
Abstract
Electrochemical systems play a decisive role in, e.g. clean energy conversion but understanding their complex chemistry remains an outstanding challenge. Constant potential and grand canonical ensemble (GCE) simulations are indispensable for unraveling the properties of electrochemical processes as a function of the electrode potential. Currently, GCE calculations performed at the density functional theory (DFT) level require fixing the Fermi level within the simulation cell. Here, we illustrate that this method is inadequate when modeling outer sphere reactions and a biased two-electrode cell. For these systems, the Fermi level obtained from DFT calculations does not accurately present the experimentally controlled electrode potential or describe the thermodynamic independent variable in GCE-DFT. To address this limitation, we developed and implemented a constant inner potential (CIP) method offering a more robust and general approach to conducting GCE-DFT simulations of electrochemical systems under constant potential or bias conditions. The primary advantage of CIP is that it uses the local electrode inner potential as the thermodynamic parameter for the electrode potential, as opposed to the global Fermi level. Through numerical and analytical studies, we demonstrate that the CIP and Fermi level GCE-DFT approaches are equivalent for metallic electrodes and inner-sphere reactions. However, CIP proves to be more versatile, as it can be applied to outer-sphere and two-electrode systems, addressing the limitations of the constant Fermi-level approach in these scenarios. Altogether, the CIP approach stands out as a general and efficient GCE-DFT method simulating electrochemical interfaces from first principles.
Similar content being viewed by others


Introduction
Electrochemical systems and reactions are ubiquitous in modern energy conversion applications, electrosynthesis, and sensors, to name but a few1,2. Their key property is the ability to control reaction thermodynamics and kinetics through the application of an electrode potential. In experiments, the potential of a working electrode is controlled from the backside of an electrode through connections to an external voltage source as shown in Fig. 1. This provides a direct way to manipulate the electrode potential, i.e. the electrochemical potential of electrons within the bulk of the electrode material2,3. External connections and a working electrode (M) are physically distinct solid phases, which can be viewed as a bath and a system in statistical mechanics, respectively. In this context, constant electrode potential experiments correspond to controlling the electrochemical potential, \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\), of an electron reservoir and the system potential, \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\), responds to the change in \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\). At thermodynamic equilibrium, electrochemical potentials of the connections and the working electrode are equal and electrochemical balance, \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}={\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\), is satisfied3,4. In general, the electrochemical potential of electrons in a given phase i can be written as
where the electron chemical potential \({\mu }_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}={\mu }_{{{{\rm{e}}}}}^{{{{\rm{0,i}}}}}+{k}_{{{{\rm{B}}}}}T\ln [{a}_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}]\) is an intrinsic material property and depends on the standard state chemical potential \({\mu }_{{{{\rm{e}}}}}^{{{{\rm{0,i}}}}}\) and activity \({a}_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}\), which is, for example, particularly important for semiconductor electrodes. ϕi is the inner potential, i.e. the electrostatic potential of phase i, z is the electron charge, and F is the Faraday constant. Below, we express all energies and electrostatic potentials in eV and Eq. (1) reads \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}={\mu }_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}-{\phi }^{{{{\rm{i}}}}}\). Given that \({\mu }_{{{{\rm{e}}}}}^{{{{\rm{i}}}}}\) is constant for a given phase and material, the inner potential of a given phase determines the applied electrode potential of either a bath or a working electrode, M4. The electrochemical potential of electrons within the electrode M is also directly proportional to the Fermi level of the given phase as \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}={E}_{{{{\rm{F}}}}}\)3.

The dotted rectangle shows the system explicitly treated in (GCE-)DFT simulations.
While experiments are routinely performed under constant potential conditions and referenced against well-defined reference electrodes, realizing constant potential atomistic simulations has been very challenging and several schemes for controlling the applied potential have been developed5,6,7. The current methods to simulate the applied electrode potential can be roughly divided into three categories: electronic structure methods within the grand-canonical ensemble density functional theory (GCE-DFT)8,9, classical force field methods10, and finite-field methods, which can be used either with DFT or classical potentials11,12,13,14.
GCE-DFT simulations typically consider a single electrode and work on the single-electrode potential scale to fix \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\) providing an exact framework for treating systems at constant electrochemical potential15. In particular, the grand canonical free energy as well as other (thermodynamic) expectation values from GCE-DFT are formally exact and unique for given external (electrode) potential with an explicit dependence on \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\)15,16,17. As discussed in detail in the section “The inner potential as the basic variable in GCE-DFT”, \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\) is proportional to the electrode work function or Fermi level3 readily available from any periodic DFT implementation. Given this generality and ability to work with just a single electrode, the Fermi level-based GCE-DFT has become widely adopted for modeling electrochemical thermodynamics15,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32 and kinetics7,28,31,33,34,35 at fixed \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\). Essentially, all GCE-DFT approaches can be seen as finite-size corrections needed to fix the electrode potential in small simulation cells. When employing a sufficiently large simulation cell, the canonical and GCE-DFT simulations are equally applicable to electrochemical simulations.
Classical force field and finite field methods adopt a different approach to constant potential simulations and describe the applied electrode potential in terms of the inner potential (ϕ) difference between two electrodes without any electronic structure information10,11,12,13,14,36,37,38,39,40,41. This attractive feature, however, comes with a cost, as two electrode interfaces need to be simulated to build an electrode potential difference in a cell and to keep the simulation cell neutral. As one is typically interested in the properties of just a single electrode, i.e. the working electrode, the second electrode acts as a passive counter electrode. While the treatment of a passive electrode is justifiable for low-cost analytic force field potentials, this does not hold for electronic structure methods or even machine learning potentials as their computational cost is two or three orders-of-magnitude larger than that of analytic classical potentials42. Therefore, future studies of electrochemical interfaces with machine learning potentials need new approaches to enable constant potential calculations of single electrodes without explicit electronic structure information.
So far, both GCE-DFT and classical force field methods have overlooked the question which of the electrochemical potentials, either \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\) or \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\), serve the relevant constant quantity in GCE-DFT simulations. While this has not been considered in the context of computational electrochemistry, recent work by Islas-Vargas et al.43 addressed this issue in detail for semiconductor–water interfaces. They emphasized that the relevant thermodynamically independent parameter for defining the electrode potential is the bath chemical potential (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\)) rather than the system chemical potential (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\)). Hence, in GCE-DFT simulations, one should fix the electron reservoir (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\)), which is controlled experimentally, rather than the electrochemical potential of the simulation cell (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\)). This distinction is not always clear in current GCE-DFT approaches and our results in the next sections demonstrate the limitations of present constant Fermi level simulations.
In this work, we present a more general GCE-DFT method, in which \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\) rather than \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) is explicitly controlled. To achieve this, we introduce the constant inner potential (CIP) DFT method, illustrating that it offers a versatile and theoretically rigorous approach for conducting constant potential ab initio simulations for metallic systems. Importantly, the CIP method provides direct control over \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\) and enables GCE-DFT simulations for systems in which the constant Fermi level methods fail to correctly mimic the experimentally applied potential. CIP-DFT emerges as a universal approach for simulating a wide variety of electrochemical systems and expands the scope of the GCE-DFT simulations from a single metallic electrode and inner-sphere reactions to outer-sphere reactions and biased two-electrode cells.
Results
The inner potential as the basic variable in GCE-DFT
CIP-DFT uses the electrode’s inner potential as the basic variable to control the applied potential in GCE-DFT simulations. Preceding GCE-DFT implementations have controlled EFDFT to monitor the electrode potential on the single or absolute electrode scale6 but this is not the only available option. There are at least four different single potential definitions3 each with different scales and reference systems, but all of them can be presented formally using the reduced absolute potential3,44
where \({\mu }_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\) is an intrinsic material property and independent of the electrode potential. \({{{\Delta }}}_{{{{\rm{S}}}}}^{{{{\rm{M}}}}}\phi ={\phi }^{{{{\rm{M}}}}}-{\phi }^{{{{\rm{S}}}}}\) stands for the inner potential difference, i.e. the electrostatic potential difference between the bulk of electrode (M) and solution (S) as shown in Fig. 2. K is a constant and depends on the chosen scale, which in DFT simulations is typically chosen6 as the electrostatic potential in vacuum or the inner potential of the implicit solvent, in which case \({U}_{{{{\rm{vac}}}}}^{{{{\rm{M}}}}}({{{\rm{abs}}}})=-{\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}=-{E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)3,45.

The inner potential within the electrode (ϕM) can be controlled and fixed to a specified value. The solution inner potential ϕS is always fixed at zero.
The CIP method uses a different reference electrode scale, a free electron in solution3,44 in which case \(K={\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{R,M}}}}}\)44 for the reference electrode (R) is made of the same material as M. In its original form44, this choice of K allows writing the absolute potential using properties of just a single electrode. Herein, it enables writing Eq. (2) as \({U}_{{{{\rm{sol}}}}}^{{{{\rm{M}}}}}({{{\rm{abs}}}})={{{\Delta }}}_{{{{\rm{S}}}}}^{{{{\rm{M}}}}}\phi -{\mu }_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}+{\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{E}}}}}={{{\Delta }}}_{{{{\rm{S}}}}}^{{{{\rm{M}}}}}\phi -{\phi }^{{{{\rm{M,R}}}}}\), which, in turn, allows direct control of \({{{\Delta }}}_{{{{\rm{S}}}}}^{{{{\rm{M}}}}}\phi\) without explicitly introducing the Fermi level after choosing ϕM,R. In practice, we pick ϕM,R to correspond to the potential zero charge of the electrode M: as shown in Supplementary Eq. (1) this choice enables controlling the applied potential by controlling the inner potential ϕM. While this reference scale is not experimentally viable3,44, it can be used in simulations44 as demonstrated very recently by the application of density-potential functional theory to electrochemical interfaces46,47. Supplementary Methods 2 further shows that the inner potential scale is natural for (GCE-)DFT simulations and leads to CIP-DFT.
In experiments performed under thermodynamic equilibrium between the connections and working electrode, it is guaranteed that the electrochemical potentials of the system and bath are equal to the Fermi level (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}={\tilde{\mu }}_{{{{\rm{M}}}}}^{{{{\rm{bath}}}}}={E}_{{{{\rm{F}}}}}\)) because both M and bath phases are explicitly present (see Fig. 1). Because the two chemical potentials and Fermi level must be equal under equilibrium, Eq. (2) ensures that the experimentally specified electrode potential corresponds to inner potential changes within either the bulk phase M or bath3,4,44. This in turn means that changes in the bath inner potential (Δϕbath) uniquely define the response of the system inner potential (ΔϕM) in equilibrium because both potentials are inherent bulk properties48; it follows that the applied electrode potential directly modifies ϕM.
The discussion above holds for both metallic and semiconducting electrodes3,4,48, and even the presence of the Schottky barrier49 between the metallic contact and the semiconductor electrode does not change the picture: \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}={\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}={E}_{{{{\rm{F}}}}}\) condition must remain at equilibrium. However, in semiconductors, it is essential to consider the finite number of charge carriers or dopants, their spatial dopant concentrations, and their effect on the electrostatics50,51. Nevertheless, the applied electrode potential, ΔU, can be expressed in terms of local electrostatic potentials and material-dependent constants for both metallic and semiconducting materials4,50,51. This applied potential is directly proportional to the bulk electrostatic potential difference, specifically the inner potentials at different charge states4,50,51,52:
This equation shows that the applied potential of any electrode is equal to the changes in the bulk electrostatic potential as long as the material, and hence \({\mu }_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\), remains unchanged. Combined with Eq. (2), this equation shows that the applied potential of any electrode can be simulated by fixing the inner potential.
The discussion above highlights the central role of electrode inner potentials in determining the applied electrode potential in electrochemical systems. This is a direct consequence of the general theory of single electrode potentials in Eq. (2), which shows that ϕM is linearly related to the single electrode potential, regardless of the chosen reference scale. From a computational perspective, it is important to note that unlike \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\), the inner potential is a local quantity defined within the bulk electrode and allows controlling the electrode potential between two bulk phases (either solid, liquid, or gas). As shown by the various examples in the following sections “Inner-sphere interaction, CIP molecular dynamics, Outer-sphere reactions, and Biased two-electrode setups within DFT”, fixing the inner potential enables modeling the applied electrode potential within the electrode in accordance with the general definition given in Eq. (2). Hence, performing simulations at constant ϕM ensures that the experimentally applied potential, i.e. \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\), is accurately represented while avoiding the need to account for the electronic quantities (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}={E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)) inherent to the used simulation cell43.
To control the electrode inner potential in GCE-DFT simulations, we introduce the constant inner potential (CIP) approach illustrated in Fig. 2. The CIP method has been implemented in the GPAW53,54 software by modifying the solvent jellium model (SJM)28. The original SJM model controls the work function, while CIP-DFT expands upon SJM by fixing the inner potential ϕM relative to the solution’s inner potential ϕS. Using Dirichlet boundary conditions, the solution’s inner potential is set to zero (ϕS = 0) far from the electrode. As displayed in Fig. 2, controlling ϕM while keeping ϕS = 0 yields a well-defined electrode potential and a constant reference point. There is no need to introduce \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) to define the absolute potential as the inner potential difference uniquely defines the electrode potential and ϕM can be converted to any experimental scale. A more detailed description of the CIP-DFT method is available in Supplementary Methods 1 and 2. In the following sections, we demonstrate the performance of CIP-DFT for simulating a variety of electrochemical systems ranging from inner-sphere reactions on metallic electrodes to outer-sphere reactions and the establishment of a biased two-electrode setup. Furthermore, we illustrate the method’s compatibility with both explicit and implicit solvent models.
Thermodynamics of GCE-DFT and CIP-DFT
Although employing ϕM is both physically and computationally well-motivated, using it as a thermodynamic variable is a new proposition. Up until now, all GCE-DFT calculations have utilized \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\) as the thermodynamic variable in constant electrode potential simulations. In order to demonstrate that ϕM can serve as a thermodynamically independent variable in GCE-DFT simulations, we have carried out an extensive thermodynamic analysis detailed in Supplementary Methods 3. This analysis shows that, for metallic electrodes, using either \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}\) or ϕM leads to identical results as illustrated in Supplementary Methods 3.2 and numerically demonstrated in the section “Inner-sphere interactions”.
Inner-sphere interactions
We first apply the CIP approach to study the pristine and oxygen-covered Au(111) surfaces at different potentials and surface charges. Oxygen adsorption presents a typical electrochemical inner-sphere reaction, where a metal electrode strongly interacts with an adsorbate. Figure 3 displays the electrostatic potentials and Fermi levels for bare and oxygen-covered gold surfaces at different surface charges/electrode potentials. Comparing the differences in Fermi level and inner potential for Au shows that both methods when applied at the same surface charge densities, give identical differences in electrode potential. This is further corroborated by the detailed analysis provided in Supplementary Methods 3.2.

a xy-averaged electrostatic potentials and Fermi levels for the pure (b) and oxygen-covered (c) Au(111) surfaces at three different surface charges, the orange curve corresponds to the potential of zero charge (PZC) simulation while the green (black) curves have surface charges σ = 0.05e (−0.05e). The gray region depicts the area in which the electrode’s inner potential is computed. The insets show the electrostatic potentials within the electrode and Fermi level. c The DOS plot for the σ = 0.05e with the red dashed line indicating the Fermi level.
The density of states (DOS) analysis in Fig. 3c shows that the electronic states of oxygen-covered Au(111) at and near the Fermi level have significant contributions from gold–oxygen interactions. This means that \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) is not independent of the presence of adsorbed oxygen and that the electrode potential might not be uniquely determined by the gold bulk. However, the electrostatic potentials in Fig. 3 show that the inner potential maxima and the Fermi level coincide perfectly for both pristine and adsorbate-covered Au surfaces. This indicates that the standard chemical potential (\({\mu }_{{{{\rm{e}}}}}^{{{{\rm{0,Au}}}}}\)) and the inner potential (ϕAu) are exclusively determined by the bulk of a gold electrode. Therefore, the electrochemical potential of electrons (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{Au}}}}}={\mu }_{{{{\rm{e}}}}}^{{{{\rm{0,Au}}}}}-{\phi }^{{{{\rm{Au}}}}}\)) and electrode potential are determined by Au bulk. Hence, for this example, the system and bath electrochemical potentials coincide, and the Fermi level and CIP approaches yield results that are numerically consistent.
CIP molecular dynamics
Our second example illustrates the simulation of an explicitly solvated Au(111) interface, presented in Fig. 4, using both constant inner potential (CIP-DFT) and constant charge (canonical DFT) molecular dynamics (MD). Overall, the results given in Fig. 4 demonstrate that CIP-DFT-MD calculations can be readily performed to study the dynamics of explicitly solvated electrochemical interfaces and that even moderately large systems can be treated to address interfacial properties under constant (inner) potential conditions. By comparing the number of MD steps in constant potential and constant charge simulations against wall-time, it was observed that the constant potential simulations were ~20–25% more time-consuming. This suggests that constant potential calculations are not as prohibitively expensive as they are sometimes presumed to be55. Figure 4 shows Fermi level and inner potential variations as a function of time for the solvated Au(111) interface in fixed charge calculations. The variations are very similar and time-dependent Fermi level and inner potential fluctuations are practically indistinguishable. This indicates that the Fermi level of a solvated Au(111) is almost fully determined by the electrode even though some hybridization between the surface and water takes place. In practice, these observations indicate that both the Fermi level and inner potential approaches are equally applicable for describing the electrode potential of this system. While this is an expected result, it differs from the recent computational results56, which show that the Fermi level and inner potential difference, \({{{\Delta }}}_{{{{\rm{S}}}}}^{{{{\rm{M}}}}}\phi\) surprisingly exhibit different fluctuations in fixed charge simulations. As discussed in detail in Supplementary Methods 3, Supplementary Discussion 1, and throughout the present work, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) and \({{{\Delta }}}_{{{{\rm{S}}}}}^{M}\phi\) should yield the same (thermodynamic) expectation values, fluctuations, and electrode potentials for systems where the Fermi level is dominated by a metallic electrode.

a The system setup used for the molecular dynamics simulations of the Au(111)–water interface. b Fermi level and inner potential fluctuations as a function of the simulation time for a solvated Au(111) surface at a surface charge of −0.2e per 88 Å2. Both values are referenced against the respective quantities of the 500th frame. c CIP-DFT molecular dynamic simulations for the solvated Au(111) surface. The inner potential is fixed to −14.0 eV with ±0.1 eV fluctuations. The potential corresponds to +0.6 V vs. Au(111) PZC.
Figure 4 also demonstrates that constant potential CIP-DFT-MD simulations of an explicitly solvated interface exhibit substantial charge fluctuations. Even though most simulations focus on thermodynamic expectation values, it needs to be stressed that variations and fluctuations are fundamental properties of statistical thermodynamic systems and must be captured to ensure thermodynamic consistency. While the grand canonical and canonical simulations can give the same thermodynamic expectation values, the other quantities may deviate in the two ensembles57. For instance, time-dependent fluctuations around an equilibrium value, which determine important electrochemical quantities such as capacitance, electric conductivity, and reaction rates, are expected to be different in different ensembles58. While fluctuations and averages can be captured through MD simulations, the calculations need to be done by enforcing the relevant thermodynamics constraints, such as constant temperature and electrode potential, used experimentally. Further evidence for the importance of using the correct ensemble is provided by the sensitivity of electrochemical kinetics to the electrode potential and surface charge34,59, the role of fluctuations in adsorption energies60, or the energy gap fluctuations determining the reaction rate of Marcus-like (proton-coupled) electron transfer theories34,61. Hence, MD simulations of electrochemical systems should be performed in the GCE framework to achieve a fully consistent treatment of electrochemical interfaces and reactions.
Outer-sphere reactions
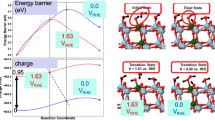
Previous GCE-DFT studies in electrochemistry have almost exclusively focused on inner-sphere reactions62,63,64, while the important class of outer-sphere reactions, where the interactions between the electrode and the redox species are weak, has gained less attention and is therefore only partially understood65. Most theoretical models of outer-sphere electron transfer assume that outer-sphere couples do not hybridize with the electrode so that the total system can be considered as a weakly interacting combination of electrode and solvent-phase subsystems66. As the electrode and redox couple belong to distinct phases, it should be possible to individually control their properties and to modulate the electrode potential independent of the solution phase as schematically shown in Fig. 5. As long as the electrode Fermi level is above the Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) reduction potential (≤−4.6 V), the electrode determines \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\). However, when the Fermi level falls below the reduction potential, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) nominally resides on the solution-phase Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) and the Fermi level cannot be controlled independently from the solution phase. This is not a problem in either adiabatic electron transfer theories66 or GCE-DFT simulations because the orbital filling depends explicitly on the electrode potential and should remain independent of other factors. However, simulations and theory of non-adiabatic electron transfer are grounded in diabatic states62, which maintain their charge state irrespective of the electrode potential. When using diabatic states and GCE-DFT to simulate electron transfer as a function of the electrode potential, it is crucial to independently control both the reactant charge state and the electrode potential. This cannot be achieved with constant Fermi level-based GCE-DFT calculations. The limitation arises from the global nature of the Fermi level, which depends on the hybridization between different parts of the total system. In addition, the Fermi level depends on the presence of the outer-sphere couple, see the results and discussion below. On the other hand, the inner potential is a local quantity measured inside the bulk of the electrode, and it enables control over both the electrode potential and reactant charge state simultaneously and independently, as shown next.

At −4.44 V, the Fermi level of the simulation cell is determined by the electrode whereas at the Au(111) PZC at −4.96 V the Fermi-level is defined by the Ru[NH3]\({}_{6}^{3+/2+}\) couple. The real redox i.e. HOMO/LUMO levels of Ru[NH3]\({}_{6}^{3+/2+}\) are shown in Fig. 6.
We consider a classical outer-sphere redox couple using a diabatic description: a Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) complex on the Au(111) surface as depicted in Fig. 6. The Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) charge remains fixed at +3, irrespective of the surface charge or \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) due to application of constrained DFT to control the Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) charge state (see the section “Methods” for computational details). To understand the variations in the electrode potential, Fermi level, and inner potential during DFT simulations of outer-sphere reactions, we modified the total charge of the simulation cell. This allowed us to investigate the potential range within ±1.0 V around the reduction potential of Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\). The molecular orbitals presented in Fig. 6a–d show that the redox couple and the electrode are strongly hybridized; this prevents separating the total system into two subsystems in GCE-DFT simulations. This is also seen in the DOS plot in Fig. 6e, which demonstrates that all electronic states near the Fermi level contain significant contributions from the Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) complex despite the correct +3 charge on the redox couple. The electronic states residing on the redox couple are visible in the DOS plot around −0.2 eV at all charge states (or electrode potentials) except when the simulation cell carries a very high negative charge (line −3.58 in Fig. 6e). This means that \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) is solely determined by the electrode, but only on highly negatively charged gold surfaces.

a–d (HOMO−1)–(HOMO+2) orbitals when EF = −4.46 eV. e Right: Density (DOS) and projected DOS (PDOS) as a function of the potential. Left: Zoom of the DOS around the Fermi level. The solid lines correspond to the DOS and the dashed lines to the PDOS of Ru[NH3]\({}_{6}^{3+}\). f Electrostatic potentials (solid lines), inner potentials (dashed line), and Fermi levels (dash-dotted lines) at different surface charges and corresponding absolute potentials.
Table 1 shows that differences in the electrode potential inferred from ϕM or \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) are small but systematically different. In particular, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) depends more sensitively on the charge state than ϕAu. This observation is attributed to the large number of electronic states residing on the redox couple at the Fermi level and means that the system Fermi level is determined by the hybridized orbitals spreading across both the electrode and solvent phase Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\). The reason for the electrode potential differences obtained from ϕAu and \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) approaches can be understood based on the results displayed in Fig. 6. The data shows that the electrostatic potential maxima within Au and the Fermi level do not coincide, but there is a non-constant difference between the inner potential maxima and \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) at the same surface charge. This non-constant shift demonstrates that \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) is not solely determined by \({\mu }_{{{{\rm{Au}}}}}^{{{{\rm{0}}}}}\) and ϕAu, but has sizable charge-dependent contributions from Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\). Instead, ϕAu is determined by the bulk of the electrode across all charge states. This is due to the charge localization on the electrode surface and the effective screening of electrostatic interactions between Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) and Au(111) by the electrode surface even when hybridization takes place.
In the Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\)-Au system, the electrode potential differences obtained from inner potential and Fermi levels are small, within 0.1 V, and correspond to ~5% differences in the applied electrode potential. For quantitatively accurate calculations this is already a notable difference as a 5% error in the over-potential leads to a factor of 7 and 3 error in the computed reaction thermodynamics and rate computed from the Butler–Volmer equation with symmetry factor 0.5, respectively. While rather small differences are observed for the Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\)-Au system, more pronounced differences are expected for systems where the surface PZC and the redox potential of the outer-sphere couple differ significantly, for electrodes with a low DOS around the redox potential such as graphene, for densely charged anionic outer-sphere species such as O\({}_{{{{\rm{2}}}}}^{-}\), and for simulations using a small slab to present the electrode. The impact of the last two instances has been demonstrated in our recent canonical DFT simulations67 of an outer-sphere electron transfer (OS-ET) step for a CO2 reduction reaction, CO2 + e− → CO\({}_{{{{\rm{2}}}}}^{-}\). Our results showed that upon electron donation from the electrode to CO2, the Fermi level of the simulation cell increases by ~0.1 eV: the electrode potential as computed from the Fermi level becomes more reducing by 0.1 V after donating an electron. This unphysical increase in the electrode potential can be explained by noting that the Fermi level of the simulation cell is primarily defined by solution-phase CO\({}_{{{{\rm{2}}}}}^{-}\), rather than the gold electrode. Overall, these numerical results provide strong support for the general proposal that the constant inner potential and Fermi level method exhibit divergent behavior in OS-ET simulations where CIP-DFT achieves a more reliable control over the applied electrode potential.
The preceding results and discussion illustrate that when outer-sphere species are present, the electrochemical potentials of the system and bath are no longer equal. This is due to the fact that the system’s electrode potential (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{M}}}}}={E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)) is influenced by contributions from the redox couple. As an outer-sphere redox couple is not directly linked to the electron bath through external connections, such species should not contribute to the electrode potential. Any changes in the electrode potential should exclusively arise from inner potential differences, in accordance with the general definition provided in Eq. (2). Therefore, the CIP approach is consistent with the general electrode potential definition, whereas electrode potentials derived from \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) do not represent the electrode material itself but instead the entire simulation system. Given that ϕM remains unaffected by the solution phase redox species and that ΔϕM = Δϕbath holds under equilibrium48, it is anticipated that the CIP method correctly replicates the influence of experimentally controlled \({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\).
Biased two-electrode setups within DFT
Our last examples pertain to the construction of a biased two-electrode electrochemical cell and a molecular junction. These configurations are frequently encountered in electrochemical experiments, in classical force field simulations of electrochemical interfaces, and in quantum transport studies2,68. However, modeling voltage bias in such configurations using DFT-based electronic structure methods presents challenges because \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) does not reflect the potential of a single electrode but rather that of the entire simulation cell. In fact, ground-state DFT is formally an equilibrium theory, which does not inherently support multiple Fermi levels within the same simulation cell69. Introducing bias between electrodes requires the utilization of more advanced techniques such as non-equilibrium Green’s function methods70,71. Recent studies have, however, shown that a Fermi level difference between two electrodes can be induced using localized molecular orbitals either within the tight-binding DFT approximation72 or the multi-space DFT (MS-DFT) approach73,74,75 without relying on Green’s function methods (see Supplementary Discussion section 2 for more discussion on MS-DFT). The practical applicability of these methods is limited by the need for localized orbitals, as solid-state DFT codes typically use either plane-wave or a real-space grid basis set to enable electron delocalizing throughout the entire simulation cell.
We apply CIP and constrained DFT (cDFT) to overcome these restrictions and model biased interfaces with real-space methods without localized basis functions or Wannier-localized orbitals. For additional computational details, please refer to the section “Methods” and Supplementary Discussion 2. Figure 7a displays a two-electrode cell comprising two Au(111) electrodes, separated by a vacuum gap. The electrode potential difference or bias between the gold surfaces was established by applying cDFT to introduce a localized external potential of 1 eV within the ’right’ electrode. Most importantly, Fig. 7a highlights that a local external cDFT potential can induce a Δϕ = −(ϕright−ϕleft) = 1.03 V bias between the two electrodes. This demonstrates that an electrode potential difference in a two-electrode cell can be realized through the use of CIP-cDFT. Figure 7a additionally illustrates that the left electrode sets the constant \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) within the simulation cell. Given that we have applied Dirichlet boundary conditions on the right-hand cell boundary, we are able to derive the absolute electrode potential for both electrodes from the inner potentials and subsequently convert them back to an experimental scale (see Supplementary Methods 2).

a The xy-averaged electrostatic potential (solid red lines), inner potentials (dashed black line) of the two gold electrodes, and the cell Fermi level (dot-dash red line). b The electrostatic potential difference (Δϕ = −(ϕDFT−ϕcDFT) black line) for a 1,4-dithiol-phenylene molecular junction under constant bias.
The use of CIP-cDFT is further demonstrated by constructing a biased two-electrode cell for a 1,4-dithiol-phenylene molecule sandwiched between two gold electrodes as depicted in Fig. 7b. In this case, we employed cDFT to introduce a +0.5 eV (−0.5 eV) external potential on the left (right) electrode. This setup results in a non-equilibrium electron density distribution between the electrodes and a bias across the molecular junction. By comparing the equilibrium and non-equilibrium electrostatic potentials (Fig. 7b) obtained from DFT and cDFT calculations, respectively, we observe that the application of cDFT leads to a 1 V bias (ΔϕDFT-cDFT) across the 1,4-dithiol-phenylene junction. This finding agrees well with the more elaborate MS-DFT method (see Fig. 2 of ref. 73). The detailed analysis of CIP-DFT and MS-DFT in the Supplementary Discussion 2, we conclude that CIP-cDFT accurately describes the bias across an electrochemical cell with similar accuracy to the more involved Green’s function or MS-DFT methods. The CIP-cDFT approach therefore provides a solution to the long-standing issue71 of simulating a finite bias in a two-electrode cell with DFT.
Discussion
Through diverse examples, we have shown that controlling ϕM rather than \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) makes the CIP-DFT approach widely applicable to computational studies of electrochemical systems under constant electrode potential conditions. This holds true even in situations where the constant \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) method fails to reproduce the applied electrode potential. The broad applicability of CIP-DFT stems from the precise definition of (single) electrode potentials given in Eq. (2). CIP-DFT directly controls the experimentally relevant electron bath electrochemical potential (\({\tilde{\mu }}_{{{{\rm{e}}}}}^{{{{\rm{bath}}}}}\)) by fixing the electrode inner potential, ϕM. Because ϕM serves as the thermodynamically relevant independent parameter in GCE-DFT (see Supplementary Methods 3), controlling in ϕM is equivalent to controlling the bath electrochemical potential48 and hence the experimentally applied electrode potential. These features distinguish CIP-DFT from all other GCE-DFT methods, where the Fermi level (\({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)) of the simulation cell is fixed. Not only is CIP-DFT more versatile than fixed EF DFT methods, but its computational cost is comparable or even lower than that of the competing approaches.
For pristine metal electrodes and inner-sphere reactions on metallic electrodes, the CIP approach yields comparable results to the widely adopted constant Fermi level description. However, a divergence arises between these two approaches when dealing with outer-sphere reactions and two-electrode setups. In outer-sphere reactions, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) ceases to be exclusively associated with the electrode and thus the Fermi level is no longer uniquely determined by the electrode. Conversely, ϕM is locally defined within the electrode, unaffected by the outer-sphere couple. As ϕM does not depend on the solvent phase, it meets the thermodynamic requirements of a reduced absolute electrode potential (Eq. (2)). On the other hand, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) does not fulfill these conditions as it depends on the presence of the redox couple, and therefore the electrode potential derived from \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) does not reflect the electrode alone. In other words, in simulations of outer-sphere reactions, \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\) is the Fermi level of the entire simulation cell, not the relevant electron bath electrochemical potential. Consequently, we consider the CIP-DFT method to be more suitable than the \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)-based GCE-DFT for constant potential ab initio calculations of outer-sphere reactions.
We have also shown that combing CIP with constrained DFT enables setting up an electrode potential difference (bias) between two electrodes in a simulation cell—this is simply impossible with \({E}_{{{{\rm{F}}}}}^{{{{\rm{DFT}}}}}\)-based GCE-DFT approaches. A particularly appealing feature of CIP-cDFT is the construction of bias potential without using local orbitals or Green’s function approaches, making the method compatible with plane-wave or real-space codes. The prospect of describing a bias between two electrodes without using Green’s function methods creates new possibilities in simulating the applied electrode potential in two-electrode electrochemical cells with DFT71,76 or addressing non-equilibrium charge transfer dynamics through single-molecule junctions (see Supplementary Discussion 2)73. In addition, we envision that CIP-cDFT can be used to, e.g. prevent unphysical and instantaneous electron transfer events77 in molecular dynamics simulations and to include the impact of explicit bias in STM simulations78. As the cDFT method is already available in several solid-state DFT codes, only minimal implementation work is needed to make CIP-cDFT widely applicable.
A possible difficulty of the CIP-DFT method is the definition of the electrode bulk region used for measuring or controlling the inner potential. While this is easy for simple metallic electrodes, defining the bulk region uniquely can be challenging for intercalation electrodes used in battery applications, porous materials found in supercapacitors, and commonly used carbon electrodes, for example. In addition, the inner potential region needs to be chosen carefully also for other complex electrodes encountered in, e.g. battery materials, such as metal/solid electrolyte interphase/electrolyte interfaces79 or electroplated dendrites80. A similar issue pertains to semiconductor electrodes. Our initial studies on a TiO2 semiconductor electrode in Supplementary Methods 3.3 show that very thick slab models need to be used for semiconductor electrodes because the bulk region can only begin after the surface charge has been completely screened by the space charge layer. The thickness of the space charge layer depends on the charge (dopant) concentration and can extend up to micrometers. For instance, the space charge thickness for a typical doping concentration of 1017 dopants per cm3 is 100 μm81. Hence, ensuring a fully screened space charge region will make any constant potential DFT calculations of semiconductors extremely costly if all nuclei and electrons are explicitly treated. A possible solution is to combine CIP-DFT with a continuum model for the bulk semiconductor to achieve screening efficiently as Campbell and Dabo82 have demonstrated. Hence, the merging of CIP-DFT with a continuum82 or e.g. a QM/MM83 model for the semiconductor bulk presents a highly attractive future approach for simulating semiconductor electrodes.
We consider that the use of inner potentials rather than Fermi levels to describe electrode potentials is conceptually appealing because this can bridge the concepts and approaches used in DFT and classical force field-based simulations of electrochemical interfaces as discussed in the section “Introduction”. Because both CIP-DFT and classical simulations model the applied potential using only the electrode and solvent inner potentials, CIP presents a unified scheme for constant potential simulations. Furthermore, ϕM rigorously contains all information needed to control the applied electrode potential and there is in principle no need to have access to the electronic structure or Fermi level; ϕM can be extracted from charge distributions obtained with either classical or quantum mechanical simulations. We consider that these features make CIP a very promising approach to include the electrode potential description in modern machine learning potentials which can take long-range interactions and atomic charges into account84,85,86,87,88. In the future, incorporating CIP in machine learning potentials may enable going beyond current QM/MM DFT/reactive force field methods89 and facilitate computational electrochemistry without explicit simulation of electrons.
Overall, we expect that our CIP-DFT methods will be broadly applied and applicable to a wide variety of interesting electrochemical systems ranging from inner-sphere to outer-sphere reactions and from single electrodes to biased two-electrode cells.
Methods
Common simulation methods
All the simulations were carried out with the GPAW code53,54,90. The wave functions were presented on a real-space grid with 0.18 Å grid spacing unless otherwise specified. The PBE functional91 was used in all the calculations. All calculations are spin-polarized. The continuum solvent was modeled using the SCMVD dielectric continuum model92 with a van der Waals radius of 2.0 Å for gold to accurately reproduce the differential capacitance15. Constant potential calculations, both in the standard Fermi level and the new constant inner potential modes, were carried out using a modified SJM implementation28. All the employed structure models were periodic in the xy-directions and non-periodic in the z-direction. Dirichlet boundary conditions were applied to achieve ϕ = 0 at the z-boundary for the considered charge states as detailed in Supplementary Methods 1. These boundary conditions uniquely define a reference electrode within the simulation cell15.
Inner sphere model
To model an inner sphere reaction, oxygen adsorption on an Au(111) hollow site was considered. The pure and oxygen-covered gold surfaces were modeled using a seven-layer thick 1 × 1 surface cell with a 10 × 10 × 1 k-point sampling.
Constant potential molecular dynamics
Constant charge and fixed potential molecular dynamics were demonstrated for an explicitly solvated Au(111) surface. The simulated system consists of a 3 × 4 × 4 Au(111) surface slab and 32 deuterated water. The simulations were started from a previously obtained pre-equilibrated structure15. Using the SCMVD model 8 Å of implicit water was added on top of the explicit water, and only the Γ-point was considered. The standard dzp-LCAO basis set93 was employed to present single-electron orbitals and density. The system was sampled at 330 K using canonical Langevin dynamics as implemented in ASE90. A time step of 1 fs and a friction coefficient of 5 ps−1 were used following the recommendations from our previous publication94. The electrode potential was referenced against the inner potential of the uncharged interface i.e. the potential of zero charge (PZC) through computing the inner potential within the two central layers of gold as shown in Fig. 4 (see also Supplementary Methods 2). The system was kept neutral by placing an SJM counter charge in the implicit solvent part (see Fig. 4).
Outer sphere simulations
As a model system for outer sphere reactions, we considered a prototypical outer-sphere redox couple, Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\). The entire simulation system, shown in Fig. 6, consists of a Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) near a five-layer thick slab of a Au(111) surface with a 5 × 5 surface cell. A 2 × 2 × 1 k-point mesh was used. The correct oxidation state (3+) on Ru[NH3]\({}_{{{{\rm{6}}}}}^{{{{\rm{3+}}}}}\) was enforced by using the recent grand canonical ensemble constrained DFT (GCE-cDFT) method at all fixed electrode potentials34,95,96. GCE-cDFT applies a spatially localized external potential to obtain an a priori chosen distribution of charges in the simulation cell, see Supplementary Discussion 2 for more discussion. The electrode potential was computed from the inner potential within the central three inner gold layers. These inner potentials were then converted to an experimental SHE scale using Au(111)’s PZC at 0.52 V vs. SHE as detailed in Supplementary Methods 2.
Semiconductor simulations
As a model system for semiconductor electrodes, we used 4–8 layer anatase (101) TiO3 slabs. To ensure a robust description of the electronic structure, the PBE + U functional with a self-consistently computed97 U = 4.5 eV affecting Ti d-orbitals was utilized. A 5 × 5 × 1 k-point mesh was used. Note, that this simple semiconductor model is chosen to demonstrate the application of CIP on semiconductor electrodes. For more realistic calculations of semiconductor electrodes also defects, structural deformations, etc., should be considered.
Two-electrode simulations
The biased two-electrode configurations consist of two gold slabs separated by vacuum (Fig. 7a) or by a 1,4-dithiol-phenylene molecular junction (Fig. 7b). The configuration in Fig. 7a has two 4 × 4 × 1 Au(111) slabs separated by 15 Å vacuum. An electrode potential difference between the electrodes was induced with cDFT to introduce a localized external potential affecting only the right-hand side electrode in Fig. 7a. The molecular junction in Fig. 7b consists of two 3 × 3 × 4 Au(111) surfaces connected by a 1,4-dithiol-phenylene molecule. Both Au slabs were enclosed by 16 Å of vacuum and only the Γ point was used. +0.5 eV (−0.5 eV) external cDFT potential was applied on the left (right) side to create a 1 V electrode potential bias across the cell. The dielectric solvent was not used in the two-electrode CIP-cDFT calculation due to extreme convergence problems caused by the finite-difference (FD) Poisson solver used by GPAW’s dielectric continuum model. The FD Poisson solver is known to be unstable for highly elongated cells and it could reach convergence for the two-electrode setup with a long and narrow cell. Hence, the two-electrode cell calculations could not be performed in the presence of the dielectric solvent. We therefore simulated the system in a vacuum using the newly implemented FastPoissonSolver, which is now the default Poisson solver in GPAW and which was observed to be stable and to provide converged results. However, FastPoissonSolver is not yet compatible with GPAW’s dielectric solvent model or the counter-charges used in SJM. For these reasons, we omitted the dielectric solvent from these two electrode simulations and kept the simulation cell charge neutral. These omissions do not, however, affect the conclusion that bias across a two-electrode simulation cell can be simulated with CIP-cDFT.
Data availability
The methodology has been implemented in the open-source GPAW code54. The scripts and data can be found at https://gitlab.jyu.fi/mamimela/cip.
References
Melander, M. M., Laurila, T. T. & Laasonen, K. Atomic-Scale Modelling of Electrochemical Systems (John Wiley and Sons, 2021).
Allen J. Bard, L. R. F. Electrochemical Methods: Fundamentals and Applications 2nd edn (John Wiley & Sons, 2001).
Trasatti, S. The absolute electrode potential: the end of the story. Electrochim. Acta 35, 269–271 (1990).
Bard, A. J., Memming, R. & Miller, B. Terminology in semiconductor electrochemistry and photoelectrochemical energy conversion (recommendations 1991). Pure Appl. Chem. 63, 569–596 (1991).
Abidi, N., Lim, K. R. G., Seh, Z. W. & Steinmann, S. N. Atomistic modeling of electrocatalysis: are we there yet? WIREs Comput. Mol. Sci. 11, e1499 (2021).
Ringe, S., Hörmann, N. G., Oberhofer, H. & Reuter, K. Implicit solvation methods for catalysis at electrified interfaces. Chem. Rev. 122, 10777–10820 (2022).
Sakaushi, K., Kumeda, T., Hammes-Schiffer, S., Melander, M. M. & Sugino, O. Advances and challenges for experiment and theory for multi-electron multi-proton transfer at electrified solid–liquid interfaces. Phys. Chem. Chem. Phys. 22, 19401–19442 (2020).
Melander, M. M. Grand canonical ensemble approach to electrochemical thermodynamics, kinetics, and model Hamiltonians. Curr. Opin. Electrochem. 29, 100749 (2021).
Schwarz, K. & Sundararaman, R. The electrochemical interface in first-principles calculations. Surf. Sci. Rep. 75, 100492 (2020).
Scalfi, L., Salanne, M. & Rotenberg, B. Molecular simulation of electrode–solution interfaces. Annu. Rev. Phys. Chem. 72, 189–212 (2021).
Deißenbeck, F., Freysoldt, C., Todorova, M., Neugebauer, J. & Wippermann, S. Dielectric properties of nanoconfined water: a canonical thermopotentiostat approach. Phys. Rev. Lett. 126, 136803 (2021).
Jia, M., Zhang, C. & Cheng, J. Origin of asymmetric electric double layers at electrified oxide/electrolyte interfaces. J. Phys. Chem. Lett. 12, 4616–4622 (2021).
Dufils, T., Jeanmairet, G., Rotenberg, B., Sprik, M. & Salanne, M. Simulating electrochemical systems by combining the finite field method with a constant potential electrode. Phys. Rev. Lett. 123, 195501 (2019).
Zhang, C. & Sprik, M. Finite field methods for the supercell modeling of charged insulator/electrolyte interfaces. Phys. Rev. B 94, 245309 (2016).
Melander, M. M., Kuisma, M. J., Christensen, T. E. K. & Honkala, K. Grand-canonical approach to density functional theory of electrocatalytic systems: thermodynamics of solid–liquid interfaces at constant ion and electrode potentials. J. Chem. Phys. 150, 041706 (2019).
Mermin, N. D. Thermal properties of the inhomogeneous electron gas. Phys. Rev. 137, A1441–A1443 (1965).
Pribram-Jones, A., Pittalis, S., Gross, E. K. U. & Burke, K. Thermal Density Functional Theory in Context 25–60 (Frontiers and Challenges in Warm Dense Matter) (Springer International Publishing, 2014).
Sundararaman, R., Goddard III, W. A. & Arias, T. A. Grand canonical electronic density-functional theory: algorithms and applications to electrochemistry. J. Chem. Phys. 146, 114104 (2017).
Taylor, C. D., Wasileski, S. A., Filhol, J.-S. & Neurock, M. First principles reaction modeling of the electrochemical interface: consideration and calculation of a tunable surface potential from atomic and electronic structure. Phys. Rev. B 73, 165402 (2006).
Goodpaster, J. D., Bell, A. T. & Head-Gordon, M. Identification of possible pathways for C-C bond formation during electrochemical reduction of CO2: new theoretical insights from an improved electrochemical model. J. Phys. Chem. Lett. 7, 1471–1477 (2016).
Otani, M. & Sugino, O. First-principles calculations of charged surfaces and interfaces: a plane-wave nonrepeated slab approach. Phys. Rev. B 73, 115407 (2006).
Jinnouchi, R. & Anderson, A. B. Electronic structure calculations of liquid–solid interfaces: combination of density functional theory and modified Poisson–Boltzmann theory. Phys. Rev. B 77, 245417 (2008).
Skulason, E. et al. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J. Phys. Chem. C 114, 18182–18197 (2010).
Letchworth-Weaver, K. & Arias, T. A. Joint density functional theory of the electrode–electrolyte interface: application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge. Phys. Rev. B 86, 075140 (2012).
Fang, Y.-H. & Liu, Z.-P. Mechanism and Tafel lines of electro-oxidation of water to oxygen on RuO2(110). J. Am. Chem. Soc. 132, 18214–18222 (2010).
Skulason, E. et al. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 9, 3241–3250 (2007).
Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. Lett. 6, 2663–2668 (2015).
Kastlunger, G., Lindgren, P. & Peterson, A. A. Controlled-potential simulation of elementary electrochemical reactions: proton discharge on metal surfaces. J. Phys. Chem. C 122, 12771–12781 (2018).
Haruyama, J., Ikeshoji, T. & Otani, M. Electrode potential from density functional theory calculations combined with implicit solvation theory. Phys. Rev. Mater. 2, 095801 (2018).
Surendralal, S., Todorova, M., Finnis, M. W. & Neugebauer, J. First-principles approach to model electrochemical reactions: understanding the fundamental mechanisms behind Mg corrosion. Phys. Rev. Lett. 120, 246801 (2018).
Huang, Y., Nielsen, R. J. & Goddard, W. A. Reaction mechanism for the hydrogen evolution reaction on the basal plane sulfur vacancy site of MoS2 using grand canonical potential kinetics. J. Am. Chem. Soc. 140, 16773–16782 (2018).
Bonnet, N., Morishita, T., Sugino, O. & Otani, M. First-principles molecular dynamics at a constant electrode potential. Phys. Rev. Lett. 109, 266101 (2012).
Ikeshoji, T. & Otani, M. Toward full simulation of the electrochemical oxygen reduction reaction on Pt using first-principles and kinetic calculations. Phys. Chem. Chem. Phys. 19, 4447–4453 (2017).
Melander, M. M. Grand canonical rate theory for electrochemical and electrocatalytic systems I: General formulation and proton-coupled electron transfer reactions. J. Electrochem. Soc. 167, 116518 (2020).
Lindgren, P., Kastlunger, G. & Peterson, A. A. A challenge to the G~ 0 interpretation of hydrogen evolution. ACS Catal. 10, 121–128 (2020).
Siepmann, J. I. & Sprik, M. Influence of surface topology and electrostatic potential on water/electrode systems. J. Chem. Phys. 102, 511–524 (1995).
Dwelle, K. A. & Willard, A. P. Constant potential, electrochemically active boundary conditions for electrochemical simulation. J. Phys. Chem. C 123, 24095–24103 (2019).
Reed, S. K., Lanning, O. J. & Madden, P. A. Electrochemical interface between an ionic liquid and a model metallic electrode. J. Chem. Phys. 126, 084704 (2007).
Takahashi, K., Nakano, H. & Sato, H. A polarizable molecular dynamics method for electrode-electrolyte interfacial electron transfer under the constant chemical-potential-difference condition on the electrode electrons. J. Chem. Phys. 153, 054126 (2020).
Petersen, M. K., Kumar, R., White, H. S. & Voth, G. A. A computationally efficient treatment of polarizable electrochemical cells held at a constant potential. J. Phys. Chem. C 116, 4903–4912 (2012).
Coretti, A. et al. Mass-zero constrained molecular dynamics for electrode charges in simulations of electrochemical systems. J. Chem. Phys. 152, 194701 (2020).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Islas-Vargas, C., Guevara-García, A. & Galván, M. Electronic structure behavior of PbO2, IrO2, and SnO2 metal oxide surfaces (110) with dissociatively adsorbed water molecules as a function of the chemical potential. J. Chem. Phys. 154, 074704 (2021).
Trasatti, S. The absolute electrode potential: an explanatory note (recommendations 1986). J. Electroanal. Chem. Interfacial Electrochem. 209, 417–428 (1986).
Cheng, J. & Sprik, M. Alignment of electronic energy levels at electrochemical interfaces. Phys. Chem. Chem. Phys. 14, 11245–11267 (2012).
Huang, J. Density-potential functional theory of electrochemical double layers: calibration on the Ag(111)-KPF6 system and parametric analysis. J. Chem. Theory Comput. 19, 1003–1013 (2023).
Huang, J. Zooming into the inner Helmholtz plane of Pt(111)–aqueous solution interfaces: chemisorbed water and partially charged ions. JACS Au 3, 550–564 (2023).
Choe, D.-H., West, D. & Zhang, S. Band alignment and the built-in potential of solids. Phys. Rev. Lett. 121, 196802 (2018).
Tung, R. T. The physics and chemistry of the Schottky barrier height. Appl. Phys. Rev. 1, 011304 (2014).
Bisquert, J., Cendula, P., Bertoluzzi, L. & Gimenez, S. Energy diagram of semiconductor/electrolyte junctions. J. Phys. Chem. Lett. 5, 205–207 (2014).
Cendula, P. et al. Calculation of the energy band diagram of a photoelectrochemical water splitting cell. J. Phys. Chem. C 118, 29599–29607 (2014).
Nozik, A. J. & Memming, R. Physical chemistry of semiconductor–liquid interfaces. J. Phys. Chem. 100, 13061–13078 (1996).
Mortensen, J. J., Hansen, L. B. & Jacobsen, K. W. Real-space grid implementation of the projector augmented wave method. Phys. Rev. B 71, 035109 (2005).
Enkovaara, J. et al. Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method. J. Phys. Condens. Matter 22, 253202 (2010).
Gross, A. Grand-canonical approaches to understand structures and processes at electrochemical interfaces from an atomistic perspective. Curr. Opin. Electrochem. 27, 100684 (2021).
Goldsmith, Z. K., Calegari Andrade, M. F. & Selloni, A. Effects of applied voltage on water at a gold electrode interface from ab initio molecular dynamics. Chem. Sci. 12, 5865–5873 (2021).
Van Vliet, C. M. Equilibrium and Non-Equilibrium Statistical Mechanics (World Scientific, 2008).
Zwanzing, R. Nonequilibrium Statistical Mechanics (Oxford University Press, 2001).
Melander, M. M. Atomic-Scale Modelling of Electrochemical Systems, chap. Constant Potential Rate Theory—General Formulation and Electrocatalysis (John Wiley and Sons, 2021).
Li, W.-L. et al. Critical role of thermal fluctuations for co binding on electrocatalytic metal surfaces. JACS Au 1, 1708–1718 (2021).
May, V. & Kühn, O. Charge and Energy Transfer Dynamics in Molecular Systems Vol. 3 (Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011).
Warburton, R. E., Soudackov, A. V. & Hammes-Schiffer, S. Theoretical modeling of electrochemical proton-coupled electron transfer. Chem. Rev. 122, 10599–10650 (2022).
Abidi, N. & Steinmann, S. N. How are transition states modeled in heterogeneous electrocatalysis? Curr. Opin. Electrochem. 33, 100940 (2022).
Domínguez-Flores, F. & Melander, M. M. Electrocatalytic rate constants from DFT simulations and theoretical models: Learning from each other. Curr. Opin. Electrochem. 36, 101110 (2022).
Liu, D.-Q. et al. Adiabatic versus non-adiabatic electron transfer at 2d electrode materials. Nat. Commun. 12, 7110 (2021).
Schmickler, W. Adiabatic and non-adiabatic electrochemical electron transfer in terms of Green’s function theory. Russ. J. Electrochem. 53, 1182–1188 (2017).
Qin, X., Hansen, H. A., Honkala, K. & Melander, M. M. Cation-induced changes in the inner- and outer-sphere mechanisms of electrocatalytic CO2 reduction. Nat. Commun. 14, 7607 (2023).
Aradhya, S. V. & Venkataraman, L. Single-molecule junctions beyond electronic transport. Nat. Nanotechnol. 8, 399–410 (2013).
Tripkovic, V., Björketun, M. E., Skúlason, E. & Rossmeisl, J. Standard hydrogen electrode and potential of zero charge in density functional calculations. Phys. Rev. B 84, 115452 (2011).
Stefanucci, G. & van Leeuwen, R. Nonequilibrium Many-Body Theory of Quantum Systems: A Modern Introduction (Cambridge University Press, 2012).
Hansen, M. H., Jin, C., Thygesen, K. S. & Rossmeisl, J. Finite bias calculations to model interface dipoles in electrochemical cells at the atomic scale. J. Phys. Chem. C 120, 13485–13491 (2016).
Oshiki, J., Nakano, H. & Sato, H. Controlling potential difference between electrodes based on self-consistent-charge density functional tight binding. J. Chem. Phys. 154, 144107 (2021).
Lee, J., Kim, H. S. & Kim, Y.-H. Multi-space excitation as an alternative to the Landauer picture for nonequilibrium quantum transport. Adv. Sci. 7, 2001038 (2020).
Kim, H. S. & Kim, Y.-H. Constrained-search density functional study of quantum transport in two-dimensional vertical heterostructures. Preprint at https://arxiv.org/abs/1808.03608 (2018).
Lee, J., Yeo, H. & Kim, Y.-H. Quasi-fermi level splitting in nanoscale junctions from ab initio. Proc. Natl Acad. Sci. USA 117, 10142–10148 (2020).
Khatib, R., Kumar, A., Sanvito, S., Sulpizi, M. & Cucinotta, C. S. The nanoscale structure of the Pt–water double layer under bias revealed. Electrochim. Acta 391, 138875 (2021).
Aierken, Y. et al. Revealing charge-transfer dynamics at electrified sulfur cathodes using constrained density functional theory. J. Phys. Chem. Lett. 12, 739–744 (2021).
Blanco, J. M., Flores, F. & Pérez, R. STM-theory: image potential, chemistry and surface relaxation. Prog. Surf. Sci. 81, 403–443 (2006).
Li, Y. & Qi, Y. Energy landscape of the charge transfer reaction at the complex Li/SEI/electrolyte interface. Energy Environ. Sci. 12, 1286–1295 (2019).
Santos, E. & Schmickler, W. The crucial role of local excess charges in dendrite growth on lithium electrodes. Angew. Chem. Int. Ed. 60, 5876–5881 (2021).
Memming, R. Semiconductor Electrochemistry (Wiley-VCH, Weinheim, 2000).
Campbell, Q. & Dabo, I. Quantum-continuum calculation of the surface states and electrical response of silicon in solution. Phys. Rev. B 95, 205308 (2017).
Stecher, T., Reuter, K. & Oberhofer, H. First-principles free-energy barriers for photoelectrochemical surface reactions: proton abstraction at TiO2(110). Phys. Rev. Lett. 117, 276001 (2016).
Grisafi, A., Nigam, J. & Ceriotti, M. Multi-scale approach for the prediction of atomic scale properties. Chem. Sci. 12, 2078–2090 (2021).
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 12, 398 (2021).
Xie, X., Persson, K. A. & Small, D. W. Incorporating electronic information into machine learning potential energy surfaces via approaching the ground-state electronic energy as a function of atom-based electronic populations. J. Chem. Theory Comput. 16, 4256–4270 (2020).
Unke, O. T. & Meuwly, M. Physnet: a neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. General-purpose machine learning potentials capturing nonlocal charge transfer. Acc. Chem. Res. 54, 808–817 (2021).
Naserifar, S., Chen, Y., Kwon, S., Xiao, H. & Goddard III, W. A. Artificial intelligence and QM/MM with a polarizable reactive force field for next-generation electrocatalysts. Matter 4, 195–216 (2021).
Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Held, A. & Walter, M. Simplified continuum solvent model with a smooth cavity based on volumetric data. J. Chem. Phys. 141, 174108 (2014).
Larsen, A. H., Vanin, M., Mortensen, J. J., Thygesen, K. S. & Jacobsen, K. W. Localized atomic basis set in the projector augmented wave method. Phys. Rev. B 80, 195112 (2009).
Korpelin, V. et al. Addressing dynamics at catalytic heterogeneous interfaces with DFT-MD: anomalous temperature distributions from commonly used thermostats. J. Chem. Phys. Lett. 13, 2644–2652 (2022).
Melander, M., Jónsson, E. O., Mortensen, J. J., Vegge, T. & García Lastra, J. M. Implementation of constrained DFT for computing charge transfer rates within the projector augmented wave method. J. Chem. Theory Comput. 12, 5367–5378 (2016).
Kaduk, B., Kowalczyk, T. & Voorhis, T. V. Constrained density functional theory. Chem. Rev. 112, 321–370 (2012).
Korpelin, V., Melander, M. M. & Honkala, K. Reducing the irreducible: dispersed metal atoms facilitate reduction of irreducible oxides. J. Phys. Chem. C 126, 933–945 (2022).
Acknowledgements
The work was supported by the Academy of Finland through projects 317739, 307853, and 338228 as well as the LACOR project funded by the Jane and Aatos Erkko Foundation. T.W. was supported by the State Scholarship Fund from the China Scholarship Council (No. 201906070128) and by the National Natural Science Foundation of China (No. 52202214). The computational resources were provided by the CSC-IT Center for Science, Espoo, Finland (https://www.csc.fi/en/) and the FGCI—Finnish Grid and Cloud Infrastructure. We also thank Dr. Laura Laverdure for critically reading the manuscript.
Author information
Authors and Affiliations
Contributions
M.M.M.: Conceptualization, methodology, software, validation, formal analysis, investigation, supervision, funding acquisition, writing—original draft, writing—review & editing. T.W.: Validation, investigation. Ti.W.: Methodology, analysis, writing—review. K.H: Formal analysis, supervision, funding acquisition, writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Melander, M.M., Wu, T., Weckman, T. et al. Constant inner potential DFT for modelling electrochemical systems under constant potential and bias. npj Comput Mater 10, 5 (2024). https://doi.org/10.1038/s41524-023-01184-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01184-4
- Springer Nature Limited
This article is cited by
-
Aqueous alternating electrolysis prolongs electrode lifespans under harsh operation conditions
Nature Communications (2024)