

Abstract
Bacteria are under immense evolutionary pressure from their viral invaders—bacteriophages. Bacteria have evolved numerous immune mechanisms, both innate and adaptive, to cope with this pressure. The discovery and exploitation of CRISPR–Cas systems have stimulated a resurgence in the identification and characterization of anti-phage mechanisms. Bacteriophages use an extensive battery of counter-defence strategies to co-exist in the presence of these diverse phage defence mechanisms. Understanding the dynamics of the interactions between these microorganisms has implications for phage-based therapies, microbial ecology and evolution, and the development of new biotechnological tools. Here we review the spectrum of anti-phage systems and highlight their evasion by bacteriophages.
Similar content being viewed by others


Main
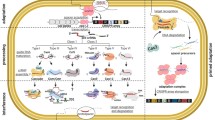
Bacteriophages (phages) are viruses that infect bacteria and it has been estimated that there are 1031 phages present in the biosphere1. Their abundance accounts for 20–40% of bacterial mortality daily2, and has a considerable impact on biogeochemical cycles3. The pressure of phage infection on bacteria has resulted in the evolution of multiple bacterial immune systems, each of which hampers different stages of the phage life cycle4,5 (Fig. 1). Unsurprisingly, phages have evolved a myriad of ways to overcome these defences5,6, which in combination with phage diversity, has contributed to the diversity of bacterial immune mechanisms.

Virulent phages replicate exclusively through the lytic cycle, whereas temperate phages may replicate through either the lytic or the lysogenic cycle. Bacteria have numerous anti-phage systems that function at different stages of the phage life cycle to prevent productive phage replication. Abortive infection mechanisms provide population protection and function at different stages of the phage life cycle, indicated by the blue dashed line.
Research interests in bacterial–phage interactions, and in particular bacterial defences, are manifold. First, although the importance of these interactions for global ecology is accepted, large sequencing efforts, such as the Tara Oceans project, are furthering our understanding by showing that phages drive rapid evolution through the daily transfer of approximately 1029 genes between bacteria7. Second, phage-based therapies are becoming feasible antibacterial treatments as alternatives to antibiotics, owing to the rise of antibiotic resistance8. For successful therapy, it is critical to understand how bacterial pathogens might become resistant to phages and, therefore, recalcitrant to treatment. Finally, phage-resistant strains are required in different industries9 and fundamental research into phage-defence mechanisms has underpinned the development of these, and other, applications, such as gene editing and diagnostics10. The importance of bacterial immune systems to these areas has led to a resurgence in the discovery and characterization of phage-resistance mechanisms. Here we focus on the diverse systems that bacteria use to resist phages and how their phage invaders can evade these immune mechanisms.
Preventing adsorption
Phages exploit at least three different lifestyles to reproduce. Virulent phages replicate exclusively through the lytic cycle, exploiting bacteria to make new phages before their release by cell lysis11 (Fig. 1). Alternatively, in addition to the lytic cycle, temperate phages can enter the lysogenic cycle and form prophages that are integrated into the bacterial chromosome or maintained extrachromosomally1 (Fig. 1). By contrast, filamentous phages cause chronic infections and are continuously secreted from the bacterium without lysis12. For infection to occur, phages must adsorb to the cell surface by binding to phage receptors, and inject their genome (Fig. 2a). To prevent adsorption, bacteria can alter or disguise receptors through surface modification (Fig. 2a). For example, receptor mutations in ompU in Vibrio cholerae confer resistance to the vibriophage ICP213. Bacteria can also use receptors as phage decoys. In this case, outer membrane vesicles (OMVs) that contain receptors bud off from Escherichia coli and Vibrio, and can bind to phages, reducing productive infections14,15 (Fig. 2a). Nonetheless, OMVs have complex effects on phage dynamics because they can also extend the host range of phages. Indeed, phage receptors were transferred by OMVs to Bacillus subtilis cells that previously lacked the receptor, rendering B. subtilis and other phage-resistant species susceptible to phages16 (Fig. 2b). Although this provides only transient susceptibility, the receptors may subsequently facilitate the transfer of receptor genes through generalized transduction, which could lead to a permanent heritable change in phage susceptibility. Inhibiting DNA entry into the bacterial cell is another defence strategy. For example, the Imm and Sp proteins of phage T4 prevent the DNA of other T-even phages from being translocated across the membrane4. However, systems that prevent DNA entry are typically encoded on prophages and inhibit infection by subsequent phages4.

a, Bacteria have developed a number of methods to prevent phage adsorption. These include altering (green), disguising (blue), modifying (red) or masking (blue circles) receptors and the use of decoy OMVs. b, Phages can co-evolve to recognize the modified receptor, through mutations, and produce extracellular-matrix-degrading enzymes. OMVs can also extend the host range of phages, by transferring receptors used by the phage to cells that previously lacked those specific phage receptors.
The fitness costs of receptor mutations have led to other strategies that impede attachment5. Phase variation enables the reversible expression of phage receptors, resulting in phage-resistant bacterial subpopulations5,17. Furthermore, receptors can be masked, preventing recognition while retaining function. For example, capsules or exopolysaccharides provide phage resistance in Staphylococcus, Pseudomonas and other species18,19 (Fig. 2a). Subtle modifications can also disguise receptors from phages, such as in Pseudomonas aeruginosa, in which pilus and O-antigen modifications and type IV pili glycosylation occludes phages20,21.
Receptor modification can select for phages that recognize the mutated, or alternative, receptors. In coevolution studies, it was shown that the receptor-binding proteins (RBPs) of phages—which bind to the bacterial receptors—are often mutated (Fig. 2b). For example, phage λ overcame a LamB receptor mutant of E. coli through tail fibre mutations, some of which caused stochastic protein folding into different forms that enabled the recognition of a new receptor (OmpF) or mutated LamB22,23. Access to a new receptor occurred through baseplate and tail fibre mutations in B. subtilis phage SPO1, which also resulted in an extended range of Bacillus hosts24. Instead of mutating, some phages—such as the E. coli phage phi92—have more than one RBP, enabling the recognition of multiple receptors25. RBP variability can be further expanded by diversity-generating retroelements that result in hypervariable RBPs. For example, the diversity-generating retroelement of the Bordetella phage BPP-1 creates variable tail fibres, changing the receptor tropism of the phage4. Finally, when receptors are masked by exopolysaccharide capsules, some phages produce depolymerases that enable them to gain access26 (Fig. 2b). Recent research on receptor interactions is driven by the goal to understand and manipulate phage–receptor interactions to extend the host range for biotechnological and medical applications27.
Restriction–modification and related defences
The biotechnological use of restriction–modification (RM) systems has led to these systems being the most well-characterized phage-resistance mechanism—they are highly diverse and ubiquitous, and are present in around 90% of bacterial genomes28. These systems distinguish self from non-self DNA to recognize and destroy phage DNA after its injection. Discrimination is due to DNA modifications at specific sequences and is characteristic of a number of anti-phage systems. Two components that are typically present in RM systems are a methyltransferase and a restriction endonuclease (Fig. 3a, b). Both recognize restriction-site sequences; the methyltransferase methylates DNA and the restriction endonuclease cleaves the unmethylated sequence. A comprehensive review of RM systems has been published previously29. A range of other phage-resistance systems have similarities to RM systems, but their functions appear to be more complex owing to the presence of additional genes.

a, Many proteins and protein domains are shared between the RM, Pgl, BREX and DISARM systems. The blue genes indicate enzymes that are responsible for DNA modification (methyltransferases (MT)), the purple gene (pgIZ) encodes a conserved protein (an alkaline phosphatase) and orange genes in the DISARM system indicate core genes. RE, restriction endonuclease. pgIW encodes a serine/threonine kinase; pgIX encodes an adenine-specific methyltransferase; pgIY encodes an ATP-binding protein; brxA encodes an RNA-binding anti-terminase; brxB encodes a protein with an unknown function; brxC encodes an ATP-binding protein; brxL encodes a protease; drmA encodes a putative helicase; drmB encodes a helicase-associated protein; drmC encodes phospholipase D/nuclease; drmD encodes an SFN2 helicase; and drmM1 encodes an N6-adenine DNA methyltransferase. b, (1) RM restricts any phage DNA that is not modified by methylation; (2) however, modified phages (green phage; see d) can replicate on RM+ strains. (3) Modified or (4) unmodified phages can replicate on an RM− strain but will lose any modifications. c, Pgl systems only restrict phages that have been previously exposed to the system. (1) A naive phage can replicate on Pgl+, (2) but upon secondary infection of a Pgl+ strain, the phage (shown in green) is restricted. (3) Modified (yellow) or (4) unmodified (grey) phages can replicate on Pgl− strains. d, Mechanisms of phages for avoiding RM and RM-like systems include methylation of DNA, removing recognition sequences from their genome and encoding a methyltransferase to methylate the phage DNA.
One RM-like phage-defence mechanism is the phage growth limitation (Pgl) system in Streptomyces coelicolor, which modifies and cleaves phage DNA30,31. Pgl has three phases and requires four genes, pglW, pglX, pglY and pglZ (Fig. 3a, c). Phages become methylated after infecting Pgl+ bacteria and, following release, these phages can infect other cells. During subsequent infections, the modified phage DNA is cleaved. Hence, although the initial infected cell does not survive phage infection, it is able to mark the phage to ‘warn’ neighbouring cells31. Genes similar to pglZ from the Pgl system were identified in six-gene clusters, including brxABCL and pglX32 (Fig. 3a). These were termed bacteriophage exclusion (BREX) genes and have been characterized in Gram-positive (B. subtilis32) and Gram-negative (E. coli33) bacteria. Akin to RM, BREX acts after DNA injection to prevent phage replication and lysogen formation, but differs as DNA cleavage was undetectable32. BREX further differs from Pgl in that it restricts phages upon first exposure32,33; however, the precise mechanism by which BREX prevents infection remains unresolved.
Another RM-like system was recently identified—termed defence island associated with restriction–modification (DISARM)34 (Fig. 3a). Class 1 and 2 DISARM share three core genes, with each class having two distinct additional genes. Class 2 DISARM includes a five-cytosine DNA methyltransferase and a system from Bacillus paralicheniformis prevented phage DNA accumulation by distinct families of double-stranded (ds)DNA phages. Notably, phages modified at a specific sequence, and therefore presumably masked from the RM-like system, were inhibited. Furthermore, DISARM offered protection against phages that lacked the sequence recognized by the RM-like system. These results indicate that the mechanism differs from classic RM systems. To add further mystery to the DISARM mechanism, the candidate nuclease was dispensable for resistance34.
Phages have amassed strategies to counteract RM, and potentially RM-like, systems6,35,36. Phage DNA can become methylated by the host methyltransferase on entry, disguising the DNA from the host restriction endonuclease. The resulting phages become phenotypically RM-insensitive; however, this epigenetic avoidance is transient and is lost following infection of methyltransferase-deficient bacteria. In addition, RM sites are mutated, underrepresented or absent in phage genomes to prevent restriction35,37,38 (Fig. 3d). Phages also exploit modified or unusual bases, such as hydroxymethylation, glycosylation, glucosylation and acetamidation to make these sites unrecognizable to the restriction endonuclease36. Specifically, coliphage 9g utilizes a deoxyarchaeosine modification to avoid restriction39. Some phage proteins (for example, Ral from λ and P22 phages) activate host methyltransferases and promote DNA modification to protect against restriction endonucleases40,41. Phages can also encode methyltransferases, which protect their DNA from restriction42,43 (Fig. 3d), such as the methyltransferase of the Bacillus phage SPR, which can modify three sites to protect against multiple nucleases42,44. Phages can also prevent degradation of their genomes using the defence against restriction (Dar) system. The Dar system of coliphage P1 limits DNA degradation by type I restriction endonucleases45,46. Dar proteins are injected along with the phage DNA and function in cis. Another successful anti-RM strategy is the direct inactivation of restriction endonucleases. The overcome classical restriction (Ocr) protein of coliphage T7 is expressed immediately after DNA injection, mimics DNA, and tightly binds and sequesters the EcoKI restriction endonuclease47,48. Routes of phage escape from the recently discovered, RM-like systems have yet to be thoroughly investigated. However, phages are likely to use similar anti-restriction mechanisms for DISARM and BREX. No phages that have escaped Pgl systems have been isolated31,49, suggesting that bacterial protection by this system may be more robust than other RM-like systems.
CRISPR–Cas adaptive immunity
The ability to cleave phage DNA in a sequence-specific manner is shared by both RM and clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein (Cas) systems. However, CRISPR–Cas provides ‘adaptive’ immunity through the generation of memories of past phage encounters that guide sequence-specific immunity50. CRISPR–Cas immunity is present in about half of sequenced bacteria and is mediated by three stages51,52,53: adaptation, expression and interference (Fig. 4a). The mechanistic diversity of CRISPR–Cas systems is considerable—currently there are two classes, six types and more than 30 subtypes54,55.

a, Schematic of the three stages of CRISPR–Cas immunity, including adaptation (stage 1), expression and maturation (stage 2), and interference (stage 3). crRNA, CRISPR RNA. b, Phages have the ability to overcome CRISPR–Cas defences through point mutations in the protospacer-adjacent motif (PAM) or protospacer, deletions or modifications of the DNA so that the DNA cannot be bound by Cas complexes. Phages can also encode protein anti-CRISPRs that can interfere with CRISPR immunity, and jumbo phages produce a nucleus-like structure that excludes Cas complexes, thus preventing DNA targeting.
Class 1 systems include types I, III and IV, which have multi-subunit Cas complexes. Various type I CRISPR–Cas subtypes have been shown to provide phage resistance56,57,58,59,60,61,62, whereas type IV systems—which are most-closely related to type I—are poorly characterized and their role in phage resistance is unknown63,64. Type III systems differ from other class 1 systems, because they target both RNA and DNA65,66. Resistance to lytic infection has been demonstrated by the type III systems of Staphylococcus epidermidis66,67,68, Lactococcus lactis69 and Streptococcus thermophilus70; however, the RNA-dependent targeting provides tolerance to prophages71. An interesting feature of type III systems is that Cas10 synthesizes intracellular signals (cyclic oligoadenylates) that bind an accessory RNase and unleash its promiscuous activity67,72,73. The RNase may have an abortive infection effect (see ‘Protecting the bacterial population’ section), adding a further layer of defence by inducing dormancy through unspecific cleavage of both host and phage transcripts74,75.
Class 2 CRISPR–Cas includes type II, V and VI systems, which are characterized by single-subunit effectors. The first direct evidence that CRISPR–Cas provides immunity against phages was provided by the type II-A system of S. thermophilus50 and was later shown in Streptococcus pyogenes76. Type II systems use Cas9 to generate dsDNA breaks, whereas type V systems use Cas1277. Although there are few studies that have investigated phage resistance by type V systems, it has been shown that the Francisella novicida system protects against phage infection in E. coli78. The dsDNA breaks induced by class 2 systems have been exploited in biotechnology, but may be less effective for clearing phages. In support of this idea, class 2 systems are less common than type I, which have a potentially more destructive DNA-shredding mechanism55. Finally, class 2 systems can recognize and cleave phage RNA. Indeed, Cas13 from the type VI system of Leptotrichia shahii cleaved phage MS2 RNA in E. coli79. Upon target recognition, Cas13 not only cuts complementary transcripts, but also becomes a promiscuous RNase79,80. This promiscuous RNase activity can cleave phage mRNAs and host RNAs, inducing dormancy and providing Cas13-mediated resistance against dsDNA phages80.
The sequence specificity of CRISPR–Cas selects for phages with mutations in targeted regions (Fig. 4b). Indeed, mutations in protospacer-adjacent motifs and spacer targets (i.e. protospacers) enable phages to overcome type I systems57,59,60,61,81 and type II systems82,83,84,85. Insertions, deletions and recombination events can also mediate phage escape50,59,81,84,85. However, type I systems have a positive feedback mechanism to restore or enhance immunity by acquiring multiple new spacers that target escape phages—a process called priming51. There is now bioinformatic and experimental evidence that priming occurs in type II systems86,87. Nevertheless, phages can evade primed strains with multiple spacers by deleting the target region81.
As type V and VI systems can also degrade non-specific single-stranded (ss)DNA (type V) or RNA (type VI), they might provide an additional layer of resistance, which may explain why escape phages are yet to be identified for these systems75. In agreement with this notion, dormancy induced by type VI systems suppressed the emergence of escape mutants and protected the bacterial population against phages80. Similar to RM system evasion, phages can modify DNA to reduce Cas complex binding and cleavage—as seen for T4 evasion of type I-E and II-A systems59,78.
Escape mutations can lead to phage fitness defects, and if essential genes are targeted, escape might be impossible. As an alternative, some phages have anti-CRISPR (Acr) proteins that inactivate CRISPR–Cas systems88,89 (Fig. 4b). Acrs have been identified for type I, II, III, V and VI systems and most interact with the Cas proteins to block activity89,90,91. Recently, an Acr has been shown to acetylate a type V system to prevent DNA binding92, and another inactivates Cas12 by triggering cleavage of CRISPR RNA bound to Cas1293. Notably, some phages must cooperate to exploit their Acrs. Acrs produced by the first phage that infects can immunosuppress the host, but may fail to fully protect the phage from CRISPR–Cas, while enabling a productive infection by successive phages94,95. It is possible that Acrs have provided a selection for CRISPR–Cas diversity, but the ecological importance of their mechanistic diversity is unclear (see Box 1).
Phage defences such as CRISPR–Cas are sometimes encoded by phages96,97. For example, CRISPR arrays occur in prophages of Clostridium difficile, which target other C. difficile phages, and CRISPR–Cas systems are also present in ‘huge’ phages98,99,100. In many phages, these systems are incomplete—lacking genes for adaptation or interference97,101. Phages that contain these incomplete systems have been proposed to co-opt the required proteins from the host, or repress transcription without cleavage, akin to RNA interference101. These phage-encoded CRISPR–Cas components may also eliminate competing phages and manipulate the hosts102. Indeed, a complete system expressed by a Vibrio phage can protect against a host defence island96. Phages can also transduce CRISPR–Cas systems between bacteria, which can provide immunity against other phages62,75. These examples highlight how some phages have manipulated CRISPR–Cas systems as a way to avoid defence systems in the host and endow them with an advantage over competing phages.
Protecting the bacterial population
In contrast to RM and adsorption inhibition, which confer individual benefits, abortive infection (Abi) anti-phage systems protect the bacterial population5. Abi is characterized by successful phage entry; however, development is interrupted, resulting in the release of few, if any, phages and the host cell dies, which prevents a phage epidemic and protects the bacterial population103. ‘Altruistic’ Abi systems are widespread in Gram-positive and Gram-negative bacteria103; however, as Abi systems are defined by phenotype, rather than genotype, their discovery has been sporadic103. Nevertheless, the presence of many Abi systems on plasmids has been used successfully to identify these systems, particularly in lactococci103. The mechanistic details of phage abortion are unknown for many systems, although disruption of essential processes, such as replication, transcription, translation and DNA packaging is common4,104.
An Abi mechanism in S. epidermidis was recently shown to involve a serine/threonine kinase (Stk)105. Activated Stk phosphorylates proteins involved in translation, transcription, cell cycle control, the stress response, central metabolism, and DNA topology and repair105. Death of infected bacteria occurs through this phosphorylation pathway, decreasing phage release and protecting the population105. The presence of serine/threonine kinases in eukaryotic viral defences suggests there are shared immune strategies between these kingdoms. Kinases also play wider roles in viral defence in bacteria, with examples in the BREX and Pgl systems30,31,32.
The phenotypic definition of Abi systems is also reflected in their mechanistic diversity. For example, E. coli lambda lysogens encode RexAB, which is activated by a poorly-characterized T4 phage protein-DNA complex104,106. When triggered, RexA activates RexB, which forms a membrane channel that leads to ATP leakage, lost membrane potential and phage exclusion106. RexAB-like systems are widespread, with their recent identification in actinobacteriophages. For example, rexAB-like genes in Mycobacterium smegmatis and Gordonia terrae prophages abort multiple phages98,107. In each host, phage escape mutants were identified and all contained mutations in the proteins that triggered RexAB activity98.
A subset of Abi systems function through a toxin–antitoxin mechanism. Toxin–antitoxin systems are composed of a toxin and an antitoxin that are usually co-transcribed. The toxins targets essential cellular processes, leading to bacterial dormancy or death. There are six types of toxin–antitoxin systems, based on the identity of the gene products (RNA or protein) and whether, and how, the toxin and antitoxin interact99. ToxIN, a type III system from Pectobacterium atrosepticum was the first example of an Abi system that was shown to function as a toxin–antitoxin mechanism100 and this has now been observed for other Abi systems108,109. Different toxin–antitoxin types can elicit phage resistance, but have not been strictly classified as Abi systems, as the outcome for the infected bacterium was not defined. Examples in E. coli include hok/sok (type I) and rnlA/rnlB (type II), which exclude phage T4110,111, and mazEF (type II), which excludes phage P1112. Many of these toxins are RNases, a characteristic shared by several Abi systems. For example, E. coli PrrC is an RNase that cleaves lysine transfer RNA (tRNALys) during infection, and only T4 phages that are able to repair this cleavage can replicate4. Thus, mutant T4 phages that lack a polynucleotide kinase or RNA ligase are aborted due to tRNALys cleavage113.
To bypass toxin–antitoxin systems, phages can encode antitoxins. For example, T4 produces Dmd, an antitoxin that inhibits E. coli RnlA and LsoA toxins114. Dmd differs from the RnlB or LsoB antitoxins, suggesting it evolved independently, which is highlighted by its different toxin neutralization mechanism114. Phages can generate diversity for escape by acquiring host genetic material through recombination. Indeed, recombination between lytic phages and resident lactococcal prophages led to Abi escape through gene loss or gain115. Recombination can also promote antitoxin acquisition by phages. For example, to escape ToxIN, phages containing a short toxI-like sequence recombined with toxIN and directly gained toxI116. Notably, in other escape phages, toxI-like sequence duplications produced pseudo-ToxI RNAs that inhibited ToxN116. Rather than encoding its own antitoxin, coliphage T7 evades a toxin–antitoxin system by producing a protein that has been proposed to prevent antitoxin degradation by the Lon protease. This ensures that the toxin remains inactive by increasing the stability of the host antitoxin117. Finally, the T4 protein Alt (an ADP-ribosyltransferase) is injected with phage DNA, which chemically modifies the MazF toxin118. ADP-ribosylated MazF has reduced cleavage activity, enabling the survival of the phage118.
Many new Abi systems await discovery and, indeed, new systems in different strains are still being uncovered. For example, Abiα was recently identified in Enterococcus faecalis and leads to asynchronous lysis119. To understand Abi responses, the phage genes involved can be revealed by isolating escape mutants. For example, ToxIN can be overcome by specific mutations in ϕM1 and T4-like phage proteins120,121. However, the often toxic and poorly characterized nature of the phage Abi-triggering proteins is a frequent challenge for mechanistic studies.
Prophage-encoded defence systems
Prophages can have immune systems that prevent subsequent phage infection of lysogens (for example, rexAB). These non-essential transcribed regions or genes within prophage genomes have been called ‘morons’ and can encode factors that benefit the host, such as defence systems122. For example, morons (or ‘immunity cassettes’) within M. smegmatis prophages provide phage defence by encoding RM and toxin–antitoxin components, and other defence systems123. These systems can be remarkably specific; for example, prophage Charlie encodes a defence system that offered protection against only one phage of many tested. A different M. smegmatis prophage encoded a (p)ppGpp synthetase similar to RelA/SpoT that is proposed to be inactivated by a prophage ‘regulator’ protein. Lytic phage replication leads to rapid dissociation of the synthetase from the regulator, (p)ppGpp accumulation, growth cessation and stalled phage development123. Another phage, Fruitloop, encodes an immunity protein that interacts with Wag31, a cell-wall synthesis protein in M. smegmatis. Fruitloop inhibits superinfection by other phages that are thought to require Wag31 for DNA injection124. Prophage-mediated phage defences are widespread. Indeed, a systematic study revealed that Pseudomonas lysogens have diverse prophage-encoded defences21. Furthermore, filamentous phages of the Inoviridae family that cause chronic infections were recently shown to have multiple toxin–antitoxin systems and superinfection systems125. As these systems are encoded by the phage, phage escape represents phage–phage coevolution. Accordingly, many genes of unknown function in prophages, especially within morons, may protect from superinfection and this knowledge may accelerate the identification of candidate resistance systems.
A new world of diverse resistance systems
Defence systems are often clustered in defence islands in bacterial genomes and unknown genes within these regions have been proposed to encode anti-phage systems54,126. This was supported by the discovery of BREX, DISARM and the Stk2 kinase31,32,34,105, and was the premise for a search that uncovered 26 broadly distributed candidate defence systems127. Nine systems have been validated as anti-phage systems, some of which protect against specific phages, whereas others provided broader defence. Although the mechanisms are undetermined, multiple protein domains have been identified that are typical for phage-defence systems (for example, helicases and nucleases), in addition to proteins that have been proposed to be repurposed for phage defence. For example, components of the Zorya system, which is proposed to be an Abi system, show homology to the MotAB proteins that form the stator of the flagella complex, and are hypothesized to form a membrane channel that results in depolarization and cell death upon phage infection127.
Prokaryotic Argonaute (Ago) proteins are also found in defence islands nearby other newly discovered and validated systems (for example, Thoeris), suggesting that they may also elicit phage defence127,128. Moreover, the eukaryote Ago proteins are key proteins in RNA-interference systems, and prokaryotic Ago proteins function as nucleic-acid-guided nucleases129. Generally, prokaryotic Ago proteins generate and associate with short-interfering DNA or RNA guides. The single-stranded guides facilitate the identification of the complementary sequence by prokaryotic Ago, which cleaves the target strand or produces double-stranded breaks130,131,132,133. Following the discovery of prokaryotic Ago proteins, further parallels are being drawn with the eukaryotic immune systems—for example, with the eukaryotic cGAS–STING pathway that senses viral DNA and activates an innate immune response. Recently, prokaryotic cGAS homologues, which cluster near defence islands, have been identified134. These cGAS-encoding genes reside in operons that include a phospholipase and two other genes that contain eukaryotic-like domains that are required for defence against some phages, but are dispensable for the defence against others. This pathway was named CBASS (cyclic-oligonucleotide-based anti-phage signalling system) and is triggered by an unidentified signal that causes cGAS to produce cyclic GMP–AMP (cGAMP). cGAMP activates the phospholipase, which aborts a range of dsDNA phages by eliciting membrane damage and cell death134. A second example is the eukaryotic-like HORMA proteins that are present in various bacteria, including E. coli135. These proteins sense unknown phage product(s) and, once activated, the HORMA domain activates a cGAS/DncV-like nucleotidyltransferase that produces the second messenger cyclic tri-AMP. Cyclic tri-AMP causes dsDNA cleavage by activating an endonuclease, which in E. coli confers λ immunity135. It is currently unknown whether this results in abortive infection or targeted destruction of the phage135,136. The discovery of eukaryotic-like defences in prokaryotes suggest that systematic searches for homologues in bacteria may uncover many new anti-phage systems.
Recently, a new type of phage defence was discovered that relies on small molecules rather than proteins137. This chemical defence is widespread in Streptomyces, a genus known for the prolific production of bioactive secondary metabolites. The metabolites block genome propagation by intercalating dsDNA. Because many secondary metabolites can diffuse and thus function outside of the cell, this has been proposed as an innate defence that protects bacteria before phage infection137. However, various aspects of the chemical defence strategy remain unclear, such as how the phage DNA is recognized as non-self.
With such a diversity of defence systems, the arms race has escalated. Indeed, jumbo phages produce nucleus-like structures inside the infected bacterium, in which phage DNA replication and transcription occur138,139. In P. aeruginosa, this nucleus-like structure protects ϕKZ from type I-C, II-A and V-A CRISPR–Cas and a type I RM system139. Moreover, in Serratia, a distinct nucleus-forming jumbo phage evades the native DNA-targeting type I-F and I-E CRISPR–Cas systems140. However, phage mRNA translated in the cytoplasm is susceptible to RNA-targeting by Cas13139 or type III-A defence140 in P. aeruginosa and Serratia, respectively. Therefore, this physical occlusion of the phage genome appears to be a widespread method to overcome anti-phage systems and this is supported by a paucity of type I spacers that target jumbo phages in nature, whereas type III-A spacers are overrepresented140.
Finally, extracellular chemicals not only engage in direct resistance against phages (for example, chemical defence137), but also facilitate communication to pre-empt bacteria to increase their immunity. Indeed, quorum sensing—cell-density-dependent signalling—upregulates bacterially encoded CRISPR–Cas and downregulates surface receptors when populations would otherwise be at increased risk of a phage epidemic141,142,143. Perhaps unsurprisingly, phages also use communication to ensure productive infection144,145. These peptide communication systems (which are also known as arbitrium) are diverse and widespread, and inform phages about host availability. Arbitrium has been proposed to limit phage-induced host decimation by determining whether phages enter the lytic or lysogenic life cycle144,145,146,147,148. Phages also encode LuxR-type proteins, which respond to Gram-negative quorum-sensing signals149,150 and quorum-sensing genes are also present in Gram-positive phages151. Although the function of phage quorum-sensing genes remains to be elucidated, they might allow phages to sense host density149. These examples of communication between phages and bacteria raise the question whether bacteria and phages engage in ‘espionage’, where either party listens in to, or interferes with, the communications of the other to manipulate the outcome for their own benefit. However, the roles and implications of phage–phage and phage–bacteria communications remain to be understood.
Perspectives
There is a clear diversity of phage-resistance mechanisms and ways that phages evade these systems. This knowledge is informing microbiology, the potential of phage-inspired therapeutics and new biotechnological tools. Despite considerable advances, we are far from understanding bacterial defences and phage counter-adaptation across scales—from molecules, single cells, communities, ecosystems and through to the global scale (Box 1). Furthermore, the recent discovery of completely new systems demonstrates that our view of the defence arsenal is incomplete, and that their identification requires more systematic approaches. Increased sequencing data will expand the success of bioinformatics strategies, but these need to be complemented by high-throughput experimental techniques. For example, phage-based positive selection of new anti-phage systems from metagenomic libraries could be exploited in a similar manner to those reported for anti-CRISPR discovery152. To advance the field, both sides of this arms race, the bacteria and the phages, must be considered.
In terms of bacterial defences, critical gaps exist in our understanding of molecular mechanisms—for both old and new systems—and new techniques should be applied to uncover their mode of action. Determining the molecular mechanisms of diverse defences will undoubtedly lead to both fundamental biological knowledge and new technologies—as exemplified by the exploitation of CRISPR–Cas and RM systems. Furthermore, most defence systems have been studied without considering other co-existing immune mechanisms. Indeed, bacteria often have multiple CRISPR–Cas systems, in addition to other innate defences. How these function together—whether redundantly or synergistically—is not well understood, but they may help bacteria to resist diverse phages and overcome escape phages153. In fact, RM and CRISPR–Cas act together to increase phage resistance, and crosstalk between CRISPR–Cas systems can provide protection against escape phages154,155. In addition, each defence system is likely to have different costs and benefits that depend on the niche inhabited and these factors may be key drivers in the evolutionary selection of defences.
Our understanding of phages is improving, in part due to the increased availability of sequencing data, but given their global abundance, we only have a tiny snapshot of this ever-changing community. Poor functional gene annotations highlight the gaps in fundamental phage biology and hinder our ability to understand their interactions with bacterial immune systems. We can focus on genes that probably influence bacterial immunity. For example, prophage-encoded defences and anti-defences are commonly found in particular genomic locations and their discovery has been facilitated by comparative genomics of phage families. Moreover, early expressed genes often have important roles in anti-defence or bacterial takeover156; however, studying these genes has been hampered by the paucity of genetic tools for phages. Reassuringly, phages are becoming genetically tractable due to CRISPR–Cas methods. To realize the ecological importance, and the therapeutic and biotechnological implications of bacterial immune systems, mechanistic studies must be complemented with evolutionary and ecological experiments to illuminate how molecular events scale to global microbial processes.
References
Fortier, L.-C. & Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365 (2013).
Suttle, C. A. Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812 (2007).
Hurwitz, B. L., Hallam, S. J. & Sullivan, M. B. Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol. 14, R123 (2013).
Dy, R. L., Richter, C., Salmond, G. P. C. & Fineran, P. C. Remarkable mechanisms in microbes to resist phage infections. Annu. Rev. Virol. 1, 307–331 (2014).
van Houte, S., Buckling, A. & Westra, E. R. Evolutionary ecology of prokaryotic immune mechanisms. Microbiol. Mol. Biol. Rev. 80, 745–763 (2016).
Samson, J. E., Magadán, A. H., Sabri, M. & Moineau, S. Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 11, 675–687 (2013).
Gregory, A. C. et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell 177, 1109–1123 (2019).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019). The first published study in which a genetically engineered phage cocktail is used to treat a bacterial infection in a human patient.
O’Sullivan, L., Bolton, D., McAuliffe, O. & Coffey, A. Bacteriophages in food applications: from foe to friend. Annu. Rev. Food Sci. Technol. 10, 151–172 (2019).
Foss, D. V., Hochstrasser, M. L. & Wilson, R. C. Clinical applications of CRISPR-based genome editing and diagnostics. Transfusion 59, 1389–1399 (2019).
Ackermann, H. W. Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 51, 135–201 (1998).
Clokie, M. R. J., Millard, A. D., Letarov, A. V. & Heaphy, S. Phages in nature. Bacteriophage 1, 31–45 (2011).
Seed, K. D. et al. Evolutionary consequences of intra-patient phage predation on microbial populations. eLife 3, e03497 (2014).
Manning, A. J. & Kuehn, M. J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11, 258 (2011).
Reyes-Robles, T. et al. Vibrio cholerae outer membrane vesicles inhibit bacteriophage infection. J. Bacteriol. 200, e00792-17 (2018).
Tzipilevich, E., Habusha, M. & Ben-Yehuda, S. Acquisition of phage sensitivity by bacteria through exchange of phage receptors. Cell 168, 186–199 (2017). This study showed that co-culturing of cells allowed for the transfer of phage receptors from sensitive bacteria to resistant bacteria.
Moxon, R., Bayliss, C. & Hood, D. Bacterial contingency loci: the role of simple sequence DNA repeats in bacterial adaptation. Annu. Rev. Genet. 40, 307–333 (2006).
Ohshima, Y., Schumacher-Perdreau, F., Peters, G. & Pulverer, G. The role of capsule as a barrier to bacteriophage adsorption in an encapsulated Staphylococcus simulans strain. Med. Microbiol. Immunol. 177, 229–233 (1988).
Scanlan, P. D. & Buckling, A. Co-evolution with lytic phage selects for the mucoid phenotype of Pseudomonas fluorescens SBW25. ISME J. 6, 1148–1158 (2012).
Harvey, H. et al. Pseudomonas aeruginosa defends against phages through type IV pilus glycosylation. Nat. Microbiol. 3, 47–52 (2018).
Bondy-Denomy, J. et al. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10, 2854–2866 (2016).
Meyer, J. R. et al. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335, 428–432 (2012).
Petrie, K. L. et al. Destabilizing mutations encode nongenetic variation that drives evolutionary innovation. Science 359, 1542–1545 (2018).
Habusha, M., Tzipilevich, E., Fiyaksel, O. & Ben-Yehuda, S. A mutant bacteriophage evolved to infect resistant bacteria gained a broader host range. Mol. Microbiol. 111, 1463–1475 (2019).
Schwarzer, D. et al. A multivalent adsorption apparatus explains the broad host range of phage phi92: a comprehensive genomic and structural analysis. J. Virol. 86, 10384–10398 (2012).
Fernandes, S. & São-José, C. Enzymes and mechanisms employed by tailed bacteriophages to breach the bacterial cell barriers. Viruses 10, 396 (2018).
Nobrega, F. L. et al. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 16, 760–773 (2018).
Oliveira, P. H., Touchon, M. & Rocha, E. P. C. The interplay of restriction–modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 42, 10618–10631 (2014).
Loenen, W. A. M., Dryden, D. T. F., Raleigh, E. A., Wilson, G. G. & Murray, N. E. Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res. 42, 3–19 (2014).
Sumby, P. & Smith, M. C. M. Genetics of the phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2). Mol. Microbiol. 44, 489–500 (2002).
Hoskisson, P. A., Sumby, P. & Smith, M. C. M. The phage growth limitation system in Streptomyces coelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology 477, 100–109 (2015).
Goldfarb, T. et al. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 34, 169–183 (2015). This study describes an RM-like system that prevents infection through a mechanism other than DNA cleavage or degradation.
Gordeeva, J. et al. BREX system of Escherichia coli distinguishes self from non-self by methylation of a specific DNA site. Nucleic Acids Res. 47, 253–265 (2019).
Ofir, G. et al. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat. Microbiol. 3, 90–98 (2018). Discovery of a type of widespread RM-like phage-resistance system.
Krüger, D. H. & Bickle, T. A. Bacteriophage survival: multiple mechanisms for avoiding the deoxyribonucleic acid restriction systems of their hosts. Microbiol. Rev. 47, 345–360 (1983).
Vasu, K. & Nagaraja, V. Diverse functions of restriction–modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77, 53–72 (2013).
Korona, R., Korona, B. & Levin, B. R. Sensitivity of naturally occurring coliphages to type I and type II restriction and modification. J. Gen. Microbiol. 139, 1283–1290 (1993).
Pleška, M. & Guet, C. C. Effects of mutations in phage restriction sites during escape from restriction–modification. Biol. Lett. 13, 20170646 (2017).
Kulikov, E. E. et al. Genomic sequencing and biological characteristics of a novel Escherichia coli bacteriophage 9g, a putative representative of a new Siphoviridae genus. Viruses 6, 5077–5092 (2014).
Loenen, W. A. M. & Murray, N. E. Modification enhancement by the restriction alleviation protein (Ral) of bacteriophage λ. J. Mol. Biol. 190, 11–22 (1986).
Semerjian, A. V., Malloy, D. C. & Poteete, A. R. Genetic structure of the bacteriophage P22 PL operon. J. Mol. Biol. 207, 1–13 (1989).
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A. & van Sinderen, D. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555 (2013).
Schlagman, S. L. & Hattman, S. Molecular cloning of a functional dam + gene coding for phage T4 DNA adenine methylase. Gene 22, 139–156 (1983).
Günthert, U. & Reiners, L. Bacillus subtilis phage SPR codes for a DNA methyltransferase with triple sequence specificity. Nucleic Acids Res. 15, 3689–3702 (1987).
Iida, S., Streiff, M. B., Bickle, T. A. & Arber, W. Two DNA antirestriction systems of bacteriophage P1, darA, and darB: characterization of darA − phages. Virology 157, 156–166 (1987).
Piya, D., Vara, L., Russell, W. K., Young, R. & Gill, J. J. The multicomponent antirestriction system of phage P1 is linked to capsid morphogenesis. Mol. Microbiol. 105, 399–412 (2017).
Atanasiu, C., Su, T. J., Sturrock, S. S. & Dryden, D. T. F. Interaction of the ocr gene 0.3 protein of bacteriophage T7 with EcoKI restriction/modification enzyme. Nucleic Acids Res. 30, 3936–3944 (2002).
Walkinshaw, M. D. et al. Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol. Cell 9, 187–194 (2002).
Laity, C., Chater, K. F., Lewis, C. G. & Buttner, M. J. Genetic analysis of the φC31-specific phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2). Mol. Microbiol. 7, 329–336 (1993).
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Jackson, S. A. et al. CRISPR–Cas: adapting to change. Science 356, eaal5056 (2017).
Hille, F. et al. The biology of CRISPR–Cas: backward and forward. Cell 172, 1239–1259 (2018).
Koonin, E. V. & Makarova, K. S. Origins and evolution of CRISPR–Cas systems. Phil. Trans. R. Soc. B 374, 20180087 (2019).
Koonin, E. V., Makarova, K. S. & Zhang, F. Diversity, classification and evolution of CRISPR–Cas systems. Curr. Opin. Microbiol. 37, 67–78 (2017).
Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Classification and nomenclature of CRISPR–Cas systems: where from here? CRISPR J. 1, 325–336 (2018).
Brouns, S. J. J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008).
Semenova, E. et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl Acad. Sci. USA 108, 10098–10103 (2011).
Datsenko, K. A. et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3, 945 (2012).
Strotskaya, A. et al. The action of Escherichia coli CRISPR–Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 45, 1946–1957 (2017).
Cady, K. C., Bondy-Denomy, J., Heussler, G. E., Davidson, A. R. & O’Toole, G. A. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 194, 5728–5738 (2012).
Westra, E. R. et al. Parasite exposure drives selective evolution of constitutive versus inducible defense. Curr. Biol. 25, 1043–1049 (2015).
Watson, B. N. J., Staals, R. H. J. & Fineran, P. C. CRISPR–Cas-mediated phage resistance enhances horizontal gene transfer by transduction. mBio 9, e02406-17 (2018).
Taylor, H. N. et al. Structural basis of type IV CRISPR RNA biogenesis by a Cas6 endoribonuclease. RNA Biol. 16, 1438–1447 (2019).
Özcan, A. et al. Type IV CRISPR RNA processing and effector complex formation in Aromatoleum aromaticum. Nat. Microbiol. 4, 89–96 (2019).
Deng, L., Garrett, R. A., Shah, S. A., Peng, X. & She, Q. A novel interference mechanism by a type IIIB CRISPR–Cmr module in Sulfolobus. Mol. Microbiol. 87, 1088–1099 (2013).
Samai, P. et al. Co-transcriptional DNA and RNA cleavage during type III CRISPR–Cas immunity. Cell 161, 1164–1174 (2015).
Jiang, W., Samai, P. & Marraffini, L. A. Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR–Cas immunity. Cell 164, 710–721 (2016).
Pyenson, N. C., Gayvert, K., Varble, A., Elemento, O. & Marraffini, L. A. Broad targeting specificity during bacterial type III CRISPR–Cas immunity constrains viral escape. Cell Host Microbe 22, 343–353 (2017).
Millen, A. M., Horvath, P., Boyaval, P. & Romero, D. A. Mobile CRISPR/Cas-mediated bacteriophage resistance in Lactococcus lactis. PLoS ONE 7, e51663 (2012).
Tamulaitis, G. et al. Programmable RNA shredding by the type III-A CRISPR–Cas system of Streptococcus thermophilus. Mol. Cell 56, 506–517 (2014).
Goldberg, G. W., Jiang, W., Bikard, D. & Marraffini, L. A. Conditional tolerance of temperate phages via transcription-dependent CRISPR–Cas targeting. Nature 514, 633–637 (2014).
Kazlauskiene, M., Kostiuk, G., Venclovas, Č., Tamulaitis, G. & Siksnys, V. A cyclic oligonucleotide signaling pathway in type III CRISPR–Cas systems. Science 357, 605–609 (2017).
Niewoehner, O. et al. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548 (2017).
Rostøl, J. T. & Marraffini, L. A. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR–Cas immunity. Nat. Microbiol. 4, 656–662 (2019).
Varble, A. & Marraffini, L. A. Three new Cs for CRISPR: collateral, communicate, cooperate. Trends Genet. 35, 446–456 (2019).
McGinn, J. & Marraffini, L. A. CRISPR–Cas systems optimize their immune response by specifying the site of spacer integration. Mol. Cell 64, 616–623 (2016).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell 163, 759–771 (2015).
Vlot, M. et al. Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR–Cas effector complexes. Nucleic Acids Res. 46, 873–885 (2018).
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016).
Meeske, A. J., Nakandakari-Higa, S. & Marraffini, L. A. Cas13-induced cellular dormancy prevents the rise of CRISPR-resistant bacteriophage. Nature 570, 241–245 (2019). This study demonstrates that a CRISPR–Cas variant can provide broad phage protection by inducing bacterial dormancy.
Watson, B. N. J. et al. Different genetic and morphological outcomes for phages targeted by single or multiple CRISPR–Cas spacers. Phil. Trans. R. Soc. B 374, 20180090 (2019).
Deveau, H. et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390–1400 (2008).
Sun, C. L. et al. Phage mutations in response to CRISPR diversification in a bacterial population. Environ. Microbiol. 15, 463–470 (2013).
Martel, B. & Moineau, S. CRISPR–Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 42, 9504–9513 (2014).
Paez-Espino, D. et al. CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. mBio 6, e00262-15 (2015).
Nicholson, T. J. et al. Bioinformatic evidence of widespread priming in type I and II CRISPR–Cas systems. RNA Biol. 16, 566–576 (2019).
Nussenzweig, P. M., McGinn, J. & Marraffini, L. A. Cas9 cleavage of viral genomes primes the acquisition of new immunological memories. Cell Host Microbe 26, 515–526 (2019).
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L. & Davidson, A. R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013). The discovery and characterization of anti-CRISPRs.
Hwang, S. & Maxwell, K. L. Meet the anti-CRISPRs: widespread protein inhibitors of CRISPR–Cas systems. CRISPR J. 2, 23–30 (2019).
Trasanidou, D. et al. Keeping CRISPR in check: diverse mechanisms of phage-encoded anti-CRISPRS. FEMS Microbiol. Lett. 366, fnz098 (2019).
Bhoobalan-Chitty, Y., Baek Johansen, T., Di Cianni, N. & Peng, X. Inhibition of type III CRISPR–Cas immunity by an archaeal virus-encoded anti-CRISPR protein. Cell 179, 448–458 (2019).
Dong, L. et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 26, 308–314 (2019).
Knott, G. J. et al. Broad-spectrum enzymatic inhibition of CRISPR–Cas12a. Nat. Struct. Mol. Biol. 26, 315–321 (2019).
Landsberger, M. et al. Anti-CRISPR phages cooperate to overcome CRISPR–Cas immunity. Cell 174, 908–916 (2018). This study demonstrates that phages cooperate through their anti-CRISPRs to immunosuppress the host CRISPR–Cas system.
Borges, A. L. et al. Bacteriophage cooperation suppresses CRISPR–Cas3 and Cas9 immunity. Cell 174, 917–925 (2018). This work describes how phages cooperate through their anti-CRISPRs to inhibit bacterial adaptive immunity.
Seed, K. D., Lazinski, D. W., Calderwood, S. B. & Camilli, A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494, 489–491 (2013).
Hargreaves, K. R., Flores, C. O., Lawley, T. D. & Clokie, M. R. J. Abundant and diverse clustered regularly interspaced short palindromic repeat spacers in Clostridium difficile strains and prophages target multiple phage types within this pathogen. mBio 5, e01045-13 (2014).
Montgomery, M. T., Guerrero Bustamante, C. A., Dedrick, R. M., Jacobs-Sera, D. & Hatfull, G. F. Yet more evidence of collusion: a new viral defense system encoded by Gordonia phage CarolAnn. mBio 10, e02417-18 (2019).
Page, R. & Peti, W. Toxin–antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214 (2016).
Fineran, P. C. et al. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl Acad. Sci. USA 106, 894–899 (2009).
Al-Shayeb, B. et al. Clades of huge phage from across Earth’s ecosystems. Preprint at bioRxiv https://doi.org/10.1101/572362 (2019).
Faure, G. et al. CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol. 17, 513–525 (2019).
Chopin, M.-C., Chopin, A. & Bidnenko, E. Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 8, 473–479 (2005).
Labrie, S. J., Samson, J. E. & Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327 (2010).
Depardieu, F. et al. A eukaryotic-like serine/threonine kinase protects Staphylococci against phages. Cell Host Microbe 20, 471–481 (2016). This study describes the discovery of an Abi system with similarities to eukaryotic defences.
Parma, D. H. et al. The Rex system of bacteriophage lambda: tolerance and altruistic cell death. Genes Dev. 6, 497–510 (1992).
Gentile, G. M. et al. More evidence of collusion: a new prophage-mediated viral defense system encoded by mycobacteriophage Sbash. mBio 10, e00196-19 (2019).
Samson, J. E., Spinelli, S., Cambillau, C. & Moineau, S. Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin–antitoxin system. Mol. Microbiol. 87, 756–768 (2013).
Dy, R. L., Przybilski, R., Semeijn, K., Salmond, G. P. C. & Fineran, P. C. A widespread bacteriophage abortive infection system functions through a type IV toxin–antitoxin mechanism. Nucleic Acids Res. 42, 4590–4605 (2014).
Pecota, D. C. & Wood, T. K. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J. Bacteriol. 178, 2044–2050 (1996).
Koga, M., Otsuka, Y., Lemire, S. & Yonesaki, T. Escherichia coli rnlA and rnlB compose a novel toxin–antitoxin system. Genetics 187, 123–130 (2011).
Hazan, R. & Engelberg-Kulka, H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol. Genet. Genomics 272, 227–234 (2004).
Snyder, L. Phage-exclusion enzymes: a bonanza of biochemical and cell biology reagents? Mol. Microbiol. 15, 415–420 (1995).
Otsuka, Y. & Yonesaki, T. Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Mol. Microbiol. 83, 669–681 (2012).
Labrie, S. J. & Moineau, S. Abortive infection mechanisms and prophage sequences significantly influence the genetic makeup of emerging lytic lactococcal phages. J. Bacteriol. 189, 1482–1487 (2007).
Blower, T. R., Evans, T. J., Przybilski, R., Fineran, P. C. & Salmond, G. P. C. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 8, e1003023 (2012).
Sberro, H. et al. Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol. Cell 50, 136–148 (2013).
Alawneh, A. M., Qi, D., Yonesaki, T. & Otsuka, Y. An ADP-ribosyltransferase Alt of bacteriophage T4 negatively regulates the Escherichia coli|MazF toxin of a toxin–antitoxin module. Mol. Microbiol. 99, 188–198 (2016).
Lossouarn, J. et al. Enterococcus faecalis Countermeasures defeat a virulent Picovirinae bacteriophage. Viruses 11, 48 (2019).
Blower, T. R. et al. Evolution of Pectobacterium bacteriophage ΦM1 to escape two bifunctional type III toxin–antitoxin and abortive infection systems through mutations in a single viral gene. Appl. Environ. Microbiol. 83, e03229-16 (2017).
Chen, B., Akusobi, C., Fang, X. & Salmond, G. P. C. Environmental T4-family bacteriophages evolve to escape abortive infection via multiple routes in a bacterial host employing ‘altruistic suicide’ through type III toxin–antitoxin systems. Front. Microbiol. 8, 127 (2018).
Cumby, N., Davidson, A. R. & Maxwell, K. L. The moron comes of age. Bacteriophage 2, e23146 (2012).
Dedrick, R. M. et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2, 16251 (2017). Demonstration that mycobacterial prophages encode diverse anti-phage defence systems and the ways that phages can evade these defences.
Ko, C.-C. & Hatfull, G. F. Mycobacteriophage Fruitloop gp52 inactivates Wag31 (DivIVA) to prevent heterotypic superinfection. Mol. Microbiol. 108, 443–460 (2018).
Roux, S. et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 4, 1895–1906 (2019).
Makarova, K. S., Wolf, Y. I., Snir, S. & Koonin, E. V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193, 6039–6056 (2011).
Doron, S. et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120–13 (2018). This study describes the identification of numerous new anti-phage systems in defence islands.
Willkomm, S., Makarova, K. S. & Grohmann, D. DNA silencing by prokaryotic Argonaute proteins adds a new layer of defense against invading nucleic acids. FEMS Microbiol. Rev. 42, 376–387 (2018).
Hegge, J. W., Swarts, D. C. & van der Oost, J. Prokaryotic Argonaute proteins: novel genome-editing tools? Nat. Rev. Microbiol. 16, 5–11 (2018).
Swarts, D. C. et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507, 258–261 (2014).
Swarts, D. C. et al. Autonomous generation and loading of DNA guides by bacterial Argonaute. Mol. Cell 65, 985–998 (2017).
Olovnikov, I., Chan, K., Sachidanandam, R., Newman, D. K. & Aravin, A. A. Bacterial Argonaute samples the transcriptome to identify foreign DNA. Mol. Cell 51, 594–605 (2013).
Zander, A. et al. Guide-independent DNA cleavage by archaeal Argonaute from Methanocaldococcus jannaschii. Nat. Microbiol. 2, 17034 (2017).
Cohen, D. et al. Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695 (2019). This study showed that a eukaryotic anti-viral signalling pathway is also present in prokaryotes, and that this pathway offers protection from a range of phages.
Ye, Q. et al. HORMA domain proteins and a Pch2-like ATPase regulate bacterial cGAS-like enzymes to mediate bacteriophage immunity. Preprint at bioRxiv https://doi.org/10.1101/694695 (2019).
Lau, R. K. et al. Structure and mechanism of a cyclic trinucleotide-activated bacterial endonuclease mediating bacteriophage immunity. Preprint at bioRxiv https://doi.org/10.1101/694703 (2019).
Kronheim, S. et al. A chemical defence against phage infection. Nature 564, 283–286 (2018). The first example of chemical defences against phages.
Chaikeeratisak, V. et al. Assembly of a nucleus-like structure during viral replication in bacteria. Science 355, 194–197 (2017). This study describes how jumbophages can produce nucleus-like protein shells inside a bacterium for phage replication.
Mendoza, S. D. et al. A bacteriophage nucleus-like compartment shields DNA from CRISPR nucleases. Nature https://doi.org/10.1038/s41586-019-1786-y (2019).
Malone, L. M. et al. A jumbo phage that forms a nucleus-like structure evades CRISPR–Cas DNA targeting but is vulnerable to type III RNA-based immunity. Nat. Microbiol. https://doi.org/10.1038/s41564-019-0612-5 (2019).
Høyland-Kroghsbo, N. M., Maerkedahl, R. B. & Svenningsen, S. L. A quorum-sensing-induced bacteriophage defense mechanism. mBio 4, e00362-12 (2013).
Høyland-Kroghsbo, N. M. et al. Quorum sensing controls the Pseudomonas aeruginosa CRISPR–Cas adaptive immune system. Proc. Natl Acad. Sci. USA 114, 131–135 (2017).
Patterson, A. G. et al. Quorum sensing controls adaptive immunity through the regulation of multiple CRISPR–Cas systems. Mol. Cell 64, 1102–1108 (2016). Demonstration that bacteria use quorum-sensing communication to coordinate multiple CRISPR–Cas systems at a population level
Erez, Z. et al. Communication between viruses guides lysis–lysogeny decisions. Nature 541, 488–493 (2017). This study reveals that phages use communication to guide the replication strategy used by progeny phages.
Stokar-Avihail, A., Tal, N., Erez, Z., Lopatina, A. & Sorek, R. Widespread utilization of peptide communication in phages infecting soil and pathogenic bacteria. Cell Host Microbe 25, 746–755 (2019).
Dou, C. et al. Structural and functional insights into the regulation of the lysis–lysogeny decision in viral communities. Nat. Microbiol. 3, 1285–1294 (2018).
Gallego del Sol, F., Penadés, J. R. & Marina, A. Deciphering the molecular mechanism underpinning phage arbitrium communication systems. Mol. Cell 74, 59–72 (2019).
Guan, Z. et al. Structural insights into DNA recognition by AimR of the arbitrium communication system in the SPbeta phage. Cell Discov. 5, 29 (2019).
Silpe, J. E. & Bassler, B. L. Phage-encoded LuxR-type receptors responsive to host-produced bacterial quorum-sensing autoinducers. mBio 10, e00638-19 (2019).
Silpe, J. E. & Bassler, B. L. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 176, 268–280 (2019).
Hargreaves, K. R., Kropinski, A. M. & Clokie, M. R. J. What does the talking?: quorum sensing signalling genes discovered in a bacteriophage genome. PLoS ONE 9, e85131 (2014).
Uribe, R. V. et al. Discovery and characterization of Cas9 inhibitors disseminated across seven bacterial phyla. Cell Host Microbe 25, 233–241 (2019).
Koonin, E. V., Makarova, K. S. & Wolf, Y. I. Evolutionary genomics of defense systems in archaea and bacteria. Annu. Rev. Microbiol. 71, 233–261 (2017).
Dupuis, M.-È. V., Villion, M., Magadán, A. H. & Moineau, S. CRISPR–Cas and restriction–modification systems are compatible and increase phage resistance. Nat. Commun. 4, 2087 (2013).
Silas, S. et al. Type III CRISPR–Cas systems can provide redundancy to counteract viral escape from type I systems. eLife 6, e27601 (2017).
Van den Bossche, A. et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. J. Proteome Res. 13, 4446–4456 (2014).
Buckling, A. & Brockhurst, M. in Evolutionary Systems Biology Vol. 751 (ed. Soyer O.) 347–370 (Springer, 2012).
Koskella, B. & Brockhurst, M. A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931 (2014).
Scanlan, P. D. Bacteria–bacteriophage coevolution in the human gut: implications for microbial diversity and functionality. Trends Microbiol. 25, 614–623 (2017).
Horne, M. T. Coevolution of Escherichia coli and bacteriophages in chemostat culture. Science 168, 992–993 (1970).
Lenski, R. E. & Levin, B. R. Constraints on the coevolution of bacteria and virulent phage: a model, some experiments, and predictions for natural communities. Am. Nat. 125, 585–602 (1985).
Buckling, A. & Rainey, P. B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. R. Soc. Lond. B 269, 931–936 (2002).
Buckling, A. & Rainey, P. B. The role of parasites in sympatric and allopatric host diversification. Nature 420, 496–499 (2002).
Brockhurst, M. A., Morgan, A. D., Fenton, A. & Buckling, A. Experimental coevolution with bacteria and phage: the Pseudomonas fluorescens—Φ2 model system. Infect. Genet. Evol. 7, 547–552 (2007).
Gómez, P. & Buckling, A. Bacteria–phage antagonistic coevolution in soil. Science 332, 106–109 (2011).
Andersson, A. F. & Banfield, J. F. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320, 1047–1050 (2008).
Stern, A., Mick, E., Tirosh, I., Sagy, O. & Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 22, 1985–1994 (2012).
Emerson, J. B. et al. Virus–host and CRISPR dynamics in Archaea-dominated hypersaline Lake Tyrrell, Victoria, Australia. Archaea 2013, 370871 (2013).
Laanto, E., Hoikkala, V., Ravantti, J. & Sundberg, L. R. Long-term genomic coevolution of host–parasite interaction in the natural environment. Nat. Commun. 8, 111 (2017).
Shmakov, S. et al. Discovery and Functional characterization of diverse class 2 CRISPR–Cas systems. Mol. Cell 60, 385–397 (2015).
Burstein, D. et al. New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237–241 (2017).
Weissman, J. L. et al. Immune loss as a driver of coexistence during host–phage coevolution. ISME J. 12, 585–597 (2018).
Jackson, S. A., Birkholz, N., Malone, L. M. & Fineran, P. C. Imprecise spacer acquisition generates CRISPR–Cas immune diversity through primed adaptation. Cell Host Microbe 25, 250–260 (2019).
Acknowledgements
Research in the Fineran laboratory on phage defence systems is supported by the Marsden Fund, Royal Society of New Zealand, the Bio-Protection Centre of Research Excellence and the University of Otago. We thank N. Birkholz for providing input on the figures and members of the Fineran laboratory for discussions and comments on the manuscript.
Author information
Authors and Affiliations
Contributions
H.G.H., B.N.J.W. and P.C.F. contributed equally to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hampton, H.G., Watson, B.N.J. & Fineran, P.C. The arms race between bacteria and their phage foes. Nature 577, 327–336 (2020). https://doi.org/10.1038/s41586-019-1894-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1894-8
- Springer Nature Limited
This article is cited by
-
Discovery of a high-performance phage-derived promoter/repressor system for probiotic lactobacillus engineering
Microbial Cell Factories (2024)
-
Viruses contribute to microbial diversification in the rumen ecosystem and are associated with certain animal production traits
Microbiome (2024)
-
Exploring the diversity and evolutionary strategies of prophages in Hyphomicrobiales, comparing animal-associated with non-animal-associated bacteria
BMC Microbiology (2024)
-
The potential use of bacteriophages as antibacterial agents against Klebsiella pneumoniae
Virology Journal (2024)
-
Inhibitors of bacterial immune systems: discovery, mechanisms and applications
Nature Reviews Genetics (2024)