
Abstract
Macrophage- and monocyte-derived cytokines are elevated in the myocardium of pressure-overloaded hearts, where they play critical roles in pathological remodeling. Caspase recruitment domain family member 9 (CARD9) regulates macrophage cytokine secretion, but its role in a transverse aortic constriction (TAC) model of pressure overload has not been evaluated. To investigate whether CARD9 may serve as a valuable therapeutic target, wild-type (WT) and CARD9-knockout mice were subjected to 3 months of TAC, and then cardiac function, hypertrophy, and fibrosis were analyzed. The expression of protein markers of myocardial autophagy and nuclear factor kappa B signaling was also investigated. At 1 month after TAC, cardiomyocyte contractile dynamics were measured in a separate cohort to further assess contractility and diastolic function. In WT but not CARD9−/− mice, TAC resulted in severe cardiomyocyte contractile dysfunction at 1 month and functional decrements in fractional shortening at 3 months in vivo. Furthermore, CARD9−/− mice did not develop cardiac fibrosis or hypertrophy. CARD9−/− mice also had decreased protein expression of inhibitor of κB kinase-α/β, decreased phosphorylation of p65, and increased expression of protein markers of autophagy. These findings suggest that CARD9 plays a role in pathological remodeling and cardiac dysfunction in mouse hearts subjected to TAC and should be investigated further.
Similar content being viewed by others


Introduction
The worldwide prevalence of hypertension is staggeringly high (20.1% of women; 24.1% of men) and is increasing in most populations, with bleak projections for the future [1]. Nearly one in five deaths globally are attributed to increased blood pressure, making it the leading metabolic risk factor in the world [2]. Nearly a million individuals died from hypertensive heart disease (HHD) alone in 2016, which did not take into account the contribution of elevated blood pressure to the development of other fatal cardiovascular diseases [3]. Interestingly, elevated blood pressure is also linked to persistent systemic inflammation [4].
Circulating monocytes [5, 6] and myocardial macrophages [7] secrete cytokines that coordinate systemic and paracrine inflammatory crosstalk among cardiomyocytes, fibroblasts, and macrophages [8]. The resulting inflammatory milieu promotes further recruitment and activation of myeloid cells, cardiac fibrosis [9], and hypertrophy. Over time, fibrotic stiffening and concentric ventricular hypertrophy result in a loss of elasticity. Dysfunctional relaxation disrupts ventricular filling and hampers cardiac output. Thickening, stiffening, and fibrosis progressively worsen as the ventricle eventually dilates, and systolic dysfunction and failure occur.
Caspase recruitment domain family member 9 (CARD9), a scaffolding protein expressed in myeloid cells, is required for the activation of the macrophage transcription factors p38 mitogen activated protein kinase and nuclear factor kappa B (NFκB) [10], which are responsible for the production of inflammatory cytokines such as tumor necrosis factor (TNF)α, pro-interleukin (IL)-1β, and IL-6 [11, 12].
Cytokine-initiated inflammation results in fibrosis, hypertrophy, and dysfunction [13] in the pressure-overloaded heart [14, 15]. Despite the fact that these cytokines are secreted in concert, most investigations have only isolated a single cytokine for targeted therapy. We posit that targeting a mutual upstream progenitor of multiple inflammatory components could potentially offer increased therapeutic benefits, and CARD9 offers such an opportunity. We have previously shown that genetic knockout (KO) of CARD9 rescues cardiovascular dysfunction in a high-fat diet model of cardiovascular disease [16]. We hypothesize that a similar protection is afforded in HHD and examined the effect of CARD9 KO on hypertrophy, fibrosis, and function in a chronic pressure overload murine model.
Methods
Animals and study design
A pair of CARD9−/− breeding mice was kindly provided by Professor Xin Lin (Department of Molecular and Cellular Oncology, University of Texas, Houston, TX) [17]. Adult C57BL/6J wild-type (WT) and CARD9−/− mice were assigned to the transverse aortic constriction (TAC) or control groups. One month after group assignment, the mice were either sacrificed for cardiomyocyte isolation or maintained for echocardiography. At 3 months, echocardiography was performed, the mice were sacrificed, and the hearts were excised, weighed, fixed in 10% neutral buffered formalin or snap frozen in liquid N2 and stored at −80 °C for further analysis.
The mice were bred, weaned, and housed in the Animal Care Facility of the University of Wyoming, College of Health Sciences. The procedures and methods were approved by the University of Wyoming Institutional Animal Care and Use Committee. The investigation complied with the Animal Welfare Act and all NIH regulations and guidelines.
TAC
A TAC model was used to induce myocardial pressure overload and provoke the maladaptive cardiac developments that also occur during hypertension [18]. Mice aged 4–6 months and weighing 25–32 g were anesthetized with ketamine (145 mg/kg bw) and xylazine (11 mg/kg bw). The mice were intubated with an endotracheal tube and ventilated (Minivent Type 845, Harvard Apparatus, MA, USA) at a respiratory volume of 260 µL and a frequency of 130 breaths per minute. Using presterilized instruments, a partial thoracotomy to the second rib was performed, followed by partial ligation (6–0 suture) around the transverse aorta against a 27-gauge needle between the left and right carotid artery bifurcations to create constriction of the aorta without complete ligation.
To ensure that the surgical procedures were comparable between groups and that differences were not due to inconsistencies in the degree of overload, color Doppler echocardiography (Vevo 2100, FUJIFILM VisualSonic Inc, ON, Canada) was used to assess trans-stenotic peak flow velocity 4 and 7 days post procedure. The peak flow velocity was then converted to estimate the pressure gradient using the modified Bernoulli equation, as previously validated [19]. TAC-induced trans-stenotic pressure gradients were similar between the WT- and CARD9−/−-TAC groups at day 4 or 7 (data not shown).
Echocardiography
Echocardiography was performed 1, 2, and 3 months post TAC. Cardiac function was evaluated in isoflurane-anesthetized (1–2%, 1 L/min flow rate) mice using 2D guided M-mode echocardiography (Philips HP Sonos 5500) with a 15–6 L linear transducer (Philips Medical Systems). Measurements were recorded from a minimum of three cardiac cycles in M-mode at similar heart rates. Left ventricular internal chamber dimensions at end diastole (LVIDd) and end systole (LVIDs) were used to calculate fractional shortening percentages using the following formula: ((LVIDd − LVIDs)/LVIDd) × 100.
Cardiomyocyte isolation
The mice were weighed and then anesthetized with ketamine (145 mg/kg bw) and xylazine (11 mg/kg bw). The hearts were excised, the aortas were cannulated, and the hearts were perfused for 5 min using a Langendorff perfusion system. The hearts were digested with collagenase (Liberase TH Research Grade, Sigma-Aldrich) (1 mg/15 mL), minced and then added to a contraction buffer as described in our previous study [16].
Contractile properties of ventricular cardiomyocytes were assessed using a MyoCam® system (IonOptix Corp., MA, USA) and edge detecting software SoftEdge (IonOptix Corp., MA, USA), and the cells were stimulated at a frequency of 0.5 Hz.
Masson’s trichrome staining
After sacrifice, the hearts were incubated in 10% neutral buffered formalin for 24–48 h at room temperature. The hearts were dehydrated, cleared, paraffinized, and then cut into 5 µm sections using a microtome.
Masson’s trichrome was used to stain collagen. Reagent preparation and staining were performed using a kit (HT15, Sigma-Aldrich) according to the manufacturer’s instructions (Procedure No. HT15). Images of the stained sections were recorded using a fluorescence microscope (Olympus, CellSens Standard). Collagen levels were quantified using a color deconvolution method as previously described [20].
Western blot analyses
Tissues were lysed in ice-cold RIPA buffer using a homogenizer (Fisher Scientific™ Model 125) and sonicated (Fisher Scientific™ CL-18). The lysate was centrifuged, the pellet was discarded, and the supernatant was retained for further preparation.
After loading equal amounts of protein in each well, proteins were separated on sodium dodecyl sulfate polyacrylamide gels (Bio-Rad Mini Protean Tetra, #1658000) and then transferred to a membrane (Bio-Rad Criterion Blotter, #1704070). The membranes were blocked in 3% bovine serum albumin and then incubated overnight with primary antibodies against p-IKKα/β and inhibitor of κB kinase (IKK)-α/β (Santa Cruz #2697 and #7607), p-p65 and p65 (CST #3033 and #8242), and light chain 3B (LC3B) I/II and p62 (CST #2775 and #5114), with GAPDH (CST #5174) as the loading control.
The membranes were incubated with horseradish peroxidase-conjugated secondary antibody (CST #7074) and then luminol, peroxide, and ddH2O (1:1:1) (LumiGlo, CST #7003). Immunoreactive bands were visualized with a Bio-Rad ChemiDoc XRS. Band intensity was quantified as the signal density using Quantity One® software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism. Two-way or mixed-model analysis of variance (ANOVA) was performed, along with Tukey’s or Sidak post tests where appropriate. When reporting overall group effects of TAC and CARD9 KO, two-way ANOVA was performed, along with Bonferroni tests where appropriate. Significance was set at p < 0.05 for all tests. The data are presented as the mean ± SEM.
Results
The hypertrophic myocardial response to pressure overload is averted by CARD9 KO
To understand the effect of pressure overload and CARD9 KO on heart size, after 1 and 3 months, the mice were sacrificed, and their hearts were weighed. After 1 month, TAC modestly increased heart weight in WT mice (WT-control 6.6 ± 0.45 mg/mm vs. WT-TAC 8.9 ± 0.52; **p < 0.01) but not CARD−/− mice (CARD−/−-control 6.8 ± 0.39 vs. CARD−/−-TAC 7.5 ± 0.36; ns). After 3 months, WT-TAC hearts were markedly enlarged (Fig. 1a, c) (WT-control 7.7 ± 0.3 mg/mm vs. WT-TAC 13.6 ± 1.1 mg/mm; ***p < 0.0001), CARD9−/− mice with TAC showed an increase in heart size compared to that of controls at month 1 and a larger increase at month 3 (CARD9−/−-control 7.1 ± 0.4 mg/mm vs. CARD9−/−-TAC 8.5 ± 0.4 mg/mm; ns), but neither time point exhibited a significant increase. Furthermore, CARD9−/−-TAC hearts were significantly smaller than their WT-TAC counterparts (CARD9−/−-TAC 8.5 ± 0.4 mg/mm vs. WT-TAC 13.6 ± 1.1 mg/mm; ###p < 0.001).

CARD9 KO prevents TAC-induced cardiac hypertrophy. a Representative images of control and TAC hearts from WT and CARD9−/− mice 1 and 3 months post TAC. b HW/TL in WT and CARD9−/− mice in the TAC or control group after 1 month. c HW/TL at 3 months post TAC. d HW/TL of the controls compared to those at 1 and 3 months post TAC. *p < 0.05, **p < 0.01, ****p < 0.0001 WT-TAC vs. Control. ###p < 0.001, CARD9−/−-TAC vs. WT-TAC. Month 1, n = 5 per group. Month 3, n = 11, 11, 7, and 6 for WT-control, WT-TAC, CARD−/−-control, and CARD−/−-TAC, respectively
CARD9−/− mice maintain in vivo cardiac function post TAC
To determine whether CARD9 plays a role in TAC-induced cardiac dysfunction in mice, echocardiography was performed, and the percentage of fractional shortening (FS%) was measured to ascertain cardiac function after 1, 2, and 3 months of TAC (Fig. 2a, b).

Echocardiographic assessment of cardiac function. a Representative 2D echocardiographic images of the LVs of WT and CARD9−/− mice in the TAC and control groups at 1 and 3 months. Statistical analysis of fractional shortening (%) (b), LVIDd (c), and LVIDs (d) in WT and CARD9−/− mice in the control and TAC groups, which were measured at 1, 2, and 3 months. *p < 0.05; **p < 0.01; ***p < 0.001 WT-TAC vs. WT-control. #p < 0.05, ##p < 0.01, ###p < 0.001, CARD9−/−-TAC vs. WT-TAC. WT-control n = 11. WT-TAC n = 10, CARD9−/−-control n = 7. CARD9−/−-TAC n = 7
After 1 month, WT-control and CARD9−/−-control mice had FS% of 46.8% and 48.1%, respectively. After 1 month of constriction, the FS% in WT mice was reduced to 36% (WT-control 46.8 ± 0.7% vs. WT-TAC 36.5 ± 2.6%; *p < 0.05), but there was no reduction in FS% in CARD9-KO mice (CARD9−/−-control 48.1 ± 0.8% vs. CARD9−/−-TAC 43.3 ± 1.2%; ns). Both TAC groups deteriorated further when assessed at 2 months. CARD9−/− mice, however, fared better than their WT counterparts (CARD9−/−-TAC 38.0 ± 1.0% vs. WT-TAC 30.0 ± 2.4%; †p = 0.052).
After 3 months, cardiac function in WT-TAC mice remained relatively stable, whereas CARD9−/− mice subjected to TAC improved slightly (WT-TAC 29.6 ± 2.3% vs. CARD9−/−-TAC 42.2 ± 1.2%; ##p < 0.01).
LVIDd expanded progressively at months 1 and 2 for all TAC mice but only significantly expanded in WT-TAC mice (WT-control 0.31 ± 0.02 cm vs. WT-TAC 0.412 ± 0.03 cm; *p < 0.05). The trend continued for WT mice at month 3 (WT-control 0.313 ± 0.02 cm vs. WT-TAC 0.414 ± 0.04 cm; **p < 0.01), whereas the effect was reversed in CARD9−/−-TAC mice (Fig. 2a, c).
LVIDs (Fig. 2a, d) increased in both TAC groups at months 1 and 2 but only continued to increase in WT-TAC mice at month 3. These increases were only statistically significant for WT mice at months 2 (WT-control 0.167 ± 0.008 cm vs. WT-TAC 0.269 ± 0.028 cm; **p < 0.01) and 3 (WT-control 0.167 ± 0.008 cm vs. WT-TAC 0.295 ± 0.034 cm; **p < 0.01). The increase only reached significance in CARD9−/− mice after 2 months (CARD9−/−-control 0.163 ± 0.012 cm vs. CARD9−/−-TAC 0.210 ± 0.015 cm; *p < 0.05).
Isolated cardiomyocytes from CARD9−/− mice maintain contractile functions post TAC
To assess the individual and combined effects of TAC and CARD9 KO on cardiomyocyte contractile dynamics, collagen was digested from excised hearts, and contractility was then measured in the isolated cardiomyocytes.
While TAC increased overall resting cell length, which accounted for 26.7% of the variance (p = 0.016), no significant differences between individual groups were observed.
TAC slowed the shortening velocity (Fig. 3b) in WT mice (WT-control 136 ± 13.7 µm/s vs. WT-TAC 86.3 ± 3.4 µm/s; *p < 0.05) but not in CARD9−/− mice. The net result was a significantly advantageous shortening velocity in cardiomyocytes from CARD9−/−-TAC mice compared to those from WT-TAC mice (CARD9−/−-TAC 138.2 ± 14.7 µm/s vs. WT-TAC 86.3 ± 3.4 µm/s; p < 0.05).

Cardiomyocyte contractile properties in WT and CARD9−/− mice 1 month after being assigned to the TAC procedure or a control group. a Resting cell length of cardiomyocytes, b shortening velocity, c peak shortening, d relengthening velocity, e time to peak shortening (TPS), and f time to 90% relengthening (TR90) *p < 0.05, **p < 0.01, ***p < 0.001, WT-TAC vs. Control. #p < 0.05, ##p < 0.01, CARD9−/−-TAC vs. WT-TAC. n = 4–7 per group
Constriction decreased peak shortening (Fig. 3c) in cardiomyocytes from WT mice (WT-control 8.16 ± 0.52 µm vs. WT-TAC 4.38 ± 0.29 µm; ***p = 0.0005) but not in those from CARD9−/− mice. CARD9−/−cardiomyocytes had considerably better peak shortening than their WT counterparts (WT-TAC 4.38 ± 0.29 µm vs. CARD9−/−-TAC 7.59 ± 0.79 µm; ##p = 0.0036).
TAC reduced relengthening velocity (Fig. 3d) in cardiomyocytes from WT mice (WT-control 117.3 ± 13.473 µm/s vs. WT-TAC 53.1 ± 4.4 µm/s; **p = 0.0017) but not in those from CARD9−/− mice. Furthermore, CARD9−/−-TAC cardiomyocytes relaxed faster than their WT counterparts (WT-TAC 53.1 ± 4.4 µm/s vs. CARD9−/−-TAC 113.5 ± 10.6 µm/s; ##p = 0.0043).
Time to peak shortening (Fig. 3e) was unaffected by TAC and CARD9 KO. The TR90 was longer in cardiomyocytes from WT-TAC mice than from WT-controls (WT-control 0.214 ± 0.013 s vs. WT-TAC ± 0.297 ± 0.019 s; **p < 0.01). TR90 was not reduced by TAC in CARD9−/− cardiomyocytes; however, these cells relengthened significantly faster than cardiomyocytes from WT-TAC mice (WT-TAC 0.297 ± 0.019 s vs. CARD9−/−-TAC 0.221 ± 0.007 s; **p < 0.01). Overall, the measures of systolic and diastolic function were diminished by pressure overload in WT mice but not CARD9−/− mice.
Hearts from CARD9−/− mice are not fibrotic after TAC
To determine the role of CARD9 in myocardial fibrotic remodeling post TAC, hearts were excised, fixed, and stained for collagen using Masson’s trichrome. Then, four fields were quantified in ImageJ and statistically analyzed.
As shown in Fig. 4a, b, hearts from WT-TAC mice exhibited severe diffusive collagen staining (WT-control 11.4% vs. WT-TAC 23.0%; ****p < 0.0001), but hearts from CARD9−/−-TAC mice did not (CARD−/−Control 2.9% vs. CARD9−/−-TAC 3.6%; ns). Indeed, the hearts from WT-TAC were significantly more fibrotic than hearts from CARD9−/−-TAC (####p < 0.0001), suggesting a detrimental effect of CARD9 in the fibrotic remodeling process during pressure overload.

Masson’s trichrome staining of myocardial sections from WT or CARD9−/− mice in the TAC and control groups at 3 months. Cardiomyocytes are stained red, nuclei are purple, and collagen is blue. Scale bar: 50 µm. a Representative histological images for each group. b Statistical analysis of Masson’s trichrome staining using color deconvolution and threshold analysis in ImageJ. ***p < 0.001, WT-TAC vs. WT-control. ###p < 0.001, CARD9−/−-TAC vs. WT-TAC. n = 4 per group
CARD9 KO is associated with altered IKKα/β signaling
IKKα/β phosphorylation can control inflammation-induced NFκB activation, linking inflammation to adverse myocardial hypertrophy and fibrosis [21]. We hypothesized that chronic TAC would increase IKKα/β phosphorylation and that CARD9 KO would prevent the increase.
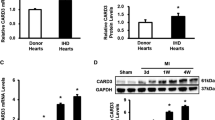
Western blot analysis showed that TAC-induced pressure overload had no effect on myocardial IKKα/β expression or phosphorylation (Fig. 5a). CARD9 KO, however, induced a significant overall decrease in myocardial IKKα/β protein expression (Fig. 5c), accounting for 67.2% of the variance (p = 0.0033). CARD9 KO upregulated phosphorylation and accounted for 47.2% of the variance in phosphorylation levels (p = 0.0262) (Fig. 5b). CARD9−/−-TAC heart lysates had significantly less overall protein expression of IKKα/β than WT-TAC heart lysates (WT-TAC 5.5 ± 0.87 vs. CARD9−/−-TAC 2.94 ± 0.5; #p < 0.05) (Fig. 5c).

Western immunoblot analysis of phosphorylated and total p65 subunit of NFκB and IKKα/β. a Representative blots showing phosphorylated and total IKKα/β, with GAPDH as a loading control. b Statistical analysis of the level of phosphorylated IKKα/β as a proportion of total IKKα/β expression and c of IKKα/β expression as a proportion of GAPDH expression. d Representative blots showing total and phosphorylated p65, with GAPDH as the loading control. e Statistical analysis of the level of phosphorylated p65 as a proportion of total p65 protein expression and of f total protein expression of p65 as a proportion of GAPDH expression. *p < 0.05, **p < 0.01 WT vs. CARD9−/−, #p < 0.05 vs. WT-TAC. n = 3 per group. ADU arbitrary densitometric units
TAC-related increases in p65 phosphorylation are blunted in CARD9−/− mouse hearts
Western blotting was used to determine the protein expression and activation of the p65 subunit of NFκB. It was hypothesized that TAC would increase the phosphorylation of p65 in WT mice but not in CARD9−/− mice. As predicted, the ratio of phosphorylated p65 increased in WT-TAC mice (WT-TAC 1.4× of WT-control; *p < 0.05) but not in their CARD9−/− counterparts (Fig. 5d, e). Total expression of the p65 subunit was unchanged in both groups (Fig. 5f).
Markers of autophagic flux are increased in the myocardium of CARD9−/− mice
Western blotting was used to measure the expression of canonical markers of autophagy in the myocardium, which we hypothesized would be altered in WT but not CARD9−/− mice subjected to pressure overload. CARD9 KO induced an increasing trend (p < 0.10) in the overall protein expression of microtubule-associated protein 1 LC3BII/I. CARD9 KO decreased the expression of Sequestosome-1 (p62) (p = 0.0017) and accounted for 68.5% of the variation. Hearts from CARD9−/−-TAC mice expressed significantly less p62 than hearts from WT-TAC mice (CARD9−/−-TAC 1.48 ± 0.28 vs. WT-TAC 3.53 ± 0.74; p < 0.05) (Fig. 6d). Taken together, these findings indicate increased autophagosome formation and clearance in CARD9−/− mice.

Western immunoblot analysis of markers of autophagy in WT and CARD9−/− hearts from mice in the TAC and control groups at 3 months. a Representative blots showing LC3BII/I expression, with GAPDH as a loading control. b Statistical analysis of the level of L3BII/I as a proportion of GAPDH expression. c Representative blots showing Sequestosome-1 (p62) and GAPDH. d Statistical analysis of the level of p62 accumulation as a proportion of GAPDH expression. *p < 0.05, **p < 0.01 WT vs. CARD9−/−, #p < 0.05 vs. WT-TAC. n = 3 per group. ADU arbitrary densitometric units
Discussion
The myocardial pressure overload caused by the increase in peripheral resistance associated with hypertension induces pathological myocardial remodeling, including fibrosis and hypertrophy, resulting in the development of cardiac dysfunction and the progression to heart failure. Inflammation plays a causative role in myocardial remodeling, but effective therapies are elusive. In response to mechanical stress, cardiomyocytes secrete cytokines and chemokines that recruit macrophages to the myocardium to further exacerbate cytokine-mediated inflammation. Rather than targeting a single cytokine, we demonstrate that mice lacking expression of CARD9, a key upstream activator of the innate immune response to pathogens and damage signals, do not exhibit the pathological cardiac effects of TAC-induced pressure overload.
There are several key findings in the current study. First, CARD9 is required for in vivo cardiac dysfunction and ex vivo cardiomyocyte contractility dysfunction after 1 month of TAC-induced pressure overload. CARD9−/− mice fully maintained cardiac function after 1 month and experienced only minor dysfunction after 2 months. Interestingly, while WT-TAC mice progressively declined, the FS% improved in CARD9−/−-TAC mice after 2 months such that cardiac function was similar between CARD9−/−-TAC and controls after 3 months, preventing the progression to heart failure and indicating a possible ongoing protective effect of CARD9 KO. The trend in the reversal in FS% in CARD9−/−-TAC mice may indicate the development of compensatory concentric hypertrophy, as suggested by a slight increase in heart weight at 3 months (Fig. 1c, d) and narrowed LVID (Fig. 2c, d) measurements.
Second, we found highly dysfunctional contraction and relaxation dynamics in isolated cardiomyocytes from WT-TAC mice (Fig. 3). Systolic dysfunction was absent in cardiomyocytes from CARD9−/− mice (Fig. 3b, c), and CARD9−/−-TAC cardiomyocytes also showed no signs of diastolic dysfunction (Fig. 3e, f), which generally precedes the development of systolic dysfunction.
Cardiomyocytes are the smallest contractile unit of the heart, and their function can be altered by chronic mechanical stressors or chemical signals, including the CARD9-mediated cytokines TNF-α [22] and IL-1β [23]. Our findings indicate that CARD9 KO may prevent diastolic and systolic dysfunction from developing at the cellular level during TAC-induced pressure overload.
Third, CARD9 may be involved in TAC-induced fibrosis and hypertrophy. Cardiac prognosis is associated with the extent of fibrosis [24], and macrophages largely drive myocardial fibrosis [25, 26]. Fibrosis and hypertrophy develop early in TAC-induced pressure overload, may precede dysfunction, and are primary risk factors that lead to cardiac failure. WT-TAC hearts were enlarged after 1 month of TAC (Fig. 1) and were markedly hypertrophied with severe fibrosis (Fig. 4) after 3 months of TAC, averaging nearly twice the mass of their controls, whereas CARD9−/−-TAC hearts were not significantly enlarged or fibrotic. Inflammation is associated with left ventricular hypertrophy in hypertension [27, 28]. As CARD9 is a potent regulator of inflammation, we hypothesized that CARD9−/− mice would be spared some degree of pathological cardiac hypertrophy.
Hypertrophy and fibrosis are independent cardiovascular risk factors in HHD, predisposing individuals to heart failure, stroke, arrhythmia, coronary events, and sudden death [29,30,31]. Despite the severe degree of fibrosis observed in WT-TAC mice, we showed that chronic pressure overload damaged cells to an extent that collagen digestion during the isolation procedure did not restore contractile function ex vivo (Fig. 3). These findings indicate that dysfunction is not limited to heart stiffening or replacement fibrosis but includes contractile dysfunction at the cardiomyocyte level.
Chronically secreted CARD9-mediated cytokines induce positive feedback inflammatory signaling in fibroblasts and cardiomyocytes, increasing transforming growth factor (TGF)-β signaling [32, 33]. Elevated TGF-β signaling stimulates the transdifferentiation of fibroblasts to myofibroblasts and alters matrix metalloproteinase/tissue inhibitor of metalloproteinase activity to favor collagen deposition and retention [26]. Given that the proliferative macrophage population in TAC is mostly a remodeling phenotype [7] and that fibrosis typifies the TAC model, we hypothesize that TGF-β-induced remodeling through fibroblasts is a likely culprit for progressive remodeling and fibrosis. Future studies of the role of CARD9 in fibrotic remodeling should investigate the effect of targeting CARD9 on the TGF-β-mediated fibrotic pathway.
Finally, CARD9−/− mice had altered myocardial IKK protein expression and phosphorylation, a blunted TAC-associated increase in p65 phosphorylation, and increased expression of markers of myocardial autophagic flux. In healthy hearts, autophagy enables the heart to maintain structure, organization, and function, whereas insufficient myocardial autophagy is disruptive, leading to hypertrophy and contractile dysfunction [34]. We have previously shown that CARD9 KO corrected deficient myocardial autophagy in a high-fat diet model of myocardial dysfunction [16]. Increased autophagy may be adaptive in pressure overload [34]; therefore, we examined the effect of CARD9 KO on markers of autophagy during TAC. After 3 months of TAC, at which point dysfunction and hypertrophy were extensive, markers of autophagic activity were similar between WT-control and -TAC mice. At 3 months, these markers indicated an increase in autophagy in CARD9−/− hearts (Fig. 6), which were not fibrotic or markedly hypertrophied. Unaltered autophagy in the face of such dramatic stress may reflect an inability of the autophagic machinery to adapt to increased autophagic requirements. By clearing damaged proteins and organelles and maintaining structural organization, autophagy could contribute to the beneficial effects of CARD9 KO on myocardial contractility. Mechanistic experiments are needed to clarify if this is the case, however.
We hypothesized that inflammation due to TAC would increase inflammatory cell signaling in the myocardium and that CARD9 KO would normalize these effects. NFκB activation is primarily determined by phosphorylation of the p65 subunit, which translocates to the cell nucleus and mediates inflammatory myocardial responses. We demonstrated here that after 3 months of TAC, IKKα/β phosphorylation as similar to that of the controls in WT and CARD9−/− mice, but CARD9 KO decreased IKKα/β expression, resulting in an increased proportion of phosphorylated IKKα/β in CARD9−/− mice. This finding potentially contradicts our hypothesis regarding recruitment of the NFκB pathway during pressure overload. We examined phosphorylation of the downstream target p65 subunit and determined that TAC increased the proportion of phosphorylated p65 in WT mouse hearts but not CARD9−/− hearts (Fig. 5e) without affecting the overall p65 protein expression (Fig. 5f). The dissimilar pattern of phosphorylation between IKK and p65 may indicate that noncanonical control is exerted over p65 during pressure overload, perhaps as described in an ischemia-reperfusion model [35].
In hypertension, pathological myocardial remodeling can result from an inflammatory positive feedback loop mediated by macrophages, cardiomyocytes, and cardiac fibroblasts. Rather than a single inflammatory element, this process involves general amplification of the inflammatory environment, including numerous chemokines and cytokines. Suitable interventional approaches should broadly reverse the inflammatory profile of HHD. Therefore, targeting upstream progenitors of multiple cytokines should logically be more impactful than targeting a single cytokine or transcription factor. It is worth noting that the TAC model only facilitates the study of the heart’s response to increased afterload. This model does not allow for investigation of the pathological changes in peripheral vascular resistance that precede the development of increased blood pressure in a hypertensive patient. This line of inquiry would need to be addressed separately using a different model. The current investigation shows that mice lacking expression of CARD9, a critical signal transducer in macrophages that is necessary for cytokine production, maintained cardiac function at multiple time points during pressure overload without developing significant cardiac hypertrophy, fibrosis, or cardiomyocyte contractile dysfunction.
References
The World Health Organization. Health statistics and information systenms: disease burden and mortality estimates. Geneva: World Health Organization; 2020.
GBD 2015 Risk Factors Collaborators Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1659–724. https://doi.org/10.1016/S0140-6736(16)31679-8.
World Health Organization. Health statistics and information systems: disease burden and mortality estimates. Geneva: World Health Organization; 2018.
Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–54. https://doi.org/10.1038/sj.jhh.1001785.
Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, et al. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–7.
Parissis JT, Korovesis S, Giazitzoglou E, Kalivas P, Katritsis D. Plasma profiles of peripheral monocyte-related inflammatory markers in patients with arterial hypertension. Correlations with plasma endothelin-1. Int J Cardiol. 2002;83:13–21.
Weisheit C, Zhang Y, Faron A, Kopke O, Weisheit G, Steinstrasser A, et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS One. 2014;9:e112710.
Mewhort HE, Lipon BD, Svystonyuk DA, Teng G, Guzzardi DG, Silva C, et al. Monocytes increase human cardiac myofibroblast-mediated extracellular matrix remodeling through TGF-beta1. Am J Physiol Heart Circ Physiol. 2016;310:H716–24.
Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–81.
Bertin J, Guo Y, Wang L, Srinivasula SM, Jacobson MD, Poyet JL, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J Biol Chem. 2000;275:41082–6.
Roth S, Ruland J. Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation. Trends Immunol. 2013;34:243–50. https://doi.org/10.1016/j.it.2013.02.006.
LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nature Immunology. 2007;8:630–8.
Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62:637–40. https://doi.org/10.1007/s00011-013-0625-0.
Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL, et al. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–31.
Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–407.
Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, et al. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J Mol Cell Cardiol. 2016;92:185–95.
Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8:198–205.
deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010. https://doi.org/10.3791/1729.
Sasson Z, Yock PG, Hatle LK, Alderman EL, Popp RL. Doppler echocardiographic determination of the pressure gradient in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1988;11:752–6. https://doi.org/10.1016/0735-1097(88)90207-0.
Chen Y, Xu C-B. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int J Clnical Exp Med. 2017;10:14904–10.
Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, et al. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci USA. 2012;109:11794–9.
Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor-necrosis-factor-alpha in the adult mammalian heart. Journal of Clinical Investigation. 1993;92:2303–12.
Liu SJ, Zhou WG, Kennedy RH. Suppression of beta-adrenergic responsiveness of L-type Ca2+ current by IL-1 beta in rat ventricular myocytes. Am J Physiol. 1999;276:H141–8.
Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharm Toxicol. 2005;45:657–87. https://doi.org/10.1146/annurev.pharmtox.45.120403.095802.
Kain D, Amit U, Yagil C, Landa N, Naftali-Shani N, Molotski N, et al. Macrophages dictate the progression and manifestation of hypertensive heart disease. Int J Cardiol. 2016;203:381–95.
Kapur NK. Transforming growth factor-beta governing the transition from inflammation to fibrosis in heart failure with preserved left ventricular function. Circ Heart Fail. 2011;4:5–7. https://doi.org/10.1161/circheartfailure.110.960054.
Salles GF, Fiszman R, Cardoso CRL, Muxfeldt ES. Relation of left ventricular hypertrophy with systemic inflammation and endothelial damage in resistant hypertension. Hypertension. 2007;50:723–8. https://doi.org/10.1161/hypertensionaha.107.093120.
Mehta SK, Rame JE, Khera A, Murphy SA, Canham RM, et al. Left ventricular hypertrophy, subclinical atherosclerosis, and inflammation. Hypertension. 2007;49:1385–91.
Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–9.
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl J Med. 1990;322:1561–6. https://doi.org/10.1056/NEJM199005313222203.
Kannel WB. Left ventricular hypertrophy as a risk factor in arterial hypertension. Eur Heart J. 1992;13 Suppl D:82–8.
Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One. 2012;7:e35144.
Sakata Y, Chancey AL, Divakaran VG, Sekiguchi K, Sivasubramanian N, Mann DL. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res Cardiol. 2008;103:60–8.
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24.
Zhang L, Ma Y, Zhang J, Cheng J, Du J. A new cellular signaling mechanism for angiotensin II activation of NF-kappaB: an IkappaB-independent, RSK-mediated phosphorylation of p65. Arterioscler Thromb Vasc Biol. 2005;25:1148–53. https://doi.org/10.1161/01.ATV.0000164624.00099.e7.
Acknowledgements
The project described was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant No. 2P20GM103432 and American Heart Association Grant No. 18TPA34170232 to GH. Prof. Xin Lin (Department of Molecular and Cellular Oncology, University of Texas, Houston, TX) provided the original CARD9−/− breeding mice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peterson, M.R., Getiye, Y., Bosch, L. et al. A potential role of caspase recruitment domain family member 9 (Card9) in transverse aortic constriction-induced cardiac dysfunction, fibrosis, and hypertrophy. Hypertens Res 43, 1375–1384 (2020). https://doi.org/10.1038/s41440-020-0507-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-020-0507-0
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
CARD9 regulates myocardial inflammation, oxidative stress, and vascular dysfunction in hypertensive rats by activating MAPK/p38 pathway in combination with NOD2
Molecular & Cellular Toxicology (2024)
-
Macrophage CARD9 mediates cardiac injury following myocardial infarction through regulation of lipocalin 2 expression
Signal Transduction and Targeted Therapy (2023)
-
Annual reports on hypertension research 2020
Hypertension Research (2022)
-
CARD9: key player or bystander in cardiac remodeling under hypertension?
Hypertension Research (2020)