
Abstract
Dysregulation of Wnt signaling has been implicated in developmental defects and in the pathogenesis of many diseases such as osteoarthritis; however, the underlying mechanisms are poorly understood. Here, we report that non-canonical Wnt signaling induced loss of chondrocyte phenotype through activation of Fz-6/DVL-2/SYND4/CaMKIIα/B-raf/ERK1/2 cascade. We show that in response to Wnt-3a, Frizzled 6 (Fz-6) triggers the docking of CaMKIIα to syndecan 4 (SYND4) and that of B-raf to DVL-2, leading to the phosphorylation of B-raf by CaMKIIα and activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2) signaling, which leads to chondrocyte de-differentiation. We demonstrate that CaMKIIα associates and phosphorylates B-raf in vitro and in vivo. Our study reveals the mechanism by which non-canonical Wnt activates ERK1/2 signaling that induces loss of chondrocyte phenotype, and demonstrates a direct functional relationship between CaMKIIα and B-raf during chondrocyte de-differentiation. The identification of Fz-6, SYND4, and B-raf as novel physiological regulators of chondrocyte phenotype may provide new potential anti-osteoarthritic targets.
Similar content being viewed by others

Introduction
Wnts signal through multiple pathways [1, 2], the best characterized of which is the Wnt/β-catenin signaling, also known as the “canonical” pathway. Canonical Wnt signaling is initiated by binding of Wnt proteins to a Frizzled-LRP5/6 receptor complex [3]. This leads to activation of the protein Disheveled (Dsh/DVL) and inhibition of phosphorylation events that otherwise target β-catenin for proteasomal destruction, resulting in its stabilization in the cytoplasm. The stabilized β-catenin accumulates and translocates to the nucleus, where it interacts with transcription factors of the T-cell-specific transcription factor/lymphoid enhancer-binding factor family to activate the transcription of target genes [4]. In contrast to the above mechanism, Wnts can instead activate other signaling pathways referred to as “non-canonical”, which does not involve stabilization of β-catenin such as the planar cell polarity pathway [5] and the Ca2+/CaMKII pathways [2, 6]. Recently, several studies connect Wnt signaling with bone disease, osteoarthritis (OA), and cancer [7,8,9]; therefore, understanding their complex signaling mechanisms and biological roles is of wide interest.
Besides Wnts, the syndecans (SYNDs) are a family of four (transmembrane) TM proteoglycans, which control cell proliferation, adhesion, and migration [10, 11]. Of mammalian SYND family members, SYND4 acts as co-receptors for growth factors and transduces signals via the intracellular domain, by interacting with numerous effectors [11].
In the cartilage, SYND4 seems to be induced specifically in hypertrophic chondrocytes and its expression is elevated in human OA cartilage [12, 13]. In addition, SYND4-null mice were shown to develop less severe OA-like cartilage destruction [13].
In this study, we report that non-canonical Wnt-3a signaling induces loss of chondrocyte phenotype through activation of Fz-6/DVL-2/SYND4/CaMKIIα/B-raf/ERK1/2 cascade. We show that SYND4 docks CaMKIIα and DVL-2 interacts with B-raf in a Wnt-dependent manner, leading to activation of B-raf by CaMKIIα and to de-differentiation of chondrocytes. To our knowledge, this is the first report describing the partners and the interactions between them and other components of the non-canonical Wnt/ERK1/2 pathway and provides molecular insights for non-canonical Wnt-driven chondrocyte de-differentiation. It also reveals a physiological function of SYND4 and CaMKIIα in this process.
Results
Non-canonical Wnt/ERK1/2 signaling mediates chondrocyte de-differentiation
Wnt-3a is expressed in the cartilage and has an established role in OA [14]. We first investigated the effect of Wnt-3a on the expression of chondrocyte-lineage markers COL2A1, SOX9, and aggrecan in human primary chondrocytes and articular cartilage explants. The expression of COL2A1, SOX9, and aggrecan was decreased by Wnt-3a in cartilage explants (Fig. 1a). We also found that the expression of the hypertrophy marker RUNX2 was increased (Fig. 1a). Similar results were obtained with human primary chondrocytes (data not shown). These results indicate that Wnt-3a activates de-differentiation of articular chondrocytes. Accordingly, treatment of human primary chondrocytes with Wnt-3a produced a shift from round and polygonal cells to fibroblast-like cells (Fig. 1b). We next investigated whether Wnt-3a mediates loss of chondrocyte phenotype through canonical Wnt pathway. Knockdown of β-catenin failed to rescue the Wnt-3a down-regulation of COL2A1 (Fig. 1c), indicating that Wnt-3a regulates COL2A1 through β-catenin-independent pathway and suggests that down-regulation of COL2A1 expression and loss of chondrocyte phenotype by Wnt-3a is mediated through non-canonical Wnt signaling pathway in human primary chondrocytes.

Wnt-3a induces loss of chondrocyte phenotype via a β-catenin-independent pathway. a Wnt-3a down-regulates the expression of COL2A1, aggrecan, and SOX9 and up-regulates that of RUNX2 in human cartilage explants (n = 5), as evaluated by Q-PCR. Values were normalized for the housekeeping gene ribosomal protein S29 and are expressed as relative expression compared with control. b Primary human chondrocyte morphology under invert microscope (×10). With Wnt-3a treatment, round and polygonal chondrocytes were shifted to fibroblast-like cells. c Fold changes of COL2A1 mRNA expression normalized to control in primary chondrocytes transfected with siRNA to β-catenin or siRNA control (Si-control) and treated with either Wnt-3a or vehicle (n = 3). d Detection of phosphorylated and total ERK1/2 in human primary chondrocytes treated with either Wnt-3a or vehicle for different time points. β-Actin was used as a loading control (n = 3). e Detection of phosphorylated and total ERK1/2 in human primary chondrocytes treated with Wnt-3a or vehicle, in the presence or absence of MEK1/2 inhibitor U0126. β-Actin was used as a loading control (n = 3). f Fold changes of COL2A1 mRNA expression normalized to control in primary chondrocytes pre-treated with the MEK1/2 inhibitor U0126 and treated with either Wnt-3a or vehicle (n = 3). g Detection of MAPK1 and MAPK3 in primary chondrocytes transfected with siRNA specific to MAPK1 (Si-MAPK1), MAPK3 (Si-MAPK3), or siRNA control (Si-control). β-Actin was used as a loading control (n = 3). h Fold changes of COL2A1 mRNA expression normalized to control in primary chondrocytes transfected with siRNA specific to MAPK1, MAPK3, or siRNA control and treated with either Wnt-3a or vehicle (n = 4). One representative blot of three independent experiments is shown. Data are expressed as mean ± S.D. of separate experiments. Statistical analysis was performed with an unpaired Student’s t test (*p < 0.05; **p < 0.01)
It has been shown that Wnt-3a also signals through β-catenin-independent mechanisms to mediate distinct physiologically relevant responses [15, 16]. Wnt-3a activates extracellular signal-regulated kinase 1 and 2 (ERK1/2) signaling in primary chondrocytes. As shown in Fig. 1d, the phospho-ERK (p-ERK) level was increased in a time-dependent manner starting within 5 min of the treatment with Wnt-3a and last over 2 h. Co-treatment with the MEK inhibitor, U0126, abrogated the activation of ERK1/2 pathway by Wnt-3a (Fig. 1e). To determine whether non-canonical Wnt/ERK1/2 pathway mediates Wnt-3a-induced down-regulation of COL2A1, we analyzed the effect of inhibition of Wnt-3a-induced activation of ERK1/2 on COL2A1 expression. Interestingly, inhibition of ERK1/2 pathway rescued Wnt-3a-induced down-regulation of COL2A1 (Fig. 1f), thus revealing that Wnt-3a inhibits the expression of COL2A1 through activation of ERK1/2 pathway. Consistent with this finding, knockdown of ERK2 (Fig. 1g) reverted the effect of Wnt-3a on the expression of COL2A1 (Fig. 1h), confirming the key role of ERK1/2 in Wnt-3a-induced down-regulation of COL2A1 and chondrocyte de-differentiation.
DVL-2 docks B-raf to activate Wnt/ERK1/2 signaling
To investigate the molecular mechanisms involved in the activation of ERK1/2 by Wnt-3a, we explored the role of DVLs. DVL is a key component of both canonical and non-canonical Wnt signaling pathways and, depending on the cellular context, specifically activates each Wnt signaling branch [17]. Mammalian DVLs include three isoforms, DVL-1, DVL-2, and DVL-3 [18,19,20]. Analysis of their expression by Western blotting showed that DVL-3 is the most abundant DVL expressed in human primary chondrocytes, whereas DVL-1 is the least abundant (Fig. 2a). In order to determine the role of DVLs in Wnt-3a-induced de-differentiation of chondrocytes, we knocked down each of DVLs individually (Fig. 2b–d) and evaluated the effect on Wnt-3a-induced decrease in the expression of COL2A1 and activation of ERK1/2 signaling. DVL-2 knockdown not only prevented down-regulation of COL2A1 by Wnt-3a but also promoted its expression (Fig. 2e). The knockdown of DVL-1 and DVL-3 did not prevent Wnt-3a-induced down-regulation of COL2A1, instead silencing of DVL-1 potentiates it (Fig. 2e). These results indicate that DVL-2 mediates Wnt-3a down-regulation of COL2A1 and suggests that, in chondrocytes, non-canonical Wnt pathway signal through DVL-2. Next, we asked whether DVL-2 silencing affects activation of ERK1/2 in response to Wnt-3a. Although Wnt-3a induces the activation of ERK1/2 signaling, it failed to stimulate this pathway in chondrocytes made deficient in DVL-2 (Fig. 2f). The knockdown of DVL-2 provoked a decline, rather than increase in ERK1/2 activation in response to Wnt-3a, indicating that DVL-2 is essential for the activation of ERK1/2 signaling by Wnt-3a.

DVL-2 mediates non-canonical Wnt-induced ERK1/2 activation and down-regulation of COL2A1 expression. a Detection of total DVL subtypes. β-actin was used as a loading control (n = 3). b Detection of DVL-1, c DVL-2 and d DVL-3 in human primary chondrocytes with or without transfection with siRNA specific to DVL-1 (Si-DVL-1), DVL-2 (Si-DVL-2), DVL-3 (Si-DVL-3), or siRNA control (Si-control). β-Actin was used as a loading control (n = 3). e Fold changes of COL2A1 expression normalized to control in primary chondrocytes transfected with siRNA specific to DVL-1, DVL-2, DVL-3, or siRNA control and treated with Wnt-3a (n = 3). f Detection of phosphorylated and total ERK1/2 in primary chondrocytes transfected with siRNA specific to DVL-1, DVL-2, DVL-3, or siRNA control and treated or not with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3). Q-PCR values were normalized for the housekeeping gene ribosomal protein S29 and are expressed as the relative expression compared with control. One representative blot of three independent experiments is shown. Data are expressed as mean ± S.D. Statistical analysis was performed with an unpaired Student’s t test (*p < 0.05; **p < 0.01)
DVL-2 is the scaffold protein that relays Wnt signaling by bridging receptor and adaptor proteins [21, 22]; therefore, we performed co-immunoprecipitation assays to analyze whether DVL-2 interacts with B-raf which transduces signals to MEK and ERK1/2 MAPK. Co-immunoprecipitation with anti-B-raf-specific antibodies showed that DVL-2 associates with B-raf and this occurs only in the presence of Wnt-3a (Fig. 3a); therefore, revealing that B-raf interacts with DVL-2 in Wnt-dependent manner. To determine whether recruitment of DVL-2 to the membrane is required for DVL-2 interaction with B-raf, we used a DVL-2 mutant lacking the DEP domain (Flag-DVL-2∆361–736) that is important for membrane recruitment [23]. We stimulated cells containing the Flag-tagged wild-type or ∆361–736 mutant of DVL-2 with Wnt-3a and then immunoprecipitated B-raf with specific antibodies. As shown in Fig. 3b, the Wnt-3a-dependent binding between DVL-2 and B-raf was observed only in the presence of Wnt-3a in cells expressing the wild-type DVL-2, but not in the DVL-2 (∆361–736) mutant. These results indicate that DVL-2 interaction with B-raf requires the presence of the DEP domain and suggest that recruitment of DVL-2 to the membrane is critical for DVL-2 docking of B-raf. Taken together, these data strongly suggest that activation of ERK1/2 pathway by Wnt-3a is regulated through recruitment of DVL-2 to the plasma membrane and docking of B-raf. Next, we analyzed whether B-raf was activated by Wnt-3a and found that phosphorylation of B-raf was markedly increased in a time-dependent manner (Fig. 3c), indicating that Wnt-3a activates B-raf in chondrocytes. To investigate the role of B-raf in Wnt-3a-induced activation of ERK1/2 signaling pathway, we stimulated chondrocytes containing the wild-type or D594V kinase-dead mutant of B-raf with Wnt-3a and then analyzed the phosphorylation of ERK1/2. As shown in Fig. 3d, forced expression of wild-type B-raf increased the phosphorylation of ERK1/2, which was increased further in response to Wnt-3a. In contrast, expression of D594V mutant significantly reduced basal activation of ERK1/2 and prevented the activation of ERK1/2 by Wnt-3a (Fig. 3e), indicating that B-raf mediates Wnt-3a-induced activation of ERK1/2 signaling pathway.

Wnt-3a induces the docking of B-raf to DVL-2 and activates B-raf to transduce signal to ERK1/2. a Detection of DVL-2 and B-raf in cell lysates of primary chondrocytes treated with Wnt-3a or vehicle after immunoprecipitation with anti-B-raf antibodies. Total cell lysates were analyzed by immunoblot with anti-DVL-2 and anti-B-raf antibodies. β-Actin was used as a loading control (n = 3). b Detection of Flag-tagged DVL-2 and B-raf proteins in cell lysates of primary chondrocytes, transfected with expression vector for Flag-tagged wild-type DVL-2 or Flag-DVL-2(∆361–736) and treated with Wnt-3a or vehicle, after immunoprecipitation with anti-B-raf antibodies. Total cell lysates were analyzed by immunoblot with anti-Flag and anti-B-raf antibodies. β-Actin was used as a loading control (n = 3). c Detection of phosphorylated and total B-raf in primary chondrocytes treated with Wnt-3a or vehicle for different time points. β-Actin was used as a loading control (n = 3). d Detection of phosphorylated and total ERK1/2 and B-raf in primary chondrocytes transfected with either empty vector or wild-type B-raf-expressing vector and treated with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3). e Detection of phosphorylated and total ERK1/2 and B-raf in primary chondrocytes transfected with either empty vector or with the vector expressing the dead mutant of B-raf (D594V) and treated with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3). One representative blot of three independent experiments is shown
Ca2+/CaMKII mediates non-canonical Wnt activation of ERK1/2
To determine whether Wnt-3a activates the non-canonical Ca2+/CaMKII-dependent pathway in chondrocytes, we analyzed the phosphorylation status of CaMKIIα and found that treatment with Wnt-3a induced the phosphorylation of CaMKIIα (p-CaMKIIα) (Fig. 4a). To explore whether calcium mobilization is involved in the activation of ERK1/2 pathway by Wnt-3a, intracellular Ca2+ accumulation was monitored in primary chondrocytes treated or not with Wnt-3a. The results showed that Wnt-3a induced Ca2+ mobilization which was efficiently blocked by pertussis toxin (PTX), a G-protein inhibitor (Fig. 4b). Western blot analysis of the phosphorylation status of CaMKIIα and ERK1/2 showed that PTX markedly reduced Wnt-3a-induced activation of both CaMKIIα and ERK1/2 (Fig. 4c). In line with this, PTX prevented down-regulation of COL2A1 by Wnt-3a (Fig. 4d). These results suggest that Wnt-3a induces loss of chondrocyte phenotype by activation of Ca2+/CaMKIIα/B-raf/ERK1/2 signaling pathway in a G-protein-dependent manner.

Wnt-3a activates CaMKIIα and ERK1/2, and down-regulates COL2A1 in a G-protein-dependent manner. a Detection of phosphorylated and total CaMKIIα in primary chondrocytes treated with Wnt-3a or vehicle for different time points. β-Actin was used as a loading control (n = 3). b Calcium mobilization in primary chondrocytes pre-treated by the G-protein inhibitor PTX or vehicle and then with Wnt-3a (n = 3). (c) Detection of phosphorylated CaMKIIα (p-CaMKIIα) and phosphorylated ERK1/2 (p-ERK1/2) and total CaMKIIα and ERK1/2 in primary chondrocytes pre-treated with PTX or vehicle and treated with Wnt-3a. β-Actin was used as a loading control (n = 3). d Fold changes of COL2A1 expression normalized to control in primary chondrocytes pre-treated with PTX or vehicle and treated with Wnt-3a (n = 3). CaMKIIα induced the activation of B-raf/ERK1/2 signaling pathway. e Detection of p-CaMKIIα, phosphorylated B-raf (p-B-raf) and p-ERK1/2 and of total CaMKIIα, B-raf, and ERK1/2 in primary chondrocytes pre-treated with KN93, KN92, or vehicle and treated with Wnt-3a. β-Actin was used as a loading control (n = 3). f Fold changes of COL2A1 expression normalized to control in primary chondrocytes pre-treated with KN93, KN92, or vehicle and treated with Wnt-3a (n = 3). Data are representative of at least three experiments. Q-PCR values were normalized for the housekeeping gene ribosomal protein S29 and are expressed as relative expression compared with control. Data are expressed as mean ± S.D. Statistical analysis was performed with an unpaired Student’s t test (*p < 0.05; **p < 0.01)
Next, we examined the role of CaMKIIα in Wnt-3a-induced activation of B-raf. To this end, chondrocytes were pre-treated or not with CaMKII inhibitor KN93 or with KN92, an analog of KN93, which does not inhibit CaMKII, and then stimulated with Wnt-3a. Analysis of cell extracts showed that not only KN93 prevented Wnt-3a-induced activation of CaMKIIα but also prevented the activation of B-raf and almost completely abrogated the activation of ERK1/2 signaling by Wnt-3a (Fig. 4e). In contrast, KN92 did not prevent the activation by Wnt-3a of either CaMKIIα, B-raf, or ERK1/2 (Fig. 4e). These results indicate that activation of B-raf and ERK1/2 signaling is CaMKIIα-dependent, and consequently de-differentiation of chondrocytes induced by Wnt-3a is mediated by CaMKIIα. In line with this, inhibition of CaMKIIα by ΚΝ93 prevented down-regulation of COL2A1 by Wnt-3a, whereas treatment with KN92 did not (Fig. 4f).
SYND4 docks CaMKIIα and interacts with DVL-2 to induce Wnt/ERK1/2 signaling and chondrocyte de-differentiation
To determine the role of SYND4 in non-canonical Wnt pathway in primary chondrocytes, we performed loss of function (Fig. 5a) and examined whether knockdown of SYND4 affects the activation of ERK1/2 signaling by Wnt-3a. As shown in Fig. 5b, knockdown of SYND4 prevents the activation of B-raf and ERK1/2 by Wnt-3a, but did not significantly affect the phosphorylation of CaMKIIα in response to Wnt-3a. These results suggest that activation of B-raf and ERK1/2 by Wnt-3a is SYND4-dependent, whereas activation of CaMKIIα is not. Interestingly, knockdown of DVL-2 strongly reduced the activation of both B-raf/ERK1/2 and CaMKIIα by Wnt-3a (Fig. 5b) arguing for a dependence of CaMKIIα activation by Wnt-3a on DVL-2 but not on SYND4.

Syndecan 4 regulates Wnt-3a-induced activation of ERK1/2 signaling and interacts with CaMKIIα and DVL-2. a Fold changes in the mRNA expression of SYND4 in primary chondrocytes transfected with siRNA to SYND4 (Si-SYND4) or siRNA control (Si-control) evaluated by Q-PCR (n = 4). b Detection of phosphorylated B-raf (p-B-raf), phosphorylated CaMKIIα (p-CaMKIIα), and phosphorylated ERK1/2 (p-ERK1/2), and of total B-raf, CaMKIIα, and ERK1/2 in cell lysates of primary chondrocytes transfected with siRNA specific to SYND4 (Si-SYND4), DVL-2 (Si-DVL-2), or siRNA control (Si-control). β-Actin was used as a loading control (n = 3). c Detection of p-CaMKIIα and HA-SYND4 in cell lysates of primary chondrocytes, transfected with either empty vector (Native) or HA-SYND4-expressing vector and treated with Wnt-3a or vehicle, after immunoprecipitation with anti-HA antibodies. Total cell lysates were analyzed by immunoblot with anti-p-CaMKIIα and anti-HA antibodies. β-Actin was used as a loading control (n = 3). d Detection of HA-SYND4 and Flag-DVL-2 in cell lysates of primary chondrocytes, transfected with empty vector (Native) or HA-SYND4-expressing or Flag-DVL-2-expressing vectors, after immunoprecipitation with anti-Flag antibodies. Total cell lysates were analyzed by immunoblot with anti-HA and anti-Flag antibodies. β-Actin was used as a loading control (n = 3). e Detection of p-B-raf, p-ERK1/2, and p-CaMKIIα, and of total B-raf, ERK1/2, and CaMKIIα in cell lysates of primary chondrocytes transfected either with empty vector (Native) or HA-SYND4∆CT vector expressing SYND4 deleted from the CT domain and treated with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3)
Given that activation of B-raf and ERK1/2 by Wnt-3a is both SYND4- and CaMKIIα-dependent, we hypothesized that SYND4 may relay Wnt signaling to B-raf and ERK1/2 through interaction with CaMKIIα. Co-immunoprecipitation assays were performed using extracts from cells that have been transiently transfected with HA-SYND4. The results clearly show that HA-SYND4 physically interacts with p-CaMKIIα (Fig. 5c). Interestingly, the interaction between SYND4 and CaMKIIα was significantly enhanced in the presence of Wnt-3a (Fig. 5c), suggesting that SYND4 interacts with CaMKIIα in a Wnt-3a-dependent manner. On the other hand, a potential interaction between SYND4 and DVL-2 was examined using extracts from chondrocytes that have been transfected with Flag-DVL-2 and HA-SYND4. Interestingly, DVL-2 was co-immunoprecipitated with SYND4 (Fig. 5d), suggesting that SYND4 interacts with DVL-2. SYND4 contains a transmembrane (TM) domain and a cytoplasmic (CT) domain and it is known that SYND4 normally transduces signal through its CT domain [10, 24], we sought to determine whether deletion of the CT domain of SYND4 (SYND4∆CT) affects the Wnt-3a-mediated activation of B-raf/ERK1/2 and CaMKIIα. As shown in Fig. 5e, Wnt-3a failed to activate B-raf and ERK1/2 signaling in chondrocytes expressing SYND4∆CT, whereas activation of CaMKIIα is not affected. Taken together, these findings suggest that the CT domain of SYND4 is required for Wnt-3a-induced activation of B-raf and ERK1/2 through interaction of its CT domain with CaMKIIα.
CaMKIIα phosphorylates B-raf
We showed above that phosphorylation of B-raf by Wnt-3a depends on CaMKIIα. Might B-raf be substrate for CaMKIIα? To investigate this hypothesis, we examined whether CaMKIIα is able to physically interact with B-raf by performing co-immunoprecipitation experiments. We stimulated chondrocyte cells with Wnt-3a and then performed immunoprecipitation with anti-B-raf-specific antibodies. As shown in Fig. 6a, p-CaMKIIα interacts with B-raf and this interaction was markedly increased by Wnt-3a. To further confirm this interaction, we conducted co-immunoprecipitation experiments in human embryonic kidney 293 (HEK293) cells that express Flag-B-raf or transfected with empty vector and subjected both to immunoprecipitation with anti-Flag antibodies. After Western blot analysis for CaMKIIα on these immunoprecipitates, we found that endogenous CaMKIIα associates with the anti-B-raf immunoprecipitation complex from cells transfected with Flag-B-raf-expressing vector (Fig. 6b), indicating that CaMKIIα associates with B-raf.

CaMKIIα associates and phosphorylates B-raf at Ser445. a Detection of p-CaMKIIα and B-raf in cell lysates of primary chondrocytes treated with Wnt-3a or vehicle after immunoprecipitation with anti-B-raf antibodies. Total cell lysates were analyzed by immunoblot with anti-p-CaMKIIα and anti-B-raf antibodies. β-Actin was used as a loading control (n = 3). b Detection of p-CaMKIIα and B-raf in the lysate of HEK293 cells transfected with empty vector (Native) or Flag-B-raf-expressing vector after immunoprecipitation with Flag antibodies. Total cell lysates were analyzed by immunoblot with anti-Flag and anti-p-CaMKIIα antibodies. β-Actin was used as a loading control (n = 3). c Sequence of B-raf peptides (438–453) containing the CaMKIIα recognition sequence RXXS. The CaMKIIα phosphorylation site is shown in red. d Brilliant Blue protein staining of purified GST fusion wild-type and B-raf mutants expressed in E. coli. Arrow indicates non-specific protein. e Detection of GST fusion wild-type and B-raf mutant proteins purified from recombinant bacterial extracts using anti-B-raf antibodies. f Autoradiography analysis of GST fusion proteins that were incubated with activated CaMKIIα in the presence of [γ-32P]ATP (32P). g Immunoblot analysis with phospho-B-raf (S445) antibodies of GST wild-type and B-raf mutants that were phosphorylated in vitro (p-B-raf) with activated CaMKIIα, unactivated CaMKIIα was used as control. Total B-raf was detected using anti-B-raf antibodies. h Detection of phospho-B-raf and total B-raf and CaMKIIα in primary chondrocytes transfected with empty vector (Native), B-raf-expressing vector, or co-transfected with B-raf and CaMKIIα (T286D) or with B-raf S445-expressing and CaMKIIα-expressing (T286D) vectors. Immunoblots were probed with anti-phospho-B-raf (S445), B-raf, or CaMKIIα. β-Actin was used as a loading control (n = 3)
Noteworthy, the Ser445 residue of B-raf could be a potential phosphorylation site for CaMKII based on the consensus substrate recognition motif Hyd-X-R-NB-X-S/T-Hyd for CaMKII [25] (Fig. 6c). An in vitro kinase assay with a glutathione S-transferase (GST) fusion protein containing B-raf kinase domain (amino acids 417–766) expressed in E. coli and purified by immunoaffinity (Fig. 6d–e) together with CaMKIIα and [γ-32P]ATP revealed that B-raf was robustly phosphorylated by CaMKIIα, as assessed by radiography (Fig. 6f). To determine whether the Ser445 was the phosphorylation site, we generated point mutation of Ser445 and discovered that mutating Ser445 to alanine (S445A) markedly reduced phosphorylation by CaMKIIα (Fig. 6f). Of note, mutation of both the Ser445 and Ser446 residues to alanine (S445/446A) reduces further the phosphorylation of B-raf (Fig. 6f). To further confirm these results, we tested whether phospho-specific antibodies to Ser445 are able to bind to the activated B-raf. A kinase assay with CaMKIIα, ATP, and B-raf or B-raf (S445A) followed by Western blot revealed that the antibodies bind only to activated B-raf but not to B-raf mutants S445A and S445/446A or unactivated B-raf (Fig. 6g). To test whether phosphorylation of B-raf is modulated by CaMKII in intact cells, we transfected wild-type B-raf or B-raf S445A in HEK293 cells, and native vector was used as the control. Immunoblots of cell lysates probed with phospho-specific antibodies to S445 indicated that B-raf was phosphorylated at Ser445 under basal conditions (Fig. 6h). However, after co-transfection with constitutively active form of CaMKIIα (T286D), the basal-level phosphorylation of B-raf was significantly increased (Fig. 6h), whereas no signal was observable with phospho-deficient mutant (Fig. 6h). Taken together, these results suggest that B-raf is phosphorylated at Ser445 residue by CaMKIIα.
Frizzled 6-mediated non-canonical Wnt-3a/ERK1/2 signaling and de-differentiation of chondrocytes
Frizzled (Fz) is a family of G-protein-coupled receptor (GPCR) proteins that serves as receptors in the Wnt signaling pathway. To address the question of which of the known FZD genes could be involved in Wnt-3a activation of Wnt non-canonical pathway, we first performed quantitative real-time PCR (qRT-PCR) to monitor the expression pattern of all genes encoding Wnt receptors in human primary chondrocytes. The result obtained showed that Fz-6 and Fz-8 are predominantly expressed in chondrocytes compared to other Fz receptors (Fig. 7a). Given that non-canonical Wnt-3a is characterized by the activation of CaMKIIα and ERK1/2 and the inhibition of COL2A1 in the human chondrocytes, we evaluated the impact of Fz-6 deficiency on this signaling pathway. The knockdown of Fz-6 efficiently decreased its expression (Fig. 7b) and did not impair Wnt-3a-induced up-regulation of the canonical Wnt/β-catenin downstream target gene AXIN2 (Fig. 7c). However, knockdown of Fz-6 abrogated Wnt-3a-induced down-regulation of the non-canonical Wnt downstream target gene COL2A1 (Fig. 7d). These data suggest that Fz-6 is not required for the activation of Wnt-3a canonical signaling, but essential for the activation of non-canonical signaling in chondrocytes. To further confirm these results, we tested whether the knockdown of Fz-6 affects Wnt-3a-induced stabilization of β-catenin and found that it has no significant effect (Fig. 7e). Next, we examined whether silencing of Fz-6 effects the activation of non-canonical ERK1/2 signaling. Indeed, knockdown of Fz-6 prevents the activation of ERK1/2 signaling by Wnt-3a and abrogates the phosphorylation of CaMKIIα (Fig. 7f). These results indicate that activation of CaMKIIα and ERK1/2 signaling is no more modulated by Wnt-3a in chondrocytes that are Fz-6-deficient. Taken together, these observations support the conclusion that Fz-6 mediates non-canonical Wnt-3a-induced de-differentiation of chondrocytes through activation of CaMKIIα/B-raf/ERK1/2 cascade.

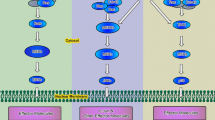
Frizzled 6 mediates non-canonical Wnt-3a-induced loss of chondrocyte phenotype. a Expression of different Fz receptors was evaluated by absolute Q-PCR. Specific DNA segment for each of the 10 Fz receptors (standards) was amplified by RT-PCR, purified on agarose gel, quantified, and used as template for Q-PCR. Absolute quantification was determined compared to standards that are run in parallel. Data are expressed as mean ± S.D. of three separate experiments. b Fold changes in the expression of Fz-6 in primary chondrocytes transfected with siRNA specific to Fz-6 (Si-Fz-6) or siRNA control (Si-control) (n = 5). c Fold changes in the expression of the Wnt canonical target gene AXIN2 (d) and of the chondrocyte phenotypic marker COL2A1 in primary chondrocytes that were transfected with siRNA specific to Fz-6 (Si-Fz-6) or siRNA control (Si-control) and treated or not with Wnt-3a (n = 3). e Western blotting for β-catenin in chondrocytes transfected with Si-Fz-6 or Si-control and treated with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3). f Detection of phosphorylated ERK1/2 (p-ERK1/2) and phosphorylated CaMKIIα (p-CaMKIIα), and total ERK1/2 and CaMKIIα in primary chondrocytes transfected with siRNA control (Si-control) or siRNA to Fz-6 (Si-Fz-6) and treated with Wnt-3a or vehicle. β-Actin was used as a loading control (n = 3). g A working model for signaling regulation by non-canonical Wnt in chondrocytes. See text for details. Q-PCR values were normalized for the housekeeping gene ribosomal protein S29 and are expressed as relative expression compared with control. One representative blot of three independent experiments is shown. Data are expressed as mean ± S.D. Statistical analysis was performed with an unpaired Student’s t test (NS, statistically not significant; *p < 0.05; **p < 0.01)
Discussion
Loss of phenotypic stability of articular chondrocytes has been proposed as an early event in the onset of OA disease [26]. De-differentiation of chondrocytes occurs through a programme with similarities to terminal differentiation of chondrocytes (hypertrophy), as can be found in the growth plate [27]. This triggers a disturbed cartilage homeostasis favoring degenerative changes [27, 28]. Wnt signaling has a pivotal role in regulating chondrocyte differentiation during endochondral ossification and was also shown to play a regulative role in OA cartilage [7, 29], leading to chondrocyte hypertrophic differentiation. Here, we demonstrated that Wnt-3a-induced loss of chondrocyte phenotype is mediated by Fz-6 through Ca2+/CaMKIIα-dependent activation of B-raf/ERK1/2 signaling pathway (Fig. 7g). Indeed, knockdown of ERK1/2 or chemical inhibition of its activation prevented Wnt-3a-induced hypertrophic de-differentiation of chondrocytes, which is associated with down-regulation of COL2A1 [30, 31], indicating that activation of ERK1/2 is a key event in Wnt-3a-induced loss of chondrocyte phenotype.
An important issue we addressed here is the mechanism of activation of ERK1/2 pathway by non-canonical Wnt signaling, which promotes chondrocyte de-differentiation. It has been shown that SYND4 is highly expressed in hypertrophic chondrocytes during embryonic development and during OA, suggesting a role in chondrocyte de-differentiation. Interestingly, loss of SYND4 in mice protects from development of OA-like disease [13]. Accordingly, we showed that knockdown of SYND4 reduced basal activation of B-raf/ERK1/2 signaling and impaired its activation by Wnt-3a in human primary chondrocytes. SYND4 has been shown to interact with different partners through its CT domain [11]. Interestingly, co-immunoprecipitation studies revealed, for the first time, that SYND4 interacts with p-CaMKIIα in a Wnt-dependent manner. On the other hand, co-immunoprecipitation studies revealed that DVL-2 docks B-raf in Wnt-dependent manner; therefore, in the presence of Wnt ligand, interaction between DVL-2 and SYND4 may promote assembly of CaMKIIα and B-raf complex necessary to relay Wnt-3a activation of ERK1/2 signaling pathway. In this case, exploring whether B-raf can directly be activated by CaMKIIα and transduces signals to MEK and ERK1/2 MAPK is an important issue. Our findings show that CaMKIIα phosphorylates B-raf in vitro, in heterologous cells and in primary chondrocytes. Moreover, overexpression of constitutively active CaMKIIα in heterologous cells results in a substantial increase in B-raf phosphorylation. Therefore, revealing for the first time that CaMKIIα phosphorylates B-raf.
Wnt-3a is known to activate both canonical and non-canonical Wnt signaling. One of the key questions is whether individual Fz receptors can activate both pathways, or if the simultaneous activation of canonical and CaMKIIα/ERK1/2 pathways is mediated through Fz receptors with different signaling specificity. We found that Fz-6 and Fz-8 are the most expressed Fz receptors in human chondrocytes. It has been shown that Fz-6 did not activate the canonical Wnt pathway following exposure to various Wnts, including Wnt-3a in HEK293 cells [32]. We have examined the role of the Wnt signaling receptor Fz-6 and demonstrated that Fz-6 does not affect the activation of β-catenin pathway as monitored by stabilization of β-catenin and expression of the target gene AXIN2. In contrast, Fz-6 is critical for the activation of non-canonical pathway as demonstrated by the phosphorylation status of CaMKIIα and ERK1/2, and down-regulation of COL2A1 following knockdown of Fz-6.
Fz proteins possess seven TM domains and their signaling depend on heterotrimeric G proteins. Fzs are predicted to couple to members of the Gαi/o, Gαq and Gαs families of G proteins, with Fz-6 predicted to interact solely with members of Gαi/o family [33]. In mammalian cells, Wnt-3a elicited Fz-dependent guanine-nucleotide exchange on Go/i proteins, which is sensitive to PTX [34]. We showed that inactivation of members of the Gαi/o family with PTX, which decouples the Go/i proteins from their receptors, markedly reduced Wnt-3a-induced activation of CaMKIIα and ERK1/2 signaling and prevented down-regulation of COL2A1 by Wnt-3a, indicating that Fz-6 acts as Go/i-coupled GPCRs as suggested by Koval and Katanaev [34]. More recently, it has been shown that Fz-6 precouples to Gαi1 and Gαq but not GαoA, Gα12, and GαS [35]. Gαi/o-coupled GPCRs typically reduce cAMP levels through Gαi/o subunits and induce changes in intracellular calcium [36]. We found that increasing the level of cAMP by forskolin did not prevent the inhibition of COL2A1 expression by Wnt-3a (data not shown), suggesting that cAMP pathway is not the main mechanism involved in Wnt-3a-induced down-regulation of COL2A1, although we cannot exclude a role by cAMP in this process.
Healthy cartilage is extremely difficult to obtain for ethical reason. In this study, we have used cartilage samples that are histological and macroscopically normal; however, this has a limitation as we cannot exclude that they were subjected to paracrine effects from neighboring disease compartment.
In summary, our study introduces a compelling new mechanism of Wnt-induced activation of ERK1/2 signaling and chondrocyte de-differentiation, which is essential in both normal development and OA pathogenesis. As de-differentiation of chondrocyte is an important issue in OA disease, our study provides new potential targets such Fz-6, B-raf, and CaMKIIα for the development of anti-OA drugs.
Materials and methods
Chondrocytes and cartilage explants isolation and culture
Human primary chondrocytes were isolated from cartilage obtained from patients who underwent joint replacement for knee OA. Cartilage tissue was dissected from preserved areas of the femoral condyles and patellar groove avoiding subchondral bone and fibrocartilage. Cartilage was washed twice with Dulbecco's Modified Eagle's Medium/Nutrient Mixture F12 (DMEM F12) complete medium (2 mM glutamine, 100 μg/ml streptomycin, 100 IU/ml penicillin, and 10% (v/v) fetal bovine serum) and the aliquot was then digested with pronase (1 mg/ml) for 30 min at 37 °C followed by an overnight digestion in collagenase B (1 mg/ml) in a similar medium as above. The chondrocytes recovered from the digestion were seeded at a density of 25,000 cells/cm2 in DMEM F12 complete medium at 37 °C in a humidified atmosphere supplemented with 5% CO2. All experiments were performed using confluent P0 cells.
For cartilage explant isolation, cartilage specimens were dissected within 1 h of operation, and full-thickness cartilage was removed from preserved areas of the femoral condyles using a 6 mm biopsy punch. Extreme care was taken to avoid subchondral bone, osteophytic cartilage, and fibrocartilage. Explants were washed with DMEM F12 complete medium and cultured in 24-well plates in the same medium and condition as chondrocytes for 48 h before use.
Aliquot of cartilage was used for histological scoring for features of OA as described previously [37]. Only cells and explants obtained from samples with a Mankin score <4 were used for subsequent experiments. This study was approved by our local research institution (Commission de la Recherche Clinique; registration no. UF 9757, CPRC 2004, Cellules souches et chondrogénèse). The protocol conforms to the ethical guidelines of the Declaration of Helsinki, and written informed consent has been obtained from each patient.
Cell and cartilage explant treatments
Human primary chondrocytes were seeded onto six-well plates at 5 × 105 cells/well and allowed to attach overnight in standard culture conditions. The cells were then treated for 12 h with 100 ng/ml of recombinant Wnt-3a (R&D Systems, Minneapolis, MN, USA) or vehicle (0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)) in DMEM F12 medium (2 mM glutamine, 100 μg/ml streptomycin, 100 IU/ml penicillin, and 2% (v/v) fetal bovine serum) prior to gene or protein expression analyses. When co-treatments with inhibitors and Wnt-3a are indicated, cells were pre-treated for 30 min with the inhibitor, that is, U0126 (10 µM; Cell Signaling, Danvers, CO, USA), DKK1 (100 ng/ml; R&D Systems), and KN93 and KN92 (10 µM; R&D Systems) prior to treatment with Wnt-3a or vehicle.
Gene expression analysis
Total RNA from chondrocytes and cartilage explants was extracted using TRIzol (Lifetech, Carlsbad, CA, USA) and purified with RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The reverse transcription was performed using 500 ng (primary chondrocytes) or 200 ng (cartilage explants) of total RNA from each sample with iScript Ready to use cDNA supermix (Bio-Rad, Hercules, CA, USA). Quantitative PCR was performed with iTaq™ Universal SYBER Green Supermix kit (Bio-Rad, Hercules, CA, USA) using StepOnePlus™ Real-Time PCR Systems (Applied Biosystems). Primer sequences are listed in Table S1.
Plasmids and siRNA
B-raf vector was a gift from Dustin Maly (University of Washington, Seattle, WA, USA), Flag-DVL-2 and Flag-DVL-2(∆361–736) vectors were a gift from Jeff Wrana (University of Toronto, Toronto, ON, Canada), and TOP-FLASH-firefly reporter plasmid was a gift from Randall Moon (University of Washington). B-raf V600E and B-raf D594V mutants were generated by site-directed mutagenesis with B-raf as template using QuikChange Site-Directed Mutagenesis kit (Agilent, Santa Clara, CA, USA). HA-SYND4, HA-SYND4∆CT, and Flag-B-raf were generated by PCR and cloned into EcoRI and BamHI or SmaI and PstI sites of pCMV empty vector (Stratagene, Valencia, CA, USA). GST C-tail constructs were amplified with synthetic primers containing NcoI and NotI flanking regions from B-raf and mutant plasmids and subcloned into the GST fusion vector pET41 (Novagene) to generate pET-B-raf∆NT/GST, pET-B-raf∆NT-S465A, and pET-B-raf∆NT-S465/466A bacterial vectors expressing the wild-type and mutants of the B-raf kinase domain (amino acids 417–766) in fusion with GST. All the vectors were verified by sequencing. Small interfering RNA (siRNA) targeting SYND4, MAPK1, MAPK3, DVL-1, DVL-2, DVL-3, CTNNB1, Fz-6, and control siRNA were purchased from Qiagen and are listed in Table S2.
Transfection
Subconfluent human primary chondrocytes in 6-well plates were transfected in triplicate with 2 µg of plasmid using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions. For siRNA transfection, cells were transfected with 25 nM siRNAs using DharmaFECT transfection reagent (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Twenty-four hours after transfection, the medium was replaced, and at 48 h after transfection, cells were stimulated for 12 h with recombinant Wnt-3a (100 ng/ml; R&D Systems) or vehicle at 37°C with 5% CO2.
Western blotting
Total protein from chondrocytes was extracted using RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1% deoxycholate, 0.1% SDS, 1% Triton X-100) supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN, USA). Cell lysates were sonicated on ice and protein concentration of the samples was determined by the Bradford method. Proteins (50 μg/lane) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, transferred to a polyvinylidene fluoride membrane (Millipore, Eschborn, Germany), and subsequently blocked in PBS-Tween-20 containing 5% non-fat milk or 5% BSA. Membranes were then incubated overnight with primary antibodies directed against p44/42 MAPK, phospho-p44/42, β-catenin, DVL-2, DVL-3, CaMKIIα, phospho-B-raf (S445) (diluted 1:1000; Cell Signaling), DVL-1, phospho-CaMKIIα, B-raf (diluted 1:200; Texas, Santa Cruz, TX, USA), Flag (diluted 1:1000; Sigma, Saint Louis, MO, USA), or HA (diluted 1:1000; BioLegend, San Diego, CA, USA), followed by incubation with horseradish peroxidase-conjugated secondary antibodies (diluted 1:2000; Cell Signaling). Antibodies were diluted in 5% BSA/0.01% Tween-20 in PBS. The blots were then developed using Clarity Western ECL substrate (Bio-Rad) according to the manufacturer's instruction.
Immunoprecipitation
For immunoprecipitation, subconfluent primary chondrocytes in 10-cm plates were treated with Wnt-3a (100 ng/ml) or vehicle for 1 h and were lysed on ice with TNE buffer (50 mM Tris, pH 8, 1% IGEPAL® CA-630, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA)) supplemented with protease and phosphatase inhibitors (Roche Diagnostics). The lysates were sonicated and centrifuged and the resultant supernatants were used to recover B-raf, and Flag-tagged and HA-tagged proteins by incubating overnight at 4 °C with anti-B-raf, anti-Flag, and anti-HA antibodies, respectively, followed by incubation with protein A beads (Millipore, Eschborn, Germany) for 3 h at room temperature. After washing with lysis buffer, bound proteins were eluted with the loading buffer and analyzed by immunoblotting. Immunoblotting was performed as described previously with antibodies against Flag, HA, B-raf, DVL-2, and p-CaMKIIα.
Intracellular calcium ion changes
Calcium mobilization induced by Wnt-3a was performed with human primary chondrocytes using the Fluo-4 NW Calcium Assay kit (Molecular Probes/Invitrogen, Carlsbad, CA, USA). Primary chondrocytes were grown on 24-well plates in the presence or absence of PTX (Sigma-Aldrich) overnight. The calcium-sensitive dye, Fluo-4 AM (2 μM), was added to the cultured chondrocytes and allowed to incorporate for 1 h (30 min at room temperature and 30 min at 37 °C). After 5 min of treatment with either vehicle (control) or with Wnt-3a, the fluorescence was measured with a Wallac/Perkin-Elmer Victor V2 fluorescent plate reader. Data were subtracted from background and presented as the mean of three separate experiments.
GST fusion protein production and in vitro kinase assay
GST fusion proteins were purified from BL21 bacterial cells transformed with pET-B-raf∆NT/GST, pET-B-raf∆NT-S465A, or pET-B-raf∆NT-S465/466A using GE Healthcare protocol. Fifty milliliters of cultures was inoculated with 0.5 ml of overnight cultures and grown at 37 °C until an absorbance at 600 nm (A600) of 0.6–0.8. Then, an osmotic stress and heat shock were induced by the addition of 0.5 M NaCl and 2 mM betaine, and incubated at 47 °C for 1 h. To induce protein expression, 1 mM isopropyl β-d-1-thiogalactopyranoside was added to the cultures, and they were grown at 20 °C overnight. The cells were then lysed with a sonicator in a Tris-buffered saline containing protease inhibitors (Roche), 1 mM EDTA, and 5% (v/v) glycerol. The sonicated lysate was incubated with a 5:1 ratio of glutathione-sepharose 4B (GE Healthcare) overnight at 4 °C and washed extensively with PBS. For CaMKII in vitro phosphorylation, B-raf fusion proteins were phosphorylated in 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 0.2 mM dithiothreitol, 0.1 mM EDTA, 1.2 µM calmodulin, 2 mM CaCl2, 200 µM ATP, and 1 pmol of [γ-32P]ATP (3000 Ci/mmol) with 200 U of recombinant activated CaMKIIα (Νew England Biolabs). The in vitro kinase assays were performed at 30 °C for 30 min. The reactions were terminated with the addition of SDS-PAGE sample buffer and incubation at 65 °C for 5 min. The proteins were resolved by SDS-PAGE and visualized by autoradiography. For in vitro kinase assays analyzed by immunoreactivity, SDS-PAGE separated proteins were transferred to polyvinylidene difluoride membranes and assayed for phosphorylation (or total protein) by immunoblotting using anti-phospho-B-raf or anti-B-raf antibodies.
Data analysis and statistical procedures
Each experiment was repeated at least three times independently. Quantitative data were expressed as mean ± S.D. Statistical analysis was performed with an unpaired two-tailed Student’s t test, and effects were considered statistically significant at *p < 0.05. One representative immunoblot of three independent experiments was shown in results.
References
MacDonald BT, Semenov MV, He X. SnapShot: Wnt/beta-catenin signaling. Cell. 2007;131:1204–1204.
Semenov MV, Habas R, MacDonald BT, He X. SnapShot: non-canonical Wnt signaling pathways. Cell. 2007;131:1378.
MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26.
Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127:469–80.
Gao B. Wnt regulation of planar cell polarity (PCP). Curr Top Dev Biol. 2012;101:263–95.
Kühl M, Sheldahl LC, Malbon CC, Moon RT. Ca2+/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem. 2000;275:12701–11.
Nalesso G, Sherwood J, Bertrand J, Pap T, Ramachandran M, De Bari C, et al. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and non-canonical pathways. J Cell Biol. 2011;193:551–64.
Vermeulen L, Felipe De Sousa EM, Van Der Heijden M, Cameron K, De Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76.
Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ. Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492:1–18.
Leonova E, Galzitskaya O. Structure and functions of syndecans in vertebrates. Biochemistry (Mosc). 2013;78:1071–85.
Elfenbein A, Simons M. Syndecan-4 signaling at a glance. J Cell Sci. 2013;126:3799–804.
Barre PE, Redini F, Boumediene K, Vielpeau C, Pujol JP. Semiquantitative reverse transcription-polymerase chain reaction analysis of syndecan-1 and-4 messages in cartilage and cultured chondrocytes from osteoarthritic joints. Osteoarthr Cartil. 2000;8:34–43.
Echtermeyer F, Bertrand J, Dreier R, Meinecke I, Neugebauer K, Fuerst M, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med. 2009;15:1072–6.
Nakamura Y, Nawata M, Wakitani S. Expression profiles and functional analyses of Wnt-related genes in human joint disorders. Am J Pathol. 2005;167:97–105.
Yun MS, Kim S-E, Jeon SH, Lee JS, Choi K-Y. Both ERK and Wnt/β-catenin pathways are involved in Wnt3a-induced proliferation. J Cell Sci. 2005;118:313–22.
Halleskog C, Schulte G. Pertussis toxin-sensitive heterotrimeric G αi/o proteins mediate Wnt/β-catenin and WNT/ERK1/2 signaling in mouse primary microglia stimulated with purified WNT-3A. Cell Signal. 2013;25:822–8.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
Hamblet NS, Lijam N, Ruiz-Lozano P, Wang J, Yang Y, Luo Z, et al. Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002;129:5827–38.
Lee YN, Gao Y, Wang HY. Differential mediation of the Wnt canonical pathway by mammalian dishevelleds-1, -2, and -3. Cell Signal. 2008;20:443–52.
Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, et al. Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet. 2008;4:e1000259
Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–18.
Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132:4421–36.
Simons M, Gault WJ, Gotthardt D, Rohatgi R, Klein TJ, Shao Y, et al. Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat Cell Biol. 2009;11:286–94.
Horowitz A, Simons M. Phosphorylation of the cytoplasmic tail of syndecan-4 regulates activation of protein kinase Cα. J Biol Chem. 1998;273:25548–51.
White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem. 1998;273:3166–72.
Haag J, Gebhard PM, Aigner T. SOX gene expression in human osteoarthritic cartilage. Pathobiology. 2008;75:195–9.
Dreier R. Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res Ther. 2010;12:216.
Pap T, Korb-Pap A. Cartilage damage in osteoarthritis and rheumatoid arthritis—two unequal siblings. Nat Rev Rheumatol. 2015;11:606–15.
Ryu JH, Chun JS. Opposing roles of WNT-5A and WNT-11 in interleukin-1β regulation of type II collagen expression in articular chondrocytes. J Biol Chem. 2006;281:22039–47.
Lefebvre V, Li P, De Crombrugghe B. A new long form of Sox5 (L‐Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998;17:5718–33.
Taschner MJ, Rafigh M, Lampert F, Schnaiter S, Hartmann C. Ca2+/Calmodulin-dependent kinase II signaling causes skeletal overgrowth and premature chondrocyte maturation. Dev Biol. 2008;317:132–46.
Golan T, Yaniv A, Bafico A, Liu G, Gazit A. The human Frizzled 6 (HFz6) acts as a negative regulator of the canonical Wnt/β-catenin signaling cascade. J Biol Chem. 2004;279:14879–88.
Möller S, Vilo J, Croning MD. Prediction of the coupling specificity of G protein coupled receptors to their G proteins. Bioinformatics. 2001;17:S174–181.
Koval A, Katanaev VL. Wnt3a stimulation elicits G-protein-coupled receptor properties of mammalian Frizzled proteins. Biochem J. 2011;433:435–40.
Kilander MBC, Petersen J, Andressen KW, Ganji RS, Levy FO, Schuster J, et al. Disheveled regulates precoupling of heterotrimeric G proteins to Frizzled 6. FASEB J. 2014;28:2293–305.
Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94.
Mankin HJ, Dorfman H, Lippiello L, Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. J Bone Jt Surg Am. 1971;53:523–37.
Acknowledgements
This work was supported by Agence Nationale de la Recherche (ANR: Solv-CDG) and by Région Lorraine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Xie, Z., Khair, M., Shaukat, I. et al. Non-canonical Wnt induces chondrocyte de-differentiation through Frizzled 6 and DVL-2/B-raf/CaMKIIα/syndecan 4 axis. Cell Death Differ 25, 1442–1456 (2018). https://doi.org/10.1038/s41418-017-0050-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0050-y
- Springer Nature Limited