

Abstract
Potassium-sensitive hypokalaemic and normokalaemic periodic paralysis are inherited skeletal muscle diseases characterized by episodes of flaccid muscle weakness1,2. They are caused by single mutations in positively charged residues (‘gating charges’) in the S4 transmembrane segment of the voltage sensor of the voltage-gated sodium channel Nav1.4 or the calcium channel Cav1.11,2. Mutations of the outermost gating charges (R1 and R2) cause hypokalaemic periodic paralysis1,2 by creating a pathogenic gating pore in the voltage sensor through which cations leak in the resting state3,4. Mutations of the third gating charge (R3) cause normokalaemic periodic paralysis5 owing to cation leak in both activated and inactivated states6. Here we present high-resolution structures of the model bacterial sodium channel NavAb with the analogous gating-charge mutations7,8, which have similar functional effects as in the human channels. The R2G and R3G mutations have no effect on the backbone structures of the voltage sensor, but they create an aqueous cavity near the hydrophobic constriction site that controls gating charge movement through the voltage sensor. The R3G mutation extends the extracellular aqueous cleft through the entire length of the activated voltage sensor, creating an aqueous path through the membrane. Conversely, molecular modelling shows that the R2G mutation creates a continuous aqueous path through the membrane only in the resting state. Crystal structures of NavAb(R2G) in complex with guanidinium define a potential drug target site. Molecular dynamics simulations illustrate the mechanism of Na+ permeation through the mutant gating pore in concert with conformational fluctuations of the gating charge R4. Our results reveal pathogenic mechanisms of periodic paralysis at the atomic level and suggest designs of drugs that may prevent ionic leak and provide symptomatic relief from hypokalaemic and normokalaemic periodic paralysis.
Similar content being viewed by others


Main
Nav1.4 channels generate action potentials that initiate muscle contraction9. They are complexes of a pore-forming α-subunit and auxiliary β1 subunits9,10,11. The α-subunit contains four homologous domains (I–IV), each with six transmembrane segments (S1–S6). Segments S1–S4 form the voltage sensor, and every third residue in S4 is positively charged. Upon depolarization, S4 moves outward through a narrow gating pore formed by S1–S3, catalysed by interactions with negative or polar residues in S2 and S312. The voltage sensor has an hourglass shape, with a narrow hydrophobic constriction site (HCS) that separates extracellular and intracellular compartments6,11. Water-filled crevices on either side of the HCS focus the membrane electric field, assuring efficient coupling of voltage to conformational changes that open the central pore12,13. Mutations in the arginine gating charges that occupy the HCS cause state-dependent cation leak through the voltage sensor, which we term ‘gating pore current’14,15.
Missense mutations of arginine gating charges in S4 of Nav1.4 cause hypokalaemic periodic paralysis and normokalaemic periodic paralysis1,2,16,17. Mutations of R1 in domains I or III to H or Q, or mutation of R2 in domains I, II and III to W, G, Q or S cause hypokalaemic periodic paralysis1,2,16,17. Mutations of R3 in domain II to G, Q or W, or of R3 in domain III to H or C cause normokalaemic periodic paralysis4,16,17. All these mutations result in non-selective gating pore current through the voltage sensor3,4,16,17,18,19. Increased inward leak leads to Na+ overload, sustained depolarization and action potential failure, which paralyze skeletal muscles3,16,17,18,19. These pathophysiological effects suggest that mutations that cause hypokalaemic periodic paralysis result in an open aqueous pathway for ion movement in the resting state of the voltage sensor, but not in the activated state, and mutations that cause normokalaemic periodic paralysis result in an open aqueous pathway in the activated state, but not in the resting state. Molecular models and mutagenesis studies support this hypothesis20,21,22. To provide direct structural evidence for this pathophysiological mechanism, we introduced mutations known to cause periodic paralysis into NavAb, a voltage-gated Na+ channel from Arcobacter butzleri, the structure of which has been solved at high resolution6,7. We characterized the resulting gating pore currents, solved the structures of mutant gating pores without and with a bound permeant ion, and investigated molecular dynamics23 of ion movement through the gating pores.
To reconstitute pathogenic hypokalaemic periodic paralysis gating pore currents in NavAb, we mutated R2 to S (R2S, analogous to Nav1.4(R672S)) and expressed the mutant in Trichopulsia ni insect cells. Transfected cells were voltage-clamped to −200 mV and depolarized in 10-mV steps to record Na+ currents. Half-maximal activation of central pore currents was observed at Va = −105 ± 0.6 mV (Fig. 1a). To measure gating pore currents, cells were held at −100 mV, at which NavAb is in the slow-inactivated state and exhibits no central pore current. Gating pore current was examined by applying pulses from + 100 to −200 mV in −10 mV steps. A nonlinear leak current component was observed in the resting state, beginning at −110 mV and increasing to −200 mV (Fig. 1b, c).

a, Central pore Na+ currents (inset) and conductance–voltage (G–V) curve for NavAb(R2S) during 200-ms depolarizations from −200 mV to the indicated potentials. Va (the voltage for half-maximal activation) = −105 ± 0.6 mV; slope factor, k = 10 ± 0.9. n = 4. b, c, Gating pore Na+ currents (Igp) and current–voltage (I–V) curves for NavAb(R2S) (blue) or NavAb(WT) (black) during test pulses from −100 mV to the indicated potentials. n = 10. d, Central pore Na+ currents (inset) and G–V curve for NavAb(R3G) during depolarizations from −160 mV to the indicated potentials (filled circles; Va = −24.8 ± 1.1 mV, k = 9 ± 1; n = 4). Voltage dependence of steady-state inactivation (open circles) for NavAb(R3G) (Vh = −47.7 ± 0.4 mV, k = 7.5 ± 0.3 (n = 4); Vh, the voltage for half-maximal inactivation)). e, f, Gating pore Na+ currents and I–V curves for NavAb(R3G) (red) or NavAb(WT) (black) for voltage steps from 0 mV to the indicated potentials. n = 11. g, Gating pore current through NavAb(R2S) for Cs+ (n = 5), K+ (n = 7), Na+ (n = 5), N-methyl-d-glucamine (NMDG, n = 5), guanidinium (G, n = 7), methylguanidinium (MG, n = 5) and ethylguanidinium (EG, n = 5) at −200 mV. ***P = 0.00029. h, Gating pore current through NavAb(R3C) for Cs+ (n = 4), Na+ (n = 6), K+ (n = 6), guanidinium (n = 4) and NMDG (n = 4) at +100 mV. **P = 0.0011. Student’s t-test, two-sided.
Mutations of the gating charge R3 that cause normokalaemic periodic paralysis (Nav1.4(R675G/Q/W)) induce outward gating pore current in activated but not in resting states6. In NavAb(R3G), central pore current was activated between −50 mV and 0 mV (Fig. 1d; Va = −24.8 ± 1.1 mV). Steady-state inactivation was observed from −90 mV to −10 mV with half maximal inactivation at Vh = −47.7 ± 0.4 mV (Fig. 1d). NavAb(R3G) conducted outward gating pore current in both activated and inactivated states at potentials more positive than −60 mV (Fig. 1e, f). These physiological studies demonstrate that NavAb provides an accurate model of Nav1.4, because gating pore current is observed only in the resting state for NavAb(R2S) and only in the activated and inactivated states for NavAb(R3G).
The pathogenic effects of gating pore mutations depend on inward leak of Na+. The R2S mutant gating pore was not significantly selective among Cs+, K+ or Na+ (Fig. 1g, P > 0.7). As is the case for Nav1.424, the gating pore of NavAb(R2S) was exceptionally permeant to guanidinium (about 28-fold greater than Na+), but it was less permeant to methylguanidinium and ethylguanidinium (Fig. 1g). The outward gating pore currents conducted by NavAb(R3G) were higher for Cs+ than for K+ or Na+, which were similar to each other (Fig. 1h). However, NavAb(R3G) was less permeant to guanidinium than to Na+ (Fig. 1h), and it was more than 16-fold less permeant to guanidinium than NavAb(R2S). The weak selectivity of R2S and R3G mutants for different inorganic cations and the high guanidinium permeability through the R2S mutant are characteristic of the corresponding mutations in Nav1.42, further supporting the validity of NavAb as a model for structural studies of gating pore mutations.
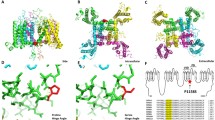
To elucidate the structure of a pathogenic gating pore in its conductive conformation in an activated voltage sensor, we solved the structure of a NavAb analogue of a normokalaemic periodic paralysis-causing mutation, NavAb(R3G), at 2.7 Å resolution (Fig. 2). Voltage-gated sodium channels have a central pore module surrounded by four symmetrically located voltage sensors (Fig. 2a). The voltage sensors of NavAb and Nav1.4 are very similar in amino acid sequence and structure (Extended Data Figs. 1 and 2). The voltage sensors of wild-type (WT) NavAb and NavAb(R3G) crystallize in the same conformation, with a Cα root mean square deviation (r.m.s.d.) of 0.39 Å (Fig. 2b, Extended Data Fig. 3). These results indicate that the R3G mutation does not perturb the overall structure of the voltage sensor and, therefore, that its pathogenic effects are caused by the loss of the R3 side chain. These channels crystallize with an activated voltage sensor6,7 (Fig. 2c), as would be expected at 0 mV. In NavAb(WT), R1, R2 and R3 are located extracellularly relative to the HCS, and their side chains point outward, toward the extracellular milieu (Fig. 2c). By contrast, R4 is located intracellularly relative to the HCS and its side chain points inward towards the cytosol (Fig. 2c). When viewed from the extracellular side, there is no water-accessible path into the cell through the wild-type voltage sensor (Fig. 2d); however, we observed a deep solvent-accessible cleft extending down to the R4 side chain in NavAb(R3G) (Fig. 2g).

a, Structure of NavAb(R3G) in top view. b, Comparison of the conformations of NavAb(WT) (grey) and NavAb(R3G) (rainbow) voltage sensor in side view. c–e, Structures of NavAb(WT) voltage sensor. c, Side view highlighting gating charges in sticks. d, Top view in space-filling format. e, MOLE2 analysis of water-accessible space in magenta. f–h, Structures of NavAb(R3G) voltage sensor. f, Side view highlighting gating charges. g, Top view in space-filling format. h, MOLE2 analysis of water-filled space in magenta. Green spheres in f and h indicate the positions of the missing side chain of R3. In d and g, the dotted red line circles the position where the gating pore would be in the activated state and the solid red line circles the open gating pore, respectively. See Extended Data Table 1 for details.
Analysis of the structure of chain B of NavAb(WT) using the MOLE2 algorithm revealed an incomplete water-accessible path extending part of the way through the voltage sensor from both extracellular and intracellular sides, which is interrupted at the HCS by R3 (Fig. 2e). Strikingly, in NavAb(R3G), the water-accessible path continues all the way through the voltage sensor, and has a diameter of 2 Å at its narrowest point, similar to the size of Na+ (Fig. 2h). By contrast, in chain A, R4 was captured in a rotamer conformation in which the arginine side chain partially blocks the inner end of the gating pore in NavAb(R3G) (Extended Data Fig. 4a). Previously reported structures of NavAb in the slow-inactivated state show that R4 adopts four slightly different rotamer conformations, with the most open having a diameter of 3 Å25 (Extended Data Fig. 4b). These results elucidate the molecular mechanism by which mutations in S4 cause pathogenic gating pore currents and suggest that ion permeation through the gating pore is controlled dynamically by the state of the voltage sensor and by rotamer conformations of R4.
In contrast to voltage-gated sodium-channel mutations that cause normokalaemic periodic paralysis, those that cause hypokalaemic periodic paralysis result in a channel that conducts gating pore current in the resting state but is closed in the activated state (Fig. 1). Therefore, we hypothesized that NavAb(R2G) would not have a continuous water-accessible path through its gating pore in the activated state. Analysis of the 2.9 Å structure of NavAb(R2G) revealed a gap with additional solvent-accessible area in the extracellular aqueous cleft in comparison to the wild-type channel, but no change in the backbone conformation (Fig. 3a, Extended Data Fig. 3). Although the increased opening of the aqueous cleft in the voltage sensor is evident in space-filling models (Fig. 3b), the R3 and R4 side chains seal the voltage sensor in this activated state, interrupting the transmembrane path and preventing ion conductance. The solvent-accessible area penetrates about 21 Å into the membrane from the extracellular side (Fig. 3c), more than 7 Å deeper than in NavAb(WT) (Fig. 2e), but it does not reach the cytosolic side. This structure illustrates why NavAb(R2G) does not conduct gating pore current in the activated state (Fig. 1).

a–c, Structures of the activated voltage sensor of NavAb(R2G). a, Side view with gating charges highlighted in sticks. b, Top view in space-filling format. The dashed red line indicates the position of the closed gating pore. c, MOLE2 analysis of water-filled space in magenta. d–f, Rosetta structural models of resting state 2 of the voltage sensor were re-optimized with the amino-acid sequence of NavAb for NavAb(WT) (d), NavAb(R2G) (e) and NavAb(R3G) (f). The perspective is rotated approximately 180° around the vertical axis to better illustrate the arginine gating charges in resting state 2. Green spheres represent missing arginine side chains of R2 and R3, respectively. Magenta blobs represent solvent-accessible volume modelled with MOLE2. g, Top view of NavAb(R2G) with one guanidinium bound to each voltage sensor. h, 2mFo−DFc electron density map (blue mesh) of residues around the guanidinium binding site at 1σ. i, Interaction network between guanidinium and amino acids in the voltage sensor of NavAb(R2G). Grey dashed lines show interatomic distances shorter than 4 Å. See Extended Data Table 1 for details.
There are no crystal structures of the voltage sensor of a voltage-gated sodium channel in the resting state, because the resting state is only accessible at negative membrane potentials. However, we developed models of three resting states using disulfide locking of substituted cysteine residues and structure prediction with the Rosetta algorithm26; these are now considered consensus models of the actual resting states27,28. To model an open gating pore with the voltage sensor in the resting state, we introduced the R2G and R3G mutations into these resting-state models and analysed the resulting structures with the MOLE2 algorithm (Fig. 3d–f). There is no continuous path through the voltage sensor in the wild-type resting-state structure (Fig. 3d), whereas the resting state of the NavAb(R2G) voltage sensor contains a continuous water-accessible path through the membrane (Fig. 3e). Loss of the R2 side chain leaves a gap at the HCS that is large enough for Na+ to pass through (Fig. 3e). By contrast, the transmembrane pathway is incomplete in NavAb(R3G) because the R2 side chain occupies the HCS and blocks the gating pore (Fig. 3f). These structural models illustrate how R2 charge mutations that cause hypokalaemic periodic paralysis result in gating pore current in the resting state.
The NavAb(R2S) mutant channel is much more permeant than NavAb(R3G) to guanidinium ions24 (Fig. 1). Guanidinium ions are chemically similar to the distal moiety of the arginine side chain, and guanidine compounds with hydrophobic substituents can block mutant gating pores24. We probed our gating pore structures for guanidinium-binding sites by soaking crystals of NavAb(R2G) and NavAb(R3G) with guanidinium and methylguanidinium to determine whether they would bind in place of the missing side chain of R2 or R3. The crystal structures did not show guanidinium binding to NavAb(R3G). However, crystals of NavAb(R2G) soaked with guanidinium or methylguanidinium diffracted to 2.7 Å and 2.5 Å resolution, respectively, and unambiguous electron density was observed in place of each R2 side chain (Fig. 3g–i, Extended Data Fig. 5a, b). Bound guanidinium is clearly seen in 2Fo−Fc maps (Fig. 3h). E32 and M29 from S1, N49 from S2, R1 and R3 from S4, and Q150 from an adjacent subunit form the binding site for guanidinium (Fig. 3i). M29 and R3 each bind guanidinium through hydrogen bonds (Fig. 3h, i). The carbonyl group of E32 and the carbonyl oxygen of R1 further lock guanidinium in place (Fig. 3h, i). The binding site is flanked by hydrogen bonds from N49 and Q150 that stabilize guanidinium from opposite sides (Fig. 3h, i). The binding site for methylguanidinium is almost identical (Extended Data Fig. 5c, d). These structures capture guanidinium bound at a specific site in the closed R2G gating pore. The amino acid residues responsible for guanidinium binding are highly conserved in NavAb, Nav1.4 and Cav1.1 (Extended Data Fig. 1). Substituted guanidinium ions can block gating pore current without major effects on Nav1.4 function24, suggesting that guanidinium-containing compounds specific for this binding site could provide a basis for structure-based drug design and be used therapeutically to relieve the symptoms of hypokalaemic periodic paralysis.
To examine relationships among structural fluctuations of the gating pore, ionic hydration and Na+ leakage, we performed molecular dynamics simulations of wild-type and R3G mutant voltage sensors in a hydrated lipid bilayer (Fig. 4). Multiple unbiased simulation repeats, with a total duration of 30 µs, show that the overall structures are conserved. Analysis of axial distributions of water molecules revealed a narrow region (−5 Å < z < 5 Å) that is more hydrated in the R3G mutant than in the wild-type voltage sensor, owing to the larger size of the lumen in the mutant (Fig. 4a–c, yellow; P < 0.002, see Extended Data Table 2). The average count of water molecules within the HCS was 3.9 ± 0.8 and 5.3 ± 0.4 for the wild type and R3G mutant, respectively (Fig. 4e). We performed umbrella-sampling simulations to compute the free energy of Na+ permeation along the principal axis of the voltage sensor. When Na+ was within the HCS, the number of water molecules in the HCS increased to 8.4 ± 0.3 in the wild type and 9.0 ± 0.3 in the R3G mutant, respectively. The free-energy profile for Na+ translocation forms a broad barrier spanning the HCS, centred at Cα of R3. The R3G mutation significantly decreases the height of this barrier from 18 ± 0.8 to 11 ± 1.4 kcal mol−1 (Fig. 4e). These values are consistent with the undetectable gating-pore conductance in the wild type and an upper limit of around 0.1 pS in the R3G mutant29. Analysis of ionic coordination shows that, at the extracellular edge of the barrier, the first solvation shell of Na+ is almost exclusively composed of water, consistent with the hydrophobic nature of the bottleneck in the voltage sensor (Fig. 4f). The total coordination number of 5.81 ± 0.02 in bulk water drops to 4.88 ± 0.04 at the peak of the free-energy barrier, suggesting a large desolvation penalty for Na+ that is partly alleviated by the cavity created in the absence of the R3 side chain. Charge–charge repulsion is also likely to contribute substantially to the higher energy barrier to Na+ permeation in the wild type, impeding the gating pore leakage observed in the R3G mutant.

a, Probability (prob.) distribution of water along the domain axis for NavAb(WT) (black) and NavAb(R3G) (red). b, c, Representation of voltage sensor from NavAb(WT) and NavAb(R3G) simulations where Na+ (blue sphere) is restrained at z = −5 Å. The S2 segment (residues 45–65) is omitted for clarity. Arrows indicate the positions of R108. d, Axial distribution of gating charge Cα for NavAb(WT) and NavAb(R3G). The axial position in the crystallographic structure is shown as a vertical line. e, Inset, probability distribution of water in the HCS (−5 Å to 5 Å) across all simulations of NavAb(WT) (black) and NavAb(R3G) (red). The total probability is separated into frames where Na+ occupies the hydrophobic constriction (solid) or is outside this region (cross-hatched). Nwat, number of water molecules. Main panel, Potential of mean force for Na+ conduction within the NavAb(WT) (black) and NavAb(R3G) (red) pore computed using umbrella sampling. The HCS is highlighted in yellow. f, Average coordination of Na+ as a function of ionic position along the principal axis of the voltage sensor, for NavAb(WT) (solid lines) and NavAb(R3G) (dashed lines). The first coordination shell of Na+ is partitioned for coordination to protein (green), water (blue), lipid head groups (orange) and counterions (purple). g–i, Representative snapshots from simulations of NavAb(R3G) depicting conformational isomerization of R4. *P < 0.002, n = 60; see Extended Data Table 2 for details. Arrow in f indicates the point of minimum coordination of Na+ by water.
The location of R4 coincides with a secondary shoulder in the free-energy profiles (Fig. 4d, R108), indicating that movement of Na+ past R4 is not rate-limiting for permeation, even though transit of Na+ past R4 causes the largest displacement of water by protein ligands (Fig. 4e). Spontaneous disruption of the R4–E59 salt bridge in 3 ± 1% of simulation frames for the wild type and R3G mutant opens the inner end of the gating pore with sufficient frequency to support gating pore current (Fig. 4g–i). Na+ often makes direct contacts with the anionic side chains of D80 and E59 (Fig. 4f, g), and its movement is coupled to dynamic rearrangements of the R4 salt-bridge network.
Overall, our results provide an unprecedented high-resolution view of functional effects of ion channel mutations that cause periodic paralysis and define the structural basis for pathogenesis of this ion channelopathy. R2G and R3G mutations do not perturb the backbone structure of the voltage sensor, suggesting that the aberrant gating pore currents are not caused by conformational changes in transmembrane alpha helices. Instead, the absence of the positively charged R2 and R3 side chains opens an aqueous gating pore that allows diffusion of Na+ into the cell, depending on the functional state of the voltage sensor. Our structural studies show how this pathogenic gating pore current is gated in resting and activated states by transmembrane movements of the S4 segment. Although our studies of R2G and R3G mutants suggest a straightforward explanation for the pathogenic gating pore current, mutations that cause hypokalaemic periodic paralysis and normokalaemic periodic paralysis that substitute large side chains such as tryptophan also cause gating pore currents4,16,17, perhaps by perturbing the local structure of the voltage sensor and thereby opening a pore across the membrane.
Our structures reveal the binding pose of a highly permeant ion, guanidinium, in the closed gating pore of the activated voltage sensor of NavAb(R2G). Substituted guanidinium derivatives can block gating pore current without impairing voltage sensor function in Nav1.424. Therefore, our high-resolution structural models may provide molecular templates for design and development of drugs that would mimic guanidinium, block gating pore current and provide symptomatic relief of periodic paralysis.
Methods
Electrophysiology
All experiments were performed using T. ni insect cells (High Five Cells, Thermofisher). Molecular biology and patch-clamp measurements were performed as described previously15,30. All constructs showed high level expression that enabled us to measure ionic current and gating pore currents 48 h after infection. Whole-cell sodium currents were recorded using an amplifier (Axopatch 200; Molecular Devices) with glass micropipettes (2–4 MΩ). The intracellular pipette solution contained (mM): 35 NaCl, 105 CsF, 10 EGTA and 10 HEPES, pH 7.4 (adjusted with CsOH). The extracellular solution contained (mM): 140 NaCl, 2 CaCl2, 1.8 MgCl2 and 10 HEPES, pH 7.4 (adjusted with NaOH).
For NavAb(R2S), the standard clamp protocol for measuring central pore currents consisted of steps from a holding potential of −200 mV to voltages ranging from −180 to 0 mV in 10 mV steps. For NavAb(R3G), cells were held at −160 mV and 10 mV voltage steps ranging from −140 mV to +50 mV were applied. A P/−10 or P/−4 leak-subtraction protocol was used to subtract linear leak and capacitive currents from holding potentials of −200 or −160 mV, respectively.
To measure gating pore currents in NavAb(R2S), cells were held at −200 mV for ~1 min to allow recovery from slow inactivation. Then, the cells were held at −100 mV for gating pore current measurements, which inactivates the central pore current. Depolarizing pulses in 10-mV steps were applied from −200 mV up to +50 mV. The intracellular pipette solution contained (mM): 140 CsF, 10 EGTA, and 10 HEPES, pH 7.4 (adjusted with CsOH). The extracellular solution contained (mM): 140 NaCl, 2 CaCl2, 1.8 MgCl2 and 10 HEPES, pH 7.4 (adjusted with NaOH). To test gating pore selectivity for different cations, NaCl was replaced by an equimolar concentration of KCl, CsCl, LiCl, NMDG or 40 mM guanidinium sulfate +100 mM NMDG, 40 mM methylguanidinium sulfate +100 mM NMDG, or 40 mM ethylguanidinium sulfate +100 mM NMDG.
To measure gating pore currents in NavAb(R3G), cells were held at 0 mV for a few min to induce slow inactivation. Then, 10-mV pulses were applied from −200 mV up to +50 mV. To measure ion selectivity of R3G, the composition of external solution was in (mM): 140 NMDG-MS, 2 CaCl2, 10 HEPES. The intracellular solution contains either 140 mM NaF, 140 mM KF or 140 mM CsF, in addition to 10 mM HEPES, 10 mM EGTA.
No online leak subtraction protocols were used during measuring of gating pore currents. Linear leak subtraction was done offline by generating a linear fit to the I–V curves at voltage ranges +100 mV to 0 mV for NavAb(R2S) and between −200 mV and 0 mV NavAb(R3G). Voltage-clamp pulses were generated and currents were recorded using Pulse software controlling an Instrutech ITC18 interface (HEKA). Data were analysed using Igor Pro 6.37 software (WaveMetrics).
Protein purification and crystallization
R2G, R2S or R3G mutations were introduced into NavAb/I217C by site-directed mutagenesis (QuikChange; Agilent) and confirmed by sequencing. Protein was expressed and purified as described6. In brief, recombinant baculovirus was generated by using the Bac-to-Bac system (Invitrogen), and T. ni cells (High Five Cells, Thermofisher) were infected for protein production. Protein was extracted with 1% digitonin (EMD Biosciences). After centrifugation, the supernatant was agitated with anti-Flag M2–agarose resin (Sigma). Flag resin was washed and eluted with Flag peptide, and the purified protein was analysed by SDS–PAGE (Extended Data Fig. 6). Purified protein was then loaded onto a Superdex 200 column (GE Healthcare) in 10 mM Tris-HCl pH 8.0, 100 mM NaCl and 0.12% digitonin. The peak fraction was concentrated to ~17 mg ml−1 and reconstituted into DMPC:CHAPSO (Anatrace) bicelles. The protein–bicelle preparation was mixed in a 1:1 ratio and set in a hanging-drop vapour-diffusion format over a well solution containing 1.8–2.0 M ammonium sulfate, 100 mM Na-citrate pH 4.8–5.2. Crystals grew to full size in a week. Crystals were cryoprotected in well solution supplemented with 28% glucose (w/v) in increments of 7% glucose during harvesting. Guanidinium- or methylguanidinium-bound crystals were cryoprotected by soaking in the same cryoprotection solution plus 10 mM guanidinium or methylguanidinium ions. Crystals were plunged into liquid nitrogen for data collection.
Data collection and structure determination
X-ray diffraction data was collected at Advanced Light Source (beamlines BL821 and BL822), and then integrated and scaled with the HKL2000 suite. Both NavAb(R2G) and NavAb(R3G) structures were solved by Phaser-MR using NavAb (PDB code: 3RVY) monomer as searching model. After initial phases, models were refined with PHENIX31 and manually re-built using COOT32. High-resolution density maps clearly showed no side-chain density for R2G or R3G. Simulated annealing omit maps were used to confirm the binding of guanidinium ions. The geometries of the final models were verified using MolProbity33. All solvent-accessible volume analysis in the voltage-sensing modules was generated with MOLE234.
Molecular modelling and dynamics
Molecular models of the NavAb(WT) and NavAb(R3G) channels were constructed using the NavAb(I217C) structure (PDB code: 3RVY)6. The latter model was generated by substituting R105 with G in all four voltage sensing domains. Both systems were embedded in a hydrated 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) bilayer with ~250 mM NaCl for a total of ~129,000 atoms. Embedding was performed using the alchembed protocol35 using an equilibrated rectangular CHARMM36 DMPC bilayer patch obtained from the Klauda laboratory website (https://terpconnect.umd.edu/~jbklauda/). The protein, lipids and ions were modelled with the CHARMM36 all-atom force field36,37,38 and water molecules were modelled with TIP3P39. NBFIX adjustments were made for Na+-backbone carbonyl O atom and Na+-lipid head group interactions40,41.
All simulations were performed with GROMACS 5.0.642. Electrostatic interactions were calculated using particle-mesh Ewald algorithm43,44 with a real-space cut-off distance of 1.2 nm, a grid spacing of 0.16 nm and cubic interpolation. Lennard–Jones interactions were cut off at 1.2 nm. Nonbonded interactions were calculated using Verlet neighbour lists45. All simulations were performed at constant temperature (300 K) using the Nosé–Hoover thermostat46,47 with temperature coupling of 0.5 ps and at constant pressure (1 atm) with the Parrinello–Rahman barostat48,49 with a time constant of 2 ps. All chemical bonds were constrained using the LINCS algorithm50. The integration timestep was 2 fs.
Because the channel and voltage sensor were initially devoid of water molecules and ions, a protein-restrained equilibration period of 30 ns was used to reduce the systematic sampling bias induced by the initial conditions (10 ns with protein heavy-atom restraints, 10 ns with backbone restraints, and 10 ns with Cα restraints, all with a force constant of 2.39 kcal mol−1 Å−2). Unbiased production simulations of 15 replicas of ‘WT’ and ‘R3G’ systems were conducted for 1,000 ns each, resulting in aggregate sampling of 15 μs for each tetramer (4 × 15 µs = 60 µs for WT and R3G voltage sensors).
Simulation snapshots beyond t = 100 ns were extracted from unbiased simulations and used as initial conditions for biased simulations, using the entire tetramer. Umbrella sampling51,52 was used to compute the free energy or potential of mean force (PMF) profile for the movement of Na+ through voltage sensing domain. The range of the reaction coordinate, −2.0 to 2.0 nm with respect to the centre of the hydrophobic constriction, was discretized into ~130 unevenly spaced windows. For each window, biased simulations were initiated with a water molecule exchanged for Na+ in all four voltage sensors. Production simulations were performed for 70–100 ns per window with a harmonic restraining potential force constant of 2.39 kcal mol−1 Å−2 and a flat-bottom cylindrical position restraint for all four Na+ ions simultaneously. The axial position of the permeating Na+ ion, z, was stored every 10 fs and the data from each of the four voltage sensors were used separately to generate four independent PMF profiles using g_wham53, enforcing cyclic periodicity of the PMF in the bulk (at z = −2.5 nm). The initial 10 ns were excluded from each umbrella sampling run. We report the mean PMF over the four voltage sensors with error bars computed using the standard error of mean over all four PMFs. The total simulation time for each of the two systems (WT and R3G) was ∼11 μs, yielding a total of ∼45 μs of voltage sensor data.
Water occupancy of the voltage sensor was computed by counting the number of water oxygen atoms within a cylinder of radius 8.0 Å. We define the hydrophobic constriction centre as the geometric centre of Cα atoms of residues 22, 57, 84 and 105. The range of the HCS is defined as −5 Å to 5 Å along the axial coordinate of the voltage sensor. Coordination of Na+ to channel ligands, water, ions and lipids was performed by computing the number of protein, water and lipid O atoms, as well as Cl− ions, within the first solvation shell of Na+ (<3.0 Å). The average coordination number at a given axial position was computed over all simulation frames regardless of the subunit, but the total coordination number in bulk water and at the hydrophobic constriction reported in the text was based on the mean and standard error of mean over the four voltage sensors. Analysis of the trajectories was performed using MDTraj54 and molecular renderings were generated using Visual Molecular Dynamics55.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: NavAb(R3G), 6C1E; NavAb(R2G)–guanidinium, 6C1K; NavAb(R2G)–methylguanidinium, 6C1M; NavAb(R2G), 6C1P.
References
Venance, S. L. et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 129, 8–17 (2006).
Cannon, S. C. Handbook of Experimental Pharmacology (Springer, Berlin, 2017).
Sokolov, S., Scheuer, T. & Catterall, W. A. Gating pore current in an inherited ion channelopathy. Nature 446, 76–78 (2007).
Struyk, A. F. & Cannon, S. C. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20 (2007).
Vicart, S. et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 63, 2120–2127 (2004).
Sokolov, S., Scheuer, T. & Catterall, W. A. Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl Acad. Sci. USA 105, 19980–19985 (2008).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Catterall, W. A. & Zheng, N. Deciphering voltage-gated Na+ and Ca2+ channels by studying prokaryotic ancestors. Trends Biochem. Sci. 40, 526–534 (2015).
Catterall, W. A., Goldin, A. L. & Waxman, S. G. International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409 (2005).
Yan, Z. et al. Structure of the Nav1.4-β1 complex from electric eel. Cell 170, 470–482 (2017).
Shen, H. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355, eaal4326 (2017).
Catterall, W. A. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928 (2010).
Starace, D. M. & Bezanilla, F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427, 548–553 (2004).
Sokolov, S., Scheuer, T. & Catterall, W. A. Ion permeation through a voltage- sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47, 183–189 (2005).
Gamal El-Din, T. M., Scheuer, T. & Catterall, W. A. Tracking S4 movement by gating pore currents in the bacterial sodium channel NaChBac. J. Gen. Physiol. 144, 147–157 (2014).
Jurkat-Rott, K., Groome, J. & Lehmann-Horn, F. Pathophysiological role of omega pore current in channelopathies. Front. Pharmacol. 3, 112 (2012).
Moreau, A., Gosselin-Badaroudine, P. & Chahine, M. Biophysics, pathophysiology, and pharmacology of ion channel gating pores. Front. Pharmacol. 5, 53 (2014).
Wu, F. et al. A sodium channel knockin mutant (Nav1.4-R669H) mouse model of hypokalemic periodic paralysis. J. Clin. Invest. 121, 4082–4094 (2011).
Wu, F. et al. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J. Clin. Invest. 122, 4580–4591 (2012).
Gosselin-Badaroudine, P., Delemotte, L., Moreau, A., Klein, M. L. & Chahine, M. Gating pore currents and the resting state of Nav1.4 voltage sensor domains. Proc. Natl Acad. Sci. USA 109, 19250–19255 (2012).
Moreau, A., Gosselin-Badaroudine, P., Boutjdir, M. & Chahine, M. Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front. Pharmacol. 6, 301 (2015).
Monteleone, S. et al. Mechanisms responsible for ω-pore currents in Cav calcium channel voltage-sensing domains. Biophys. J. 113, 1485–1495 (2017).
Chakrabarti, N. et al. Catalysis of Na+ permeation in the bacterial sodium channel NavAb. Proc. Natl Acad. Sci. USA 110, 11331–11336 (2013).
Sokolov, S., Scheuer, T. & Catterall, W. A. Ion permeation and block of the gating pore in the voltage sensor of Nav1.4 channels with hypokalemic periodic paralysis mutations. J. Gen. Physiol. 136, 225–236 (2010).
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N. & Catterall, W. A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 (2012).
Yarov-Yarovoy, V. et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl Acad. Sci. USA 109, E93–E102 (2012).
Vargas, E. et al. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol. 140, 587–594 (2012).
Catterall, W. A., Wisedchaisri, G. & Zheng, N. The chemical basis for electrical signaling. Nat. Chem. Biol. 13, 455–463 (2017).
Cooper, K. E., Gates, P. Y. & Eisenberg, R. S. Diffusion theory and discrete rate constants in ion permeation. J. Membr. Biol. 106, 95–105 (1988).
Gamal El-Din, T. M., Martinez, G. Q., Payandeh, J., Scheuer, T. & Catterall, W. A. A gating charge interaction required for late slow inactivation of the bacterial sodium channel NavAb. J. Gen. Physiol. 142, 181–190 (2013).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Petřek, M., Kosinová, P., Koca, J. & Otyepka, M. MOLE: a Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure 15, 1357–1363 (2007).
Jefferys, E., Sands, Z. A., Shi, J., Sansom, M. S. P. & Fowler, P. W. Alchembed: a computational method for incorporating multiple proteins into complex lipid geometries. J. Chem. Theory Comput. 11, 2743–2754 (2015).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Noskov, S. Y. & Roux, B. Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 377, 804–818 (2008).
Venable, R. M., Luo, Y., Gawrisch, K., Roux, B. & Pastor, R. W. Simulations of anionic lipid membranes: development of interaction-specific ion parameters and validation using NMR data. J. Phys. Chem. B 117, 10183–10192 (2013).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software X 1–2, 19–25 (2015).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Verlet, L. Computer ‘experiments’ on classical fluids. I. thermodynamical properties of Lennard–Jones molecules. Phys. Rev. 159, 98–103 (1967).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 (1984).
Parrinello, M. & Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 45, 1196–1199 (1980).
Nosé, S. & Klein, M. L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076 (1983).
Hess, B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008).
Torrie, G. M. & Valleau, J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23, 187–199 (1977).
Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 91, 275–282 (1995).
Hub, J. S., de Groot, B. L. & van der Spoel, D. g_wham—a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 (2010).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). 27–28.
Acknowledgements
We thank the beamline staff at the Advanced Light Source (BL8.2.1 and BL8.2.2) for assistance during data collection and J. Li for technical and administrative assistance. This research was supported by National Institutes of Health research grants R01 NS015751 (W.A.C.) and R01 HL112808 (W.A.C. and N.Z.), by the Howard Hughes Medical Institute (N.Z.), and by Canadian Institutes of Health Research grant MOP 130461 (R.P.).
Author information
Authors and Affiliations
Contributions
D.J., T.M.G.E.-D., C.I., P.L., R.P., N.Z. and W.A.C. designed experiments. D.J., T.M.G.E.-D., C.I. and P.L. conducted experiments. D.J., T.M.G.E.-D., C.I., R.P., N.Z. and W.A.C. analysed the results. T.M.G.E.-D., C.I. and W.A.C. wrote the paper with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Sequence alignment of the voltage sensor of NavAb with those of human Nav1.4 homologous domain (D)II, Nav1.4 DIV, Cav1.1 DII and Cav1.1 DIV.
Coloured rectangles represent transmembrane helices. Black arrows indicate residues that form the guanidinium binding site, blue arrows indicate the hydrophobic constriction site and red arrows indicate the conserved intracellular negative cluster.
Extended Data Fig. 2 Superposition of the NavAb(WT) voltage sensor and the Electrophorus electricus (electric eel) Nav1.4 DIV voltage sensor.
a–b, Comparison of the conformations of NavAb(WT) voltage sensor (orange) and EeNav1.4 voltage sensor DIV (PDB code: 5XSY) (grey) in side view and top view, respectively. Arginine sensors and hydrophobic residues in the HCS are labelled and shown with side chains in sticks.
Extended Data Fig. 3 Superposition of the voltage sensors of NavAb(WT) and mutant channels.
a–b, Voltage sensor structure alignment between NavAb(WT) (grey) and NavAb(R3G) (green) in side view and top view, respectively. c–d, Voltage sensor structure alignment between NavAb(WT) (grey) and NavAb(R2G) (cyan) in side view and top view, respectively. Arginine sensors and hydrophobic residues in the HCS are labelled and shown with side chains in sticks.
Extended Data Fig. 4 R4 side chain conformational changes.
a, Different conformations of the R4 rotamer in NavAb(R3G) chain A (green) and chain B (orange). b, Different conformations of the R4 rotamer in the four subunits of NavAb in the slow-inactivated state (PDB code: 4EKW).
Extended Data Fig. 5 Electron density maps for bound guanidinium and methylguanidinium ions.
a, 2mFo−DFc electron density map (blue mesh) of residues around the methylguanidinium binding site at 1σ. b, Overlay of guanidinium binding site (green) and methylguanidinium binding site (orange). c–d, Simulated annealing map (Fo−Fc) contoured at 3σ for methylguanidinium and guanidinium, respectively.
Extended Data Fig. 6 Purification of NavAb(R3G).
a, Representative gel-filtration chromatography of NavAb(R3G); highlighted peak fractions were concentrated for crystallization. b, Concentrated sample was visualized on SDS–PAGE by Coomassie blue staining.
Supplementary information
Supplementary Discussion
This file contains Supplementary Discussion of Nomenclature for Periodic Paralyses; Functional Properties of NavAb/R2G, NavAb/R2S, and NavAb/R3G; Amplitude of Gating Pore Current; and associated references.
Rights and permissions
About this article

Cite this article
Jiang, D., Gamal El-Din, T.M., Ing, C. et al. Structural basis for gating pore current in periodic paralysis. Nature 557, 590–594 (2018). https://doi.org/10.1038/s41586-018-0120-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0120-4
- Springer Nature Limited
This article is cited by
-
An LQT2-related mutation in the voltage-sensing domain is involved in switching the gating polarity of hERG
BMC Biology (2024)
-
Structural mechanism of voltage-gated sodium channel slow inactivation
Nature Communications (2024)
-
The structural basis of divalent cation block in a tetrameric prokaryotic sodium channel
Nature Communications (2023)
-
De novo CACAN1D Ca2+ channelopathies: clinical phenotypes and molecular mechanism
Pflügers Archiv - European Journal of Physiology (2020)
-
Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation
Scientific Reports (2019)