX factors cooperate with other transcription factors such as OCT4, KLF4, PAX6, Nanog, BRN2, and PRX116, and with ATP-consuming chromatin remodellers33.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Plasmids and strains
Full-length histone sequences from Homo sapiens were incorporated into the following plasmids: pET22B-H2B, pET22b-H3.2, pET3a-H4 (kindly provided by the W. Fischle laboratory). The H2A construct was cloned into a LIC1B vector (MacroLabs) and contained an N-terminal 6×His-tag followed by a tobacco etch virus (TEV) protease cleavage site (HHHHHHENLYFQS). The SOX2 DBD construct contained residues 36–121 of the full-length SOX2 (UniProt ID P48431). The DBD sequence was codon-optimized and synthesized by IDT as a gBlock. The gBlock was inserted into a LIC1B plasmid following N-terminal 6×His-tag and a TEV protease cleavage site sequences. The SOX11 DBD with short flanking sequences contained residues 33–138 of full-length SOX11 (UniProt ID P35716). It was inserted into a LIC1B plasmid. The construct was identical to the SOX11 construct used in a previous study7. Protein constructs are schematically shown in Extended Data Fig. 2.
Protein purification
Histones were purified according to standard protocols34,35. Purified histones were flash-frozen and lyophilized. Histones were resuspended in unfolding buffer (6 M guanidine hydrochloride, 20 mM HEPES pH 7.5, 10 mM dithiothreitol (DTT)). H2A, H2B, H3 and H4 were mixed in 1.2:1.2:1:1 ratios, and dialysed against three changes of refolding buffer high (RB high: 20 mM HEPES pH 7.5, 1 mM EDTA, 2 M NaCl, 2 mM DTT). After dialysis, the sample was concentrated and loaded onto a size-exclusion chromatography column (Superdex 200 10/300 GL, GE Healthcare). A peak corresponding to the complete octamer was collected and used for nucleosome reconstitution. The SOX2 DBD was expressed in BL21 (DE3) RIL Escherichia coli cells and purified over a sequence of columns: affinity His-Trap HP, cation exchange HiTrap SP-HP and size-exclusion Superdex75 10/300 GL (GE Healthcare). The His-tag was cleaved off after the affinity purification step. The SOX11 DBD was expressed and purified exactly as the SOX2 DBD. Purified proteins in the final buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 150 mM NaCl, 1 mM DTT) were flash-frozen and stored at −80°.
DNA preparation
DNA-1 template sequence was: ATCTACACGACGCTCTTCCGATCTAATTTATGTTTGTTAGCGTTATACTATTCTAATTCTTTGTTTCGGTGGTATTGTTTATTTTGTTCCTTTGTGCGTTCAGCTTAATGCCTAACGACACTCGGAGATCGGAAGAGCACACGTGAT. This sequence was directly (no changes) adopted from the NCAP-SELEX experiment with nucleosomes and SOX transcription factor described previously7.
DNA-1a sequence with all but one of the TTGT motifs replaced by a random sequence was: ATCTACACGACGCTCTTCCGATCTAATTTATTCAGACTAGCGTTATACTATTCTAATTTCAGACTTCGGTGGTCAGACTTATCAGACTCCTTTGTGCGTTCAGCTTAATGCCTAACGACACTCGGAGATCGGAAGAGCACACGTGAT.
Widom 601 DNA template used as a control was: ATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCGAT. Three bases at each end were changed to accommodate an EcoRV restriction site.
The DNA* template used for the nucleosome–SOX* structure determination was 151 bp long and almost identical to the DNA-1: ATCCCTACACGACGCTCTTCCGATCTAATTTATGTTTGTTAGCGTTATACTATTCTAATTCTTTGTTTCGGTGGTATTGTTTATTTTGTTCCTTTGTGCGTTCAGCTTAATGCCTAACGACACTCGGAGATCGGAAGAGCACACGTCTGAT. Two additional nucleotides on each side in the DNA* template are highlighted in bold. The rational for using a slightly longer 151-bp DNA construct was the following. The H2A C-terminal tail regulates nucleosome conformation by binding to linker DNA at different locations and stabilizes the nucleosome36. When a longer DNA construct is used, the H2A C-terminal tail stabilizes the DNA ends better (in comparison with the shorter constructs), thus shifting the equilibrium towards a ‘closed’ nucleosome conformation even in presence of SOX transcription factor.
The ‘Widom+1’ DNA template had the following sequence: ATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTTCCTTTGTGCGTTATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCGAT. The DNA-1b template with all of the TTGT motifs mutated had the following sequence: ATCTACACGACGCTCTTCCGATCTAATTTATTCAGACTAGCGTTATACTATTCTAATTTCAGACTTCGGTGGTCAGACTTATCAGACTCTCAGACGCGTTCAGCTTAATGCCTAACGACACTCGGAGATCGGAAGAGCACACGTGAT.
A plasmid pMK containing four consecutive copies of a DNA template of interest separated by EcoRV restriction sites was ordered from GeneArt (Thermo Fisher). The plasmid was produced in large quantities in E. coli XL1 blue cells, and purified with a NucleoBond PC 10000 kit (Macherey Nagel). The plasmid was digested with EcoRV enzyme (NEB) overnight, and produced four copies of the insert per plasmid. The plasmid was then precipitated with PEG-600037. The insert was further purified by size-exclusion chromatography with a Superose 6 Increase column (GE Healthcare). Peak fractions were pooled and concentrated by ethanol precipitation. Alternatively, DNA templates (DNA-1b, Widom+1 and Cy3-labelled DNA-1) were amplified from a plasmid with one insert copy via PCR. PCR product was purified via anion exchange on Resource Q 6 ml column (GE Healthcare).
Sample preparation and experiments for Förster resonance energy transfer
H2A(K119C) was prepared as described above and labelled according to ref. 38 with Cy5–maleimide (GE GEPA25031). Fluorescent DNA-1 template was produced by PCR with a Cy3 5′ labelled primer (IDT). Nucleosomes containing both labelled or just the donor Cy3 as control were reconstituted. Cy3 label was located at the SHL +7 end of the nucleosome. In the buffer containing 10 mM HEPES pH 7.5, 1 mM MgCl2, 0.01 mM ZnCl2, 1 mM DTT, 10 mM NaCl, 0.5 mg/ml BSA, nucleosomes (60 nM concentration) and SOX were mixed on ice and the spectra were recorded. Excitation wavelength of 510 nm was used. Förster resonance energy transfer (FRET) efficiency was calculated using a standard formula E = 1 − IDA/ID. Four independent experiments were performed.
Nucleosome reconstitution
Nucleosomes were reconstituted from the histone octamer and DNA template with a salt gradient as previously described35. In brief, octamer and DNA were mixed in 1.2:1 ratio in RB high, transferred into Slide-A-Lyzer MINI Dialysis Units 7,000 MWCO (Thermo Fisher), and dialysed gradually over a course of 24 h from RB high into RB low (20 mM HEPES pH 7.5, 1 mM EDTA, 20 mM NaCl, 2 mM DTT). Freshly reconstituted nucleosomes were concentrated in Amicon Ultra-0.5 centrifugal filters MWCO 10,000 (Sigma Aldrich).
Cryo-EM grid preparation and data collection
Nucleosomes at 1.6 μM concentration were mixed with 20× molar excess of SOX transcription factor at 4 °C in the final buffer containing 20 mM HEPES pH 7.5, 1 mM EDTA, 30 mM NaCl, 2 mM DTT, and used for cryo-grid preparation. First, R 2/1 Cu 300 mesh grids (Quantifoil) were glow-discharged with PELCO easiGlow (Ted Pella) device for 120 s. Next, 3.5 μl of sample was applied to the grid in the Vitrobot Mark IV (FEI) chamber at 100% humidity and 16 °C. The excess of liquid was blotted away for 10 s, and the grid was vitrified by plunging into liquid ethane. Data collection was performed on a G2 Titan Krios microscope (FEI) equipped with a K2 Summit direct electron detector (Gatan). Data were collected with EPU software (Thermo Fisher), with defocus ranging from 0.9 to 3.4 μm at a nominal magnification of 130,000× and a pixel size of 1.05 Å/pixel. Data were collected with an energy filter slit set to 30 eV. The total electron dose of 45 e−/A2 was distributed over 40 movie frames. For all imaged samples at least 50% of the data were collected at 25° stage tilt to partially compensate for preferred orientation of particles on the grid, and to improve angular distribution. The quality of the reconstructions was improved compared to the zero-tilt data. Data collection was monitored on-the-fly with Warp39 and cryoSPARC 2D classification40.
Data processing and analysis
Processing details are summarized in Extended Data Table 1. For every dataset, particles were picked with gAutomatch, CTF determination was performed with Gctf41. The initial reference from the free-nucleosome set was obtained ab initio in cryoSPARC, low-pass-filtered to 40 Å and used as a starting point for the 3D classification of all datasets. For every dataset, to speed up the computation, binned particles with the pixel size of 4.2 Å were extracted and subjected to several rounds of 2D classification and 3D classification in Relion42. Classes showing high-resolution features were selected for further processing. Next, selected particles were re-extracted with a pixel size of 2.1 Å, and were 3D-classified and cleaned again. Finally, particles were re-extracted at the final pixel size of 1.05 Å and box size of 400 pixels, and subjected to 3D refinement. For all datasets, processing was performed without symmetry application (C1). Final Fourier shell correlation (FSC) curves supplied with directional FSC curves and anisotropy estimates were calculated using 3DFSC server43 (Extended Data Fig. 4). In addition, for each map local resolution was calculated in Relion (Extended Data Fig. 4).
For the free nucleosome dataset, after CTF refinement and 3D refinement, final maps were sharpened using B-factor of −75. The final dataset contained 368,270 particles. When classified, this dataset showed typical levels of partial DNA unwrapping (about 10 bp) at the nucleosome entry or exit sites in around 15% of the data (similarly to ref. 26); however, the overwhelming majority of particles contributed to a fully wrapped nucleosome reconstruction.
In case of the nucleosome–SOX2 dataset, classes that showed additional densities were selected after 3D classification (with global soft mask applied). Next, a selected subset was subjected to a round of focused classification with a small soft spherical mask centred at the additional density near SHL +2 of the nucleosome. A class showing strong additional density was selected and further refined. The final dataset was CTF-refined to compensate for local defocus variations. As a final step, the dataset was subjected to non-uniform refinement in cryoSPARC, which led to an improved local resolution distribution in the 3D reconstruction. The final map was sharpened using a B-factor of −100. The final dataset contained 32,301 particles. For an overview of the processing pipelines for both nucleosome–SOX11 datasets, see Extended Data Figs. 9, 10. The starting number of particles (several million) was similar for nucleosome–SOX2 and for nucleosome–SOX11 datasets; however, nucleosome–SOX2 yielded a smaller number of ‘good’ particles that resulted in a final reconstruction.
The nucleosome–SOX11 dataset was processed in a similar way. The final dataset after initial steps of coarse cleaning was classified into four classes, out of which two were of high quality. One of the classes (202,142 particles) showed a clear additional density at SHL +2 and detached terminal DNA. The corresponding final map was sharpened using a B-factor of −100. Another class with two additional densities (nucleosome–SOX112) contained 114,104 particles. It was refined and sharpened using a B-factor of −120. In this nucleosome–SOX112 structure, the SOX factor molecules are located at SHL +2 and SHL −2, but are not related by the two-fold pseudo-symmetry of the nucleosome. This confirms that the density for the second SOX factor is not an artefact of particle misalignment during data processing. Lower occupancy of SOX11 at SHL −2 may be due to the presence of a weaker binding motif TTCT in that position. The local curvature induced by SOX binding at SHL −2 is not as pronounced as at SHL +2, possibly also owing to a weaker binding motif.
The nucleosome–SOX11* dataset resulted in two distinct classes. The first class (130,870 particles) resulted in a map virtually identical to the nucleosome–SOX structure, but with slightly lower resolution (about 4.0 Å). The remaining 63,821 particles resulted in the nucleosome–SOX* map. The final nucleosome–SOX* map was sharpened using B-factor of −100.
Model fitting and refinement
To model the free nucleosome structure, we started from a canonical nucleosome structure obtained by cryo-EM (PDB code 6FQ5)44 and altered the DNA sequence to correspond to the DNA-1 template using Chimera45. Several amino acid residues in the Xenopus laevis histones were substituted with ones corresponding to the H. sapiens histones in Coot46. Next, the model was fitted into the corresponding sharpened cryo-EM map of the free nucleosome and refined in real space using Phenix47.
The refined model of the free nucleosome was used to generate models for the nucleosome–SOX complex structures. In case of the nucleosome–SOX2 complex, both the nucleosome model and the SOX2 structure (PDB code 1O4X) were placed into the cryo-EM map, nucleosome DNA regions outside of the map were removed and the model was refined in real space using Phenix. For the nucleosome–SOX11 models, the nucleosome and the X-ray structure of SOX11 (determined in this study) were placed into the density and refined in real space using Phenix. In both cases, extra reference model restraints (σ = 1) were imposed to keep the model close to the available higher-resolution X-ray structure. In addition, base-pair and base-stacking restraints were used during refinements, excluding the region near the SOX transcription-factor binding site because strong local DNA distortion was evident in this region of the map. An equivalent procedure was used for modelling the other structures described here.
Electrophoretic mobility shift assay
Nucleosomes at a final concentration of 1.1 nM were mixed with purified proteins (SOX2 or SOX11 DBD). The final buffer contained 10 mM HEPES pH 7.5, 1 mM MgCl2, 0.01 mM ZnCl2, 1 mM DTT, 10 mM NaCl, 0.5 mg/ml BSA, 5% glycerol as in ref. 12 (Extended Data Fig. 3). Samples were incubated at 10 min at room temperature, mixed with Novex Hi-Density TBE sample buffer (Thermo Fisher), and loaded onto a 6% TBE PAGE. Electrophoresis was performed at 4 °C at 100 V in 0.5× TBE buffer for 1.5–2 h. Gels were stained with SYBR Gold dye (Thermo Fisher), washed, and imaged with Typhoon 9500 FLA Imager (GE Healthcare Life Sciences).
Electrophoretic mobility shift assays (EMSAs) (Extended Data Fig. 6) were performed identically to the procedure described above, but with higher final glycerol concentration to better observe the effects of point mutations of SOX11 on the nucleosome-binding properties of SOX11 in a wider range of apparent affinities. A control EMSA in the 12% glycerol buffer is shown in Extended Data Fig. 6a, b.
Digestion assay
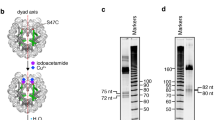
Two hundred and fifty nanograms of nucleosome or DNA was mixed on ice with increasing amounts of SOX11 in digestion buffer (20 mM HEPES pH 7.5, 30 mM NaCl, 10 mM magnesium acetate, 0.1 mg/ml BSA). Then, 0.125 units of restriction enzyme BfuCI (NEB) was added to each reaction. Samples were incubated at 37 °C for 30 min, and the enzyme was inactivated by incubating at 65 °C for 20 min. Samples were then incubated with proteinase K and urea, and then were loaded onto a 4–20% TBE-gel. Electrophoresis was performed at 180 V in 1× TBE buffer for 40 min. Gels were stained with SYBR Gold dye (Thermo Fisher), washed, and imaged with a Typhoon 9500 FLA Imager (GE Healthcare Life Sciences). Two independent experiments were performed both for the DNA and nucleosome digestion assays. Band intensities for the digestion product were measured in ImageJ according to standard routine48.
Crystallization and X-ray structure determination
DNA oligonucleotides (TATTGTTTATTTTGTT and AACAAAATAAACAATA) were synthesized and PAGE-purified by IDT. Complimentary oligonucleotides were annealed by heating to 95 °C and stepwise cooling to 4 °C (1° per 90 s) at a concentration of 1.5 mM. Concentrated purified SOX11 DBD and 16-mer DNA were mixed in 1:1.2 ratio and incubated on ice for 30 min. Crystallization was achieved by the hanging drop vapour diffusion method at 20 °C by mixing 1 μl of sample solution with 1 μl of reservoir solution containing 100 mM NaOAc pH 4.5, 200 mM CaOAc, 17% PEG400. Crystals were cryo-protected by 35% PEG400 (v/v) in the final storage solution and flash-frozen in liquid nitrogen.
X-ray diffraction data were collected at beamline X10SA at the Swiss Light Source using a Pilatus 6M detector. Data were indexed and integrated using XDS and scaled using XSCALE49. The structure was solved by molecular replacement with PHASER50, using the structure of the free SOX2 (PDB code 1GT051) as the search model. The crystals belonged to space group P61 and diffracted to a resolution of 2.5 Å. The asymmetric unit contained two protein–DNA complexes (Extended Data Fig. 2d). Density modification and model building was carried out with phenix.autobuild and manually completed in Coot. The model was iteratively refined with phenix.refine and outliers were fixed in Coot. The final Rfree factor was 26%. The final model contained SOX11 residues 46–122 and DNA nucleotides 1–14. Diffraction data and refinement statistics are summarized in Extended Data Table 2.
Estimation of the effect of Mg2+ on the nucleosome–SOX11 structure
Because nucleosomes are known to be sensitive to Mg2+ concentration, we wanted to test whether Mg2+ affects the nucleosome–SOX11 structure. Nucleosomes at 1.6 μM concentration were mixed with 20× molar excess of SOX11 transcription factor at 4 °C in the final buffer containing 20 mM HEPES pH 7.5, 30 mM NaCl, 1 mM DTT supplied with additional 1 mM MgCl2, 0.01 mM ZnCl2, 0.5% glycerol. Such sample buffer resembles the buffer used for our EMSAs except for glycerol and BSA, which should be avoided in cryo-EM samples. Next, we used this sample for cryo-grid preparation. We collected a dataset on the Titan Krios equipped with a K3 Gatan detector, nominal pixel size 1.07 Å. Processing was done similarly to the other datasets described here. The final set contained 93493 particles. After light 3D classification (removing low resolution classes), the cryo-EM map (at 4 Å resolution, with 0.73 sphericity) looked highly similar to our original nucleosome–SOX11 structure. The comparison is shown in the Extended Data Fig. 6g. We concluded that Mg2+ ions do not alter nucleosome–SOX structure. Overall, the Mg2+ sample looked better than the original one in terms of SOX occupancy and quality: a higher portion of particles from the original set contributed to the final reconstruction. We did not further analyse this dataset.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The cryo-EM density reconstructions and final models have been deposited with the Electron Microscopy Data Bank (EMD-10390, EMD-10391, EMD-10392, EMD-10393 and EMD-10394) and with the Protein Data Bank (PDB) (6T78, 6T79, 6T7A, 6T7B, 6T7C and 6T7D). All data are available in the Article and its Supplementary Information.
References
Iwafuchi-Doi, M. & Zaret, K. S. Cell fate control by pioneer transcription factors. Development 143, 1833–1837 (2016).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Adachi, K., Suemori, H., Yasuda, S. Y., Nakatsuji, N. & Kawase, E. Role of SOX2 in maintaining pluripotency of human embryonic stem cells. Genes Cells 15, 455–470 (2010).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Fuda, N. J., Ardehali, M. B. & Lis, J. T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192 (2009).
Wang, J. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22, 1798–1812 (2012).
Zhu, F. et al. The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81 (2018).
Cirillo, L. A. et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289 (2002).
Boller, S., Li, R. & Grosschedl, R. Defining, B cell chromatin: lessons from EBF1. Trends Genet. 34, 257–269 (2018).
Chen, J. et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285 (2014).
Velychko, S. et al. Excluding Oct4 from Yamanaka cocktail unleashes the developmental potential of iPSCs. Cell Stem Cell 25, 737–753.e4 (2019).
Soufi, A. et al. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 (2015).
Meers, M. P., Janssens, D. H. & Henikoff, S. Pioneer factor–nucleosome binding events during differentiation are motif encoded. Mol. Cell 75, 562–575.e5 (2019).
Malik, V. et al. Pluripotency reprogramming by competent and incompetent POU factors uncovers temporal dependency for Oct4 and Sox2. Nat. Commun. 10, 3477 (2019).
Biddle, J. W., Nguyen, M. & Gunawardena, J. Negative reciprocity, not ordered assembly, underlies the interaction of Sox2 and Oct4 on DNA. eLife 8, e41017 (2019).
Kamachi, Y. & Kondoh, H. Sox proteins: regulators of cell fate specification and differentiation. Development 140, 4129–4144 (2013).
Badis, G. et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723 (2009).
Jauch, R., Ng, C. K., Narasimhan, K. & Kolatkar, P. R. The crystal structure of the Sox4 HMG domain–DNA complex suggests a mechanism for positional interdependence in DNA recognition. Biochem. J. 443, 39–47 (2012).
Bergsland, M. et al. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev. 25, 2453–2464 (2011).
Wiebe, M. S., Nowling, T. K. & Rizzino, A. Identification of novel domains within Sox-2 and Sox-11 involved in autoinhibition of DNA binding and partnership specificity. J. Biol. Chem. 278, 17901–17911 (2003).
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997).
Williams, D. C., Jr, Cai, M. & Clore, G. M. Molecular basis for synergistic transcriptional activation by Oct1 and Sox2 revealed from the solution structure of the 42-kDa Oct1.Sox2.Hoxb1–DNA ternary transcription factor complex. J. Biol. Chem. 279, 1449–1457 (2004).
Voong, L. N. et al. Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell 167, 1555–1570.e15 (2016).
Li, S., Zheng, E. B., Zhao, L. & Liu, S. Nonreciprocal and conditional cooperativity directs the pioneer activity of pluripotency transcription factors. Cell Rep. 28, 2689–2703 (2019).
Hall, M. A. et al. High-resolution dynamic mapping of histone–DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129 (2009).
Bilokapic, S., Strauss, M. & Halic, M. Histone octamer rearranges to adapt to DNA unwrapping. Nat. Struct. Mol. Biol. 25, 101–108 (2018).
Donovan, B. T., Chen, H., Jipa, C., Bai, L. & Poirier, M. G. Dissociation rate compensation mechanism for budding yeast pioneer transcription factors. eLife 8, e43008 (2019).
Pepenella, S., Murphy, K. J. & Hayes, J. J. Intra- and inter-nucleosome interactions of the core histone tail domains in higher-order chromatin structure. Chromosoma 123, 3–13 (2014).
Gordon, F., Luger, K. & Hansen, J. C. The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nucleosomal arrays. J. Biol. Chem. 280, 33701–33706 (2005).
Shogren-Knaak, M. et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 (2006).
Dorigo, B., Schalch, T., Bystricky, K. & Richmond, T. J. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327, 85–96 (2003).
Song, F. et al. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344, 376–380 (2014).
Engelen, E. et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 43, 607–611 (2011).
Luger, K., Rechsteiner, T. J. & Richmond, T. J. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119, 1–16 (1999).
Dyer, P. N. et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004).
Li, Z. & Kono, H. Distinct roles of histone H3 and H2A tails in nucleosome stability. Sci. Rep. 6, 31437 (2016).
Lis, J. T. & Schleif, R. Size fractionation of double-stranded DNA by precipitation with polyethylene glycol. Nucleic Acids Res. 2, 383–390 (1975).
Shimko, J. C., North, J. A., Bruns, A. N., Poirier, M. G. & Ottesen, J. J. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 408, 187–204 (2011).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Bilokapic, S., Strauss, M. & Halic, M. Structural rearrangements of the histone octamer translocate DNA. Nat. Commun. 9, 1330 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017).
Kabsch, W. Xds. Acta Crystallogr. D 66, 125–132 (2010).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D 63, 32–41 (2007).
Reményi, A. et al. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev. 17, 2048–2059 (2003).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Klaus, M. et al. Structure and decoy-mediated inhibition of the SOX18/Prox1-DNA interaction. Nucleic Acids Res. 44, 3922–3935 (2016).
Palasingam, P., Jauch, R., Ng, C. K. & Kolatkar, P. R. The structure of Sox17 bound to DNA reveals a conserved bending topology but selective protein interaction platforms. J. Mol. Biol. 388, 619–630 (2009).
Werner, M. H., Huth, J. R., Gronenborn, A. M. & Clore, G. M. Molecular basis of human 46X,Y sex reversal revealed from the three-dimensional solution structure of the human SRY–DNA complex. Cell 81, 705–714 (1995).
Acknowledgements
We thank members of the Cramer laboratory, in particular H. Hillen, D. Tegunov, and G. Kokic for advice; the crystallization facility at our institute, in particular J. Wawrzinek and U. Steuerwald; and W. Fischle for providing histone expression constructs. Part of this work was performed at Beamline X10SA at the SLS at the PSI. S.O.D. was supported by an EMBO long-term fellowship (ALTF-949-2016). P.C. was supported by the Deutsche Forschungsgemeinschaft (EXC 2067/1-390729940), the ERC Advanced Investigator Grant TRANSREGULON (grant agreement no 693023) and the Volkswagen Foundation.
Author information
Authors and Affiliations
Contributions
S.O.D. designed and carried out all experiments and data analysis. F.Z., supported by J.T., identified the original DNA template used in the study. C.D. assisted with cryo-EM data collection. P.C. designed and supervised research. S.O.D. and P.C. interpreted the data and wrote the manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 DNA constructs and motif positions.
Related to Fig. 1. a, DNA-1 sequence. Two DNA strands are coloured in dark and light blue, canonical core motifs TTGT are coloured in red, TTXT motifs are shown in orange. Only motifs that allow SOX binding to the DNA minor groove are considered. The position of the third nucleotide of each motif in the DNA-1 sequence is indicated. Motifs at SHL+2 and −2 are shown in bold. b, Top views of the nucleosome. H2A, H2B, H3 and H4 are shown in yellow, red, blue and green, respectively, DNA is shown in dark and light blue. SHLs are labelled. c, DNA-1a template sequence. Only one TTGT motif is present (red). d, Structure of SOX2 (PDB code 1O4X) was aligned to each of the motifs present in nucleosome 1 and allowing binding of SOX to the minor groove. The number of clashes was calculated using ‘findclash’ command in Chimera software. SOX2 binding to motifs at SHL +2 and SHL −2 gives rise to the least amount of clashes with DNA and histones compared to other locations. The ideal position (modelled) of the SOX motif on the dyad would result in a comparably low number of clashes.
Extended Data Fig. 2 Conservation of SOX-family DBD sequence and structure, and X-ray structure of the SOX11–DNA complex.
Related to Fig. 1. a, Domain organization of the human SOX protein family. DBDs are shown as pink rectangles; unstructured functionally diverse regions are shown as wavy lines. Protein constructs used in this study are marked. The alignment of DBD sequences (produced using Clustal Omega) is shown below52. b, Structural conservation of SOX factors. Crystal and nuclear magnetic resonance structures of SOX transcription factors: SOX2 (ref. 22), SOX11 (this study), SOX4 (ref. 18), SOX9, SOX18 (ref. 53) and SOX17 (ref. 54); SRY (ref. 55) has a similar fold. Superimposition of all the structures reveals that they are virtually identical. c, DNA is engulfed by the strongly positively charged inner surface of the SOX11 DBD. d, Ribbon representation of the SOX11 X-ray structure. e, Two copies of SOX11–DNA in the asymmetric unit. The contact between the two is mediated by DNA stacking. f, Comparison of the observed DNA conformation with canonical B-DNA (green). SOX11 introduces a kink into DNA that is typical for HMG box proteins.
Extended Data Fig. 3 EMSAs of SOX2 and SOX11 in complex with DNA or nucleosomes.
Related to Figs. 1–3. EMSAs reveal formation of SOX-factor complexes with DNA (left) or nucleosomes (right). DNA or nucleosome concentration is 1.1 nM. a, EMSA of DNA-1–SOX2 and nucleosome-1–SOX2 complexes. b, EMSA of DNA-1–SOX11 and nucleosome-1–SOX11 complexes. c, EMSA of DNA-1a–SOX11 and nucleosome-1a–SOX11 complexes. d, EMSA of DNA–Widom+1 and nucleosome–Widom+1–SOX11 complexes. e, EMSA of DNA Widom 601–SOX11 and nucleosome Widom 601–SOX11 complexes. Relevant bands are labelled. All experiments were repeated at least twice with similar results. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 4 Global and local resolution of cryo-EM reconstructions.
Related to Figs. 1–3. a, Example micrograph reveals preferred orientation of nucleosomes. Scale bar, 20 nm. b, FSC plots for five reported reconstructions. c, Local resolution distribution. In some maps, the resolution varies from 3 Å (dark blue) through 4 Å (green) to 6 Å (red). The lower resolution of some DNA regions indicates flexibility. d, Angular distribution plots. Scale shows the number of particles assigned to a particular angle. Blue, a low number of particles; yellow, a high number of particles. e, Directional FSC plots for the reconstructions calculated on the 3DFSC server48. Sphericity, as the degree of anisotropy present in the reconstructions, is indicated above each plot. Histograms indicate the portion of voxels with a particular resolution. f, Visualization of different regions of the nucleosome–SOX11 map. In the nucleosome core, histone side chains are clearly visible; SOX density has a lower resolution, but helical densities are clearly distinguishable. g, Rigid-body fit of the SOX2–DNA structure (PDB code 1O4X) into the nucleosome–SOX2 cryo-EM map (left). Rigid-body fit of the SOX11–DNA X-ray structure into the nucleosome–SOX11 cryo-EM map (right). The region containing SOX and a short DNA stretch was isolated from the rest of the map for clarity.
Extended Data Fig. 6 EMSAs using SOX2, and wild-type or mutant SOX11 in complex with DNA or nucleosomes.
Related to Figs. 1–3. EMSAs reveal the formation of SOX-factor complexes with DNA or nucleosomes. All experiments were repeated at least twice with similar results. For gel source data, see Supplementary Fig. 1. a, EMSA of DNA-1 or nucleosome-1 with wild-type SOX2. b, EMSA of DNA-1 or nucleosome-1 with wild-type SOX11. c, EMSA of DNA-1 or nucleosome-1 with SOX11(H75A/S80A). d, EMSA of DNA-1 or nucleosome-1 with SOX11(Y118A). Relevant bands are labelled. To observe a wider range of the binding curve for the mutants, a larger amount of glycerol (12% final concentration) was used here (as compared to the EMSAs shown in Extended Data Fig. 3, which used 5% glycerol)—thus, the apparent affinity is higher. DNA or nucleosome concentration is 1.1 nM. e, EMSAs reveal formation of SOX–DNA or SOX–nucleosome complexes at different concentrations of salt. There is virtually no difference in binding at 10 or 30 mM NaCl, whereas binding is weaker at 150 mM NaCl. DNA or nucleosome concentration is 1.1 nM. f, EMSAs reveal formation of SOX complexes with DNA-1a or nucleosome-1a (only one canonical motif present) as compared to DNA-1b and nucleosome-1b (in which all canonical SOX motifs were mutated). SOX concentrations ranged from 0 to 200 nM, DNA concentration was 1.1 nM. g, Superimposition of two nucleosome–SOX11 cryo-EM maps obtained in the presence of 1 mM MgCl2 (grey density) or in the absence of MgCl2 (coloured). Magnesium does not influence the structure of the SOX–nucleosome complex. Cross-correlation between the two unmasked maps is 0.94 (Chimera).
Extended Data Fig. 7 Nucleosome-DNA end unwrapping.
Related to Figs. 1, 3. a, DNA-1 sequence with BfuCI digestion sites. BfuCI digestion assays (two independent experiments for each sample, shown here) of the DNA-1, nucleosome-1 or nucleosome-1b (no TTGT motifs) in the presence of increasing amounts of SOX11. The restriction site (about 20 nucleotides away from the entry and exit sites of the nucleosome) becomes more accessible with higher concentrations of SOX11. In a DNA-1 digestion assay, the amount of digestion product increases slightly in the low SOX11 concentration range, and then stays constant over a broad concentration range. For gel source data, see Supplementary Fig. 1. b, BfuCI digestion assay plot for free DNA-1 (black), nucleosome-1 (green) and mutated nucleosome-1b (orange) in the presence of increasing amounts of SOX11. Each experiment was performed independently twice (n = 2). Mean values (lines) and individual measurements (dots) are shown. Band intensity was calculated using standard routine in ImageJ52. c, Example fluorescence spectra of Cy3–Cy5-labelled nucleosome in the presence of increasing amounts of SOX11. d, FRET efficiency plot. Mean values with s.d. are shown (independent experiments, n = 4).
Extended Data Fig. 8 Repositioning of the H4 N-terminal tail.
Related to Fig. 4. Cryo-EM maps are shown with a Gaussian smoothening filter (Chimera49) applied for clarity. SOX is coloured in pink, H4 is shown in green and the repositioned H4 tail is shown in orange. a, Free nucleosome. Views of SHL +2 and SHL −2 are shown to illustrate the position of the H4 tail (a dashed line). Residue K16 is marked with a circle. b, Nucleosome–SOX11 with SOX11 located at SHL +2. On the right, a superimposition with the free nucleosome map is shown to highlight different orientations of the H4 tail. c, Nucleosome–SOX11 complex with SOX11 located at SHL −2. In this location, SOX is oriented differently and does not clash with or reposition the H4 tail. d, Nucleosome–SOX11*. The H4 N-terminal tail is repositioned compared with that in the free nucleosome. Repositioning of the H4 tail was reported in case of strong distortions in the nucleosome structure30. e, SOX binding repositions the H4 N-terminal tail and might impair nucleosome stacking. Side view of two stacking nucleosomes (from PDB code 1AOI). H4 interacts with the acidic patch on the neighbouring H2A–H2B histone dimer. H4 tail repositioning is incompatible with nucleosome stacking.
Supplementary information
Supplementary Figure 1
Raw images of gels related to Extended Data Figs 3, 6 and 7. The cropping borders are indicated with dashed lines.
Video 1: Overview of Nucleosome-Sox complexes
Pioneer factor binding remodels the nucleosome. Binding of Sox11 to the nucleosome leads to local DNA distortion and unwrapping of terminal DNA. The five cryo-EM structures reported here have been used to generate this animation. For details compare text.
Rights and permissions
About this article

Cite this article
Dodonova, S.O., Zhu, F., Dienemann, C. et al. Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672 (2020). https://doi.org/10.1038/s41586-020-2195-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2195-y
- Springer Nature Limited
This article is cited by
-
Structural basis of Integrator-dependent RNA polymerase II termination
Nature (2024)
-
Molecular and structural basis of the chromatin remodeling activity by Arabidopsis DDM1
Nature Communications (2024)
-
Structural mechanism of synergistic targeting of the CX3CR1 nucleosome by PU.1 and C/EBPα
Nature Structural & Molecular Biology (2024)
-
DNA binding redistributes activation domain ensemble and accessibility in pioneer factor Sox2
Nature Communications (2024)
-
Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications
Signal Transduction and Targeted Therapy (2024)