
Abstract
It has been widely reported that glioma stem-like cells (GSCs) serve a crucial role in the malignant progression of glioma. In particular, recent studies have reported that long non-coding RNAs (lncRNAs) are closely associated with glioma development. However, the underlying molecular regulatory mechanistic role of GSCs remains poorly understood. The present study established two highly malignant glioma stem-like cell lines from clinical surgical specimens. In these, it was found that the lncRNA growth arrest-specific 5 (GAS5) expression was downregulated in GSCs and high-grade glioma tissues, compared with normal human astrocyte cells (NHAs) and normal brain tissues, respectively, which also showed a positive correlation with patient survival. Functional assays revealed that knocking down GAS5 expression promoted the proliferation, invasion, migration, stemness, and tumorigenicity of GSGs, while suppressing their apoptosis. Mechanistically, GAS5 directly sponged miR-23a, which in turn functioned as an oncogene by inhibiting E-cadherin, through the assays of reverse transcription-quantitative PCR (RT-qPCR) and luciferase reports. In addition, rescue experiments demonstrated that GAS5 could promote the expression and function of E-cadherin in a miR-23a-dependent manner. Collectively, these data suggest that GAS5 functions as a suppressor in GSCs by targeting the miR-23a/E-cadherin axis, which may be a promising therapeutic target against glioma.
Similar content being viewed by others


Introduction
Gliomas are the most common primary intracranial tumors in the central nervous system, among which glioblastoma multiforme (GBM) is the most prevalent. GBM is highly malignant and aggressive [1]. The overall survival and prognosis of GBM are poor, although the therapeutic regimens available are being optimized continuously at a steady pace [2]. Previous reports attributed the poor prognosis from GBM to the presence of GSCs, which exhibit substantial cellular heterogeneity [3]. They have also been documented to be responsible for driving the characteristics of tumor progression, including initiation, metastasis, and treatment resistance [3]. However, the molecular mechanisms underlying GSCs function remain to be fully elucidated, which is in demand for improving the efficacy of glioma treatments [3, 4].
lncRNAs have been revealed to play critical roles in various pathophysiological processes, such as cell proliferation and apoptosis, by activating or silencing the expression of corresponding genes on both transcription and/or post-transcription levels [5]. Accumulating evidence supported the notion that lncRNAs were abnormally expressed in various malignancies, such as gliomas and hepatocellular carcinoma, where they mediated the unfavorable phenotypes [6]. GAS5, located on chromosome 1q25, was frequently reported as a potential tumor suppressor in various malignancies, such as breast cancer [7], hepatocellular carcinoma [8], and ovarian cancer [9]. It had also been found that GAS5 could inhibit the proliferation and metastasis of glioma cells, suggesting that it can be a promising therapeutic target for glioma [10]. However, little is known about the function and associated molecular mechanism of GAS5 in GSCs, which requires further investigation.
E-cadherin (CDH1) encodes the E-cadherin cell-cell adhesion protein and has been widely recognized to be a tumor suppressor involved in epithelial-to-mesenchymal transition (EMT), such as breast cancer, by inhibiting metastasis [11, 12]. EMT enables polarized epithelial cells to transform into mesenchymal cells, which develop enhanced invasive and migratory characteristics [13]. The loss of CDH1 expression is a crucial activation event during EMT, which has been shown to contribute to glioma progression [14].
Therefore, in the present study, the expression profile of GAS5 in glioma was analyzed using The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas (CGGA) databases. It was found that GAS5 expression was negatively associated with the malignancy of glioma. Based on the integrated analysis of both function assays and further molecular mechanistic assays, it was revealed that GAS5 mediated CDH1 increase by sponging miR-23a, which inhibited the malignant phenotype of GSCs.
Materials and methods
Tumor specimens and cell culture
Clinical cancer (10 cases) and adjacent normal brain tissue (10 cases) specimens derived from patients with glioma were collected from the Department of Neurosurgery, Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine (Hangzhou, China), after obtaining informed consent. The protocols were approved by the Ethics Committee of the Zhejiang University School of Medicine.
The excised glioma tissues were minced with sterile ophthalmic scissors. Papain Dissociation System (Worthington) was added to the tissue digestion at 37 °C for 20 min. Filtered the suspension through a 70 μm sterile sieve and added 5 ml of red blood cell lysis buffer (Beyotime Institute of Biotechnology) lysis for 5 min, centrifuged at 1100 rpm for 5 min, and discarded the supernatant. The cell suspension was cultured in DMEM/F12 medium designed for neural stem cell growth [15], which was included with 1× B27 Supplement (Gibco), 20 ng/ml epidermal growth factor (EGF; Gibco) and 20 ng/ml basic fibroblast growth factor (bFGF; Gibco). Primary cultured human GSC1 and GSC2 cells derived from the surgical specimen of patients with glioma were cultured in the neural stem cell medium. Glioma cell lines (T98G, LN229, and A172) and normal human astrocyte cells (NHAs) were cultured in DMEM (Hyclone; Cytiva) containing 10% fetal bovine serum (FBS; Biological Industries). All cells were cultured in the incubator at 37 °C with 5% CO2.
Immunofluorescence staining
GSCs were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.25% Triton X-100 (Beyotime Institute of Biotechnology) and incubated with a blocking solution for 60 min at room temperature to inhibit non-specific bindings. These fixation cells were then incubated first with primary antibodies against SOX2 (#3579S) and OCT4 (#2750) (1:200, Cell Signaling Technology, Inc.) at room temperature for 60 min. After washing with PBS three times, the secondary antibody (#A0516; #A0562; Beyotime Institute of Biotechnology) was applied prior to incubation for 30 min at room temperature in the dark. Finally, the cells were stained with DAPI at room temperature and observed under a fluorescence microscope (Carl Zeiss AG) at ×200 magnification.
Flow cytometry
Single-cell suspensions of GSCs were suspended in 100 μl PBS and incubated with primary antibodies against SOX2 (# 3579S) and OCT4 (# 2750) (1:200, Cell Signaling Technology, Inc.) for 60 min at room temperature, after digestion and 800 rpm centrifugation. The cells were then washed with PBS and incubated with the secondary antibody (Beyotime Institute of Biotechnology) for 60 min at room temperature. After 800 rpm centrifugation and washing with PBS 3 times, the cells were re-suspended in 200 μl PBS and finally analyzed with flow cytometry (BD, Beckman) using the Cytexpert 2.0 software.
Vector construction and cell transfection
The overexpression vector of GAS5 (LV-GAS5), two short hairpin RNAs (shRNA) targeting CDH1 (shCDH1) and GAS5 (shGAS5-1 and shGAS5-2) and the corresponding negative control, the microRNA (miR)-23a mimics, inhibitors and corresponding negative controls, were all constructed by Shanghai GenePharma Co., Ltd. The vectors and corresponding controls were transfected into GSCs according to the manufacturer’s protocol. GSCs were seeded in 24-well plates at a confluence of ~70–80% before transfection. Transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. The medium was replaced with a fresh growth medium 48 h after transfection. The sequences used for transfection were given in Table S1.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells with TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) and reverse transcribed into cDNA using a reverse transcription kit (k1622, Thermo Fisher Scientific, Inc.). qPCR was subsequently performed to measure the expression levels of GAS5, CDH1, and miR-23a. The expression of GAPDH and U6 served as the reference genes. The expression level was analyzed using the 2−ΔΔCq method [16]. The primer sequences were given in Table S2.
5-Ethynyl-20-deoxyuridine (EdU) assay
Single-cell suspensions of GSCs (5 × 104 cells per well) were seeded into 24-well plates pre-coated with Matrigel (30 min, room temperature, 50 µl; 1:100 dilution; BD Biosciences) for adherence in an incubator containing 5% CO2 at 37 °C. In total, 50 µM EdU (C10310; Guangzhou RiboBio Co., Ltd.) was added into each well for incubation for 2 h at 37 °C. The cells were then fixed in 4% polyformaldehyde for 20 min and stained using the Apollo dye solution for 25 min at room temperature. Cell nuclei were stained with DAPI for 20 min. The proportion of EdU-positive cells was finally analyzed under a fluorescence microscope at ×200 magnification.
Transwell assay
Transwell assays were performed to evaluate the invasion and migration of GSCs. Upper chambers were coated with Matrigel (37 °C; 50 µl; 1:8 dilution; BD Biosciences) in an incubator overnight for invasion assays. Otherwise, the upper chambers were pre-coated with Matrigel (50 µl; 1:100 dilution) for migration assays, in which the Matrigel was aspirated after 20 min. The subsequent protocol was performed as previously described [15].
GSC sphere formation assay
Single-cell suspensions of GSCs were seeded into 24-well plates at a density of 100 cells per well. The sphere numbers of GSCs (>50 µm) in each well were then counted under a microscope. The sphere formation efficiency of GSCs was calculated by the number of spheres formed/100 × 100%.
Cell apoptosis assay
The cell apoptosis rate of the GSCs was detected using the Annexin V-APC/7-AAD kit (AP105, Multisciences). Single-cell suspensions of GSCs were washed with PBS and re-suspended in the binding buffer. In total, 5 µl annexin V-APC and 5 µl 7-AAD were added to the suspensions of GSCs in the dark at room temperature and incubated for 15 min. The cell apoptosis rate was analyzed by flow cytometry within 60 min after staining and finally analyzed using the Cytexpert 2.0 software.
Tumorigenicity assay
Briefly, 4–6 week old athymic Balb/c nude mice (total of 25 female mice, 20–25 g) were allocated to groups with randomization and bred in the animal center under specific pathogen-free (SPF) conditions. Single cell suspensions of 1 × 106 GSCs with GAS5 overexpressed or knocked down (along with the corresponding negative controls) were injected subcutaneously into the right flank of each mouse. A total of 3 weeks later, the tumor volume was calculated using the formula length × width2 × 0.5. The animal experiments were approved by the Ethics Committee of the Zhejiang University School of Medicine.
Luciferase reporter assay
The wildtype (WT) and mutant (MUT) sequences of the 3′-untranslated region (UTR) of GAS5 or CDH1 with miR-23a binding sites were inserted into the pMIR-REPORT vectors (GenePharma Co., Ltd.). These pMIR-REPORT vectors, together with miR-23a mimics or the corresponding negative control, were then transfected into the GSCs for 48 h. The Dual-luciferase reporter assay system (Promega Corporation) was used to evaluate the luciferase activity of GSCs.
Western blotting
Total protein from GSCs was extracted using the RIPA buffer (Beyotime Institute of Biotechnology). Protein concentration was determined by the BCA Protein Assay Kit (Beyotime Institute of Biotechnology), after which 20 μg total protein per lane were separated by 10% SDS-PAGE transferred onto PVDF membranes. These membranes were then incubated with primary antibodies against CDH1 (# 3195S; 1:1000; Cell Signaling Technologies, Inc.) and GAPDH (Cell Signaling Technologies, Inc.) overnight at 4 °C. The membranes were then incubated with secondary antibodies (A0208; 1:2000; Beyotime Institute of Biotechnology) for 60 min at room temperature. The enhanced chemiluminescence (ECL) method was used for visualization.
Statistical analysis
All experiments in vitro were repeated 3 times and 5 mice per group in vivo. All data in the present study are presented as the mean ± SD and analyzed using the GraphPad Prism software (Version 8.0.2, GraphPad Software, Inc.). One-way/two-way analysis of variance (one/two-way ANOVA) was used to determine the differences among ≥ three groups. Student’s t-test was used to evaluate the differences between two groups. The variance was similar among the groups that were being statistically compared. P < 0.05 was considered to indicate a statistically significant difference.
Results
Expression of GAS5 is negatively associated with the malignancy of glioma
The expression of GAS5 was first analyzed in different glioma subtypes in The Cancer Genome Atlas (TCGA) and Chinese Glioma Genome Atlas (CGGA) databases. It was found that GAS5 expression was negatively associated with the degree of glioma malignancy and overall survival. Gliomas were divided into different subtypes based on the status of isocitrate dehydrogenase (IDH) mutation and 1p/19q codeletion. It was well known that the malignant degree of IDH mutation subtype and 1p/19q codeleted subtype was lower than IDH wildtype and 1p/19q non-codeleted subtype, respectively. The expression of GAS5 in the IDH wildtype subtype was significantly lower than that in the IDH mutant subtype according to the CGGA_693 and TCGA_GBMLGG datasets (Fig. 1A, B). In addition, the expression of GAS5 in the 1p/19q non-codeleted subtype was significantly lower than that in the 1p/19q co-deleted subtype (Fig. 1C, D). Further analysis found that the expression of GAS5 in the IDH wildtype subtype was significantly lower than that in the IDH mutant combined with 1p/19q co-deleted subtype and IDH mutant combined with 1p/19q non-codeleted subtype (Fig. 1E, F). Overall survival analysis revealed that the survival rate of glioma patients with lower GAS5 expression was significantly decreased (Fig. 1G, H). To demonstrate that GAS5 is an independent prognostic factor, we performed univariate and multivariate Cox regression analysis, incorporating grade, age, IDH mutation status, 1p/19q codeletion status and MGMT promoter status into variables, and graphed them as forest plots. It was shown that GAS5 was an independent protective factor for prognosis (Fig. S1).

The differential expression of GAS5 between the IDH mutant and IDH wildtype subtypes in (A) CGGA_693 and (B) TCGA_GBMLGG datasets. ****P < 0.0001, Student’s t-test. The differential expression of GAS5 between the 1p/19q codeleted and 1p/19q non-codeleted subtypes in (C) CGGA_693 and (D) TCGA_GBMLGG datasets. ****P < 0.0001, Student’s t-test. The differential expression of GAS5 among the IDH mutant and 1p/19q codeleted, IDH mutant and 1p/19q non-codeleted, and IDH wildtype subtypes in (E) CGGA_693 and (F) TCGA_GBMLGG datasets, respectively. ****P < 0.0001, one-way ANOVA. The overall survival rate of patients with glioma in (G) low GAS5 expression group and (H) high GAS5 expression group.
Primary culture of GSCs derived from clinical surgical specimens
Two human GSCs (GSC1 and GSC2), derived from two patients diagnosed with glioblastoma, were cultured to investigate the function of GAS5 in GSCs. GSC1 and GSC2 exhibited typical sphere-like cell clusters. They could also adherently proliferate on laminin-coated plates (Fig. 2A). To further verify that the cells were GSCs, the expression of GSCs makers was measured in GSC1 and GSC2. Immunofluorescence assays indicated that the expression of the GSCs makers SOX2 and OCT4 was positive in GSC1 and GSC2 (Fig. 2B). In addition, flow cytometry analysis showed that the positivity rates of SOX2, OCT4, CD133, Nestin and CD44 in GSC1 and GSC2 (Fig. 2C and S2). The control was negative isotype control, which was used for identifying the contribution of non-specific background to staining to reduce the risk of false-positive results.

A Primary culture of human GSCs derived from two patients diagnosed with glioblastoma. Sphere-like cell clusters and adherent growth patterns of GSCs were cultured. B Immunofluorescence assays detected the expression of GSCs makers in the two GSC cell lines. C Positivity rates of the GSC markers were analyzed by flow cytometry in the two GSC cell lines. The control was negative isotype control.
GAS5 overexpression inhibits the proliferation, invasion, migration, and stemness of GSCs while promoting apoptosis in vitro
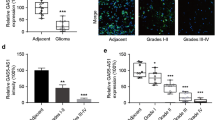
GAS5 expression was detected in GSC1 and GSC2, glioma cell lines T98G, LN229 and A172 and glioma tissues, all of which found decreased GAS5 expression in GSCs, glioma cells and tissues, compared with NHAs and normal brain tissues (Fig. 3A, B). To evaluate the possible function of GAS5 in GSCs, GSC1 and GSC2 were transfected with the GAS5 overexpression vector and the corresponding negative control. RT-qPCR verified that GAS5 expression in the groups of transfection with GAS5 overexpression vector was significantly higher than that in the group of transfection with negative control (Fig. 3C). Subsequent EdU assay data indicate that GAS5 overexpression decreased the proliferative ability of GSC1 and GSC2 (Fig. 3D, E). Transwell assays then revealed that overexpression of GAS5 resulted in significant decreases in the invasion and migration capabilities of GSC1 and GSC2 (Fig. 3F–I). In addition, sphere formation assays similarly demonstrated that overexpression of GAS5 markedly reduced the sphere formation efficiency of GSC1 and GSC2 (Fig. 3J, K). Flow cytometry then revealed that GAS5 overexpression in GSC1 and GSC2 led to marked increases in the cell apoptosis rate (Fig. 3L, M).

GAS5 overexpression inhibits the proliferation, invasion, migration and stemness of GSCs while promoting apoptosis in vitro. RT-qPCR analyzed GAS5 expression in (A) glioma tissues, B glioma cell lines and GSCs. C GAS5 expression was analyzed with RT-qPCR in GSCs transfected with GAS5 or NC. **P < 0.01 and ***P < 0.001, Student’s t-test. Cell proliferation was (D) observed and (E) quantified after GAS5 overexpression in GSCs using EdU assays. **P < 0.01, Student’s t-test. F–I The capacity of invasion and migration was assessed after GAS5 overexpression in GSCs by Transwell assays. **P < 0.01 and ***P < 0.001, Student’s t-test. Cell stemness in GSCs was (J) observed and (K) quantified after GAS5 overexpression using sphere formation assays. Cell apoptosis was (L) observed and (M) quantified using flow cytometry assays after GAS5 overexpression in GSCs.
Silencing GAS5 promotes proliferation, invasion, migration, and stemness while inhibiting apoptosis in GSCs in vitro
GAS5 knockdown was performed in GSC1 and GSC2 by transfection with shRNAs targeting GAS5 (including shGAS5-1 and shGAS5-2) and the corresponding negative control. RT-qPCR verified that GAS5 expression in the transfection groups with shRNAs was significantly lower than that in the transfection group with negative control (Fig. 4A). EdU assays revealed that GAS5 knockdown enhanced the proliferative capacity of GSC1 and GSC2 (Fig. 4B–D). Transwell assays indicated that knocking down GAS5 expression promoted the invasion and migration of GSC1 and GSC2 (Fig. 4E–H). Sphere formation assay data then showed that GAS5 knockdown significantly increased the sphere formation efficiency of GSC1 and GSC2 (Fig. 4I, J). Flow cytometry results suggested that GAS5 knockdown in GSC1 and GSC2 decreased the cell apoptosis rate (Fig. 4K, L).

A RT-qPCR analysis of GAS5 expression in GSCs after transfection with the shNC, shGAS5-1 or shGAS5-2. *P < 0.05 and **P < 0.01, one-way ANOVA. B–D Proliferation ability was evaluated after GAS5 downregulation in GSCs using EdU assays. *P < 0.05 and **P < 0.01, one-way ANOVA. E–H Cell invasion and migration capacity were measured after GAS5 knockdown in GSCs using Transwell assays. **P < 0.01 and ***P < 0.001, one-way ANOVA. Cell stemness was (I) evaluated and (J) quantified after GAS5 knockdown in GSCs using sphere formation assays. Cell apoptosis was (K) evaluated and (L) quantified using flow cytometry assays after GAS5 knockdown in GSCs.
GAS5 overexpression inhibits the growth of GSCs in vivo
Tumorigenicity assays were performed to evaluate the effects of GAS5 on GSCs growth in vivo. Subcutaneous injection of GSC1 transfected with shGAS5 or the corresponding negative control was performed. The results showed that GAS5 knockdown in GSCs led to increases in tumor volume and weight, compared with those in the control group (Fig. 5A–C). In addition, GSC2 transfected with the GAS5 overexpression vector or the corresponding negative control were subcutaneously implanted. These results showed that overexpression of GAS5 in GSCs led to lower tumor volumes and weights, compared with those in the control group (Fig. 5D–F). Besides, HE staining and immunohistochemical analysis of xenograft tumors were performed. It showed that GAS5 knockdown led to Ki67 increase and Bcl-2 reduction, compared with those in the control group; overexpression of GAS5 led to Ki67 reduction and Bcl-2 increase, compared with those in the control group (Fig. S3).

A Tumorigenicity assays were performed in nude mice following subcutaneous inoculation of GSC1 cells transfected with shNC, shGAS5-1 or shGAS5-2. Tumor (B) volume and (C) weight were measured in the shNC, shGAS5-1 and shGAS5-2 groups. ****P < 0.0001, two-way ANOVA; *P < 0.05 and **P < 0.01, one-way ANOVA. D Tumorigenicity assays were performed in nude mice after subcutaneous inoculation of GSC2 cells transfected with NC or GAS5. Tumor (E) volume and (F) weight were measured in the NC and GAS5 groups. ****P < 0.0001, two-way ANOVA; *P < 0.05, Student’s t-test.
GAS5 functions as a sponge for miR-23a in GSCs
Bioinformatics analysis was conducted to identify the possible miRNA targets of GAS5 to explore the mechanism of GAS5 in regulating the biological function of GSCs. Potential bonding sites between GAS5 and miR-23a were predicted using the StarBase online database (Fig. 6A). Further verification with RT-qPCR found higher expression of miR-23a in GSCs, glioma cell lines and glioma tissues compared with those in the NHAs and normal brain tissues (Fig. 6B, C). In addition, miR-23a expression was negatively regulated by GAS5 in GSCs (Fig. 6D, E). WT and MUT vectors of GAS5 were constructed for luciferase activity assays to assess the direct binding between miR-23a and GAS5 (Fig. 6A). It showed that overexpression of miR-23a significantly reduced the luciferase activity of GSCs transfected with GAS5-WT, whereas the efficacy was lost in terms of GAS5-MUT (Fig. 6F, G), which suggested that GAS5 could direct bind with miR-23a.

A The putative binding site between miR-23a and GAS5. Wildtype and mutant vectors of GAS5 were constructed for luciferase activity assays. B, C miR-23a expression was measured using RT-qPCR in glioma tissues, glioma cell lines and GSCs. *P < 0.05, Student’s t-test; *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA. D, E RT-qPCR assays were performed to verify the negative regulation between miR-23a and GAS5 in GSCs. *P < 0.05 and **P < 0.01, one-way ANOVA; Student’s t-test. F, G Luciferase reporter assays were performed to verify the direct binding between miR-23a and GAS5. *P < 0.05 and **P < 0.01, Student’s t-test.
miR-23a knockdown inhibits the proliferation, invasion, and migration of GSCs while promoting apoptosis by targeting CDH1
According to bioinformatics analysis, CDH1 was one of the putative downstream targets of miR-23a (Fig. 7A). CDH1-WT and CDH1-MUT vectors were constructed for luciferase activity assays to validate the direct binding between miR-23a and CDH1 (Fig. 7A). To further explore the relationship between miR-23a and CDH1, both the miR-23a inhibitor and CDH1 shRNA were transfected into GSC1 and GSC2. RT-qPCR and western blotting data revealed that miR-23a knockdown could increase CDH1 expression, which could be partially reversed by CDH1 shRNA co-transfection (Fig. 7B, C). Luciferase reporter assays then suggested that overexpression of miR-23a significantly decreased the luciferase activity of GSCs transfected with CDH1-WT compared with CDH1-MUT (Fig. 7D, E). Furthermore, EdU assays found that knocking down miR-23a expression suppressed the proliferation of GSCs, which was reversed by knocking down CDH1 expression (Fig. 7F–H). Subsequently, Transwell assays revealed that the weakened invasive and migratory capabilities of GSCs induced by miR-23a knockdown were partially reversed by knocking down CDH1 expression (Fig. 7I–L). Flow cytometry assays found that knocking down miR-23a expression in GSCs led to increases in cell apoptosis, which was reversed by knocking down CDH1 expression (Fig. 7M, N).

A The predicted binding sites of miR-23a on the CDH1 3’untranslated region. The vectors of CDH-WT and CDH1-MUT were constructed for luciferase activity assays. B Western blot and (C) RT-qPCR analyzed the CDH1 expression in GSCs transfected with the miR-23a inhibitor or miR-23a inhibitor together with CDH1 shRNA. *P < 0.05 and **P < 0.01, one-way ANOVA. D, E Luciferase reporter assays were performed to verify the direct binding between miR-23a and CDH1-WT in GSCs. *P < 0.05, Student’s t-test. F–H Cell proliferation was measured in GSCs transfected with the miR-23a inhibitor or miR-23a inhibitor together with CDH1 shRNA using EdU assays. *P < 0.05 and **P < 0.01, one-way ANOVA. I–L Invasion and migration abilities in GSCs were evaluated after transfection with the miR-23a inhibitor or miR-23a inhibitor together with CDH1 shRNA using Transwell assays. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA. M, N Cell apoptosis rate was determined in GSCs transfected with the miR-23a inhibitor or miR-23a inhibitor together with CDH1 shRNA using flow cytometry.
Knocking down GAS5 promotes the proliferation, invasion, and migration of GSCs while inhibiting apoptosis by targeting miR-23a
GSC1 and GSC2 were transfected with shGAS5 or shGAS5 together with miR-23a inhibitors to elucidate the mechanism of GAS5 downregulation promoted the malignancy of GSCs. RT-qPCR and western blot analysis found that shGAS5 transfection can decrease CDH1 expression, which was partly reversed by transfection with miR-23a inhibitors (Fig. 8A, B). EdU assays subsequently revealed that shGAS5 transfection increased the proliferative capacity of GSCs, which could be reversed by transfection with miR-23a inhibitors (Fig. 8C–E). Consistent with results from EdU assays, the increased capabilities of cell invasion and migration of GSCs induced by shGAS5 transfection was also partly reversed by transfection with miR-23a inhibitors (Fig. 8F–I). Flow cytometry analysis revealed that shGAS5 transfection inhibited the apoptosis of GSCs, which was also reversed by miR-23a inhibitor transfection (Fig. 8J, K).

A, B RT-qPCR and western blot analyses of CDH1 expression in GSCs transfected with shGAS5 or shGAS5 together with miR-23a inhibitors. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA. C–E EdU assays were performed to measure the proliferative capabilities of GSCs transfected with shGAS5 or shGAS5 together with miR-23a inhibitors. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA. F–I Cell invasion and migration were measured in GSCs transfected with shGAS5 or shGAS5 together with miR-23a inhibitors using Transwell assays. **P < 0.01, ***P < 0.001 and one-way ANOVA. J, K Flow cytometry assessed the apoptosis rate in GSCs transfected with shGAS5 or shGAS5 together with miR-23a inhibitors.
Discussion
GBM is one of the most aggressive and common forms of primary malignancies in the central nervous system [17]. GSCs, a minor sub-population within the tumor, can actively contribute to the dismal prognosis caused by tumor recurrence and chemo- and radio-therapy resistance [17]. It has been reported due to their ability to self-renew and proliferate in an unfavorable tumor microenvironment [18]. GSCs not only exhibit potent tumor-initiating and tumor-propagating characteristics but can also promote malignant invasion [19]. Therefore, therapies targeting GSCs have been proposed for improving GBM treatment and overcoming therapeutic resistance [20]. In the present study, two GSCs derived from the clinical specimens of glioma patients were constructed and identified using neural stem cell markers. Subsequently, they were used to screen for potential therapeutic targets since they appear more representative of human physiological conditions.
An increasing number of studies have suggested that lncRNAs serve an essential role in the occurrence and progression of gliomas [21, 22]. Therefore, investigating the molecular regulation mechanism of lncRNAs may reveal a promising therapeutic target in gliomas. GAS5, located on chromosome 1q25, has been reported to serve a tumor suppressor role [23, 24]. Lower expression levels of GAS5 were previously found to be associated with poorer survival and cisplatin resistance in patients with cervical cancer through regulation of programmed cell death 4 (PDCD4) via competing endogenous RNA of miR-21 [25]. In addition, GAS5 was reported to enhance the radio-sensitivity of esophageal squamous cell carcinoma cells by upregulating RECK expression [26]. It was also demonstrated that GAS5 could be a molecular switch for regulating proliferation in CD133+ cells by inhibiting glucocorticoid receptor-mediated cell cycle control in pancreatic cancer [27]. Furthermore, lung cancer cell-derived exosomal GAS5 was previously proposed as a biomarker for identifying early-stage non-small cell lung cancer [28]. It was also reported that GAS5 could effectively inhibit glioma cells’ proliferative and invasive capacities by targeting glutathione S-transferase mu 3 (GSTM3) [29]. However, the function and relative mechanisms of GAS5 in GSCs remain unclear. The present study found that GAS5 overexpression efficiently inhibited the proliferation, invasion, migration, and tumorgenicity of GSCs but promoted their apoptosis, which was mediated through the miR-23a/CDH1 axis.
miRNAs have previously emerged as potential targets in tumor progression [30]. miR-23a was previously found to be associated with regulating various cellular processes, including cell proliferation, apoptosis, and metastasis, in several types of tumors, such as oral squamous cell carcinoma [31] and non-small cell lung cancer [32]. It was reported that miR-23a expression was increased, which could promote proliferation and suppress apoptosis by targeting PDCD4 in gastric cancer [33]. In addition, overexpression of miR-23a was found to promote the progression of esophageal squamous cell carcinoma by targeting TNF receptor-associated factor 5 [34]. By contrast, miR-23a expression was downregulated and functioned as a tumor suppressor in breast cancer [35]. It was also previously demonstrated that miR-23a could promote the progression of glioma by suppressing PTEN [36]. However, the role of miR-23a in GSCs has not been reported previously. According to the present study, miR-23a knockdown could inhibit the proliferation, invasion and migration of GSCs while promoting their apoptosis by targeting CDH1.
CDH1 is a crucial component in EMT that has been widely reported to be a pivotal protein for maintaining the integrity of cell-cell junctions in the epithelial layer [37]. Downregulation of CDH1 could decrease intercellular adhesion to promote tumor invasion and metastasis [38]. It was previously reported that bone morphogenic protein 4 suppressed cell invasion by GBM by upregulating CDH1 expression [39]. Univariate Cox regression analysis in another study revealed that lower expression levels of CDH1 were associated with shorter progression-free survival from ependymoma [40]. Results of the present study suggested that GAS5 promoted the expression of CDH1 by sponging miR-23a. Actually, the binding site of miR-23a to GAS5 and CDH1 was the same sequence. Because miR-23a directly bound with GAS5 and CDH1, miR-23a overexpression could bind with more GAS5 and CDH1, resulting in their downregulation. Conversely, miR-23a knocking-down bound with fewer GAS5 and CDH1, resulting in their overexpression.
In summary, the present study not only uncovered the critical effects of GAS5 as a tumor suppressor in GSCs but also explored the molecular regulation mechanisms through which miR-23a contributed to the progression of GSCs. In addition, CDH1 was identified as a direct target, which may provide a novel target for developing potential treatment strategies targeting GSCs in glioma.
Data availability
All raw data of the study was available.
References
Liu Y, Wang X, Li J, Tang J, Li B, Zhang Y, et al. Sphingosine 1-phosphate liposomes for targeted nitric oxide delivery to mediate anticancer effects against brain glioma tumors. Adv Mater. 2021;33:e2101701.
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov. 2021;11:1192–211.
Taga T, Tabu K. Glioma progression and recurrence involving maintenance and expansion strategies of glioma stem cells by organizing self-advantageous niche microenvironments. Inflamm Regen. 2020;40:33.
Li J, Liao T, Liu H, Yuan H, Ouyang T, Wang J, et al. Hypoxic glioma stem cell-derived exosomes containing Linc01060 promote progression of glioma by regulating the MZF1/c-Myc/HIF1alpha axis. Cancer Res. 2021;81:114–28.
Luo ML, Li J, Shen L, Chu J, Guo Q, Liang G, et al. The role of APAL/ST8SIA6-AS1 lncRNA in PLK1 activation and mitotic catastrophe of tumor cells. J Natl Cancer Inst. 2020;112:356–68.
Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 2021;41:109–20.
Li G, Qian L, Tang X, Chen Y, Zhao Z, Zhang C. Long noncoding RNA growth arrestspecific 5 (GAS5) acts as a tumor suppressor by promoting autophagy in breast cancer. Mol Med Rep. 2020;22:2460–8.
Zhang WY, Zhan HL, Li MK, Wu GD, Liu Z, Wu LF. Long noncoding RNA Gas5 induces cell apoptosis and inhibits tumor growth via activating the CHOP-dependent endoplasmic reticulum stress pathway in human hepatoblastoma HepG2 cells. J Cell Biochem. 2022;123:231–47.
Long X, Song K, Hu H, Tian Q, Wang W, Dong Q, et al. Long non-coding RNA GAS5 inhibits DDP-resistance and tumor progression of epithelial ovarian cancer via GAS5-E2F4-PARP1-MAPK axis. J Exp Clin Cancer Res. 2019;38:345.
Ding Y, Wang J, Zhang H, Li H. Long noncoding RNA-GAS5 attenuates progression of glioma by eliminating microRNA-10b and Sirtuin 1 in U251 and A172 cells. Biofactors. 2020;46:487–96.
Yang Y, Zhao B, Lv L, Yang Y, Li S, Wu H. FBXL10 promotes EMT and metastasis of breast cancer cells via regulating the acetylation and transcriptional activity of SNAI1. Cell Death Discov. 2021;7:328.
Mendonsa AM, Na TY, Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene. 2018;37:4769–80.
Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–26.
Tao C, Huang K, Shi J, Hu Q, Li K, Zhu X. Genomics and prognosis analysis of epithelial-mesenchymal transition in glioma. Front Oncol. 2020;10:183.
Wang H, Li H, Jiang Q, Dong X, Li S, Cheng S, et al. HOTAIRM1 promotes malignant progression of transformed fibroblasts in glioma stem-like cells remodeled microenvironment via regulating miR-133b-3p/TGFbeta axis. Front Oncol. 2021;11:603128.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25:402–8.
Sabu A, Liu TI, Ng SS, Doong RA, Huang YF, Chiu HC. Nanomedicines targeting glioma stem cells. ACS Appl Mater Interfaces. 2022. https://doi.org/10.1021/acsami.2c03538.
Lah TT, Novak M, Breznik B. Brain malignancies: glioblastoma and brain metastases. Semin Cancer Biol. 2020;60:262–73.
Boyd NH, Tran AN, Bernstock JD, Etminan T, Jones AB, Gillespie GY, et al. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics. 2021;11:665–83.
Hou Y, Sun B, Liu W, Yu B, Shi Q, Luo F, et al. Targeting of glioma stem-like cells with a parthenolide derivative ACT001 through inhibition of AEBP1/PI3K/AKT signaling. Theranostics. 2021;11:555–66.
Peng Z, Liu C, Wu M. New insights into long noncoding RNAs and their roles in glioma. Mol Cancer. 2018;17:61.
Zeng Z, Chen Y, Geng X, Zhang Y, Wen X, Yan Q, et al. NcRNAs: multiangle participation in the regulation of glioma chemotherapy resistance (Review). Int J Oncol. 2022;60:76.
Mo R, Li J, Chen Y, Ding Y. lncRNA GAS5 promotes pyroptosis in COPD by functioning as a ceRNA to regulate the miR2233p/NLRP3 axis. Mol Med Rep. 2022;26:219.
Zhang WY, Zhan HL, Li MK, Wu GD, Liu Z, Wu LF. Long noncoding RNA Gas5 induces cell apoptosis and inhibits tumor growth via activating the CHOP-dependent endoplasmic reticulum stress pathway in human hepatoblastoma HepG2 cells. J Cell Biochem. 2022;123:231–47.
Fang X, Zhong G, Wang Y, Lin Z, Lin R, Yao T. Low GAS5 expression may predict poor survival and cisplatin resistance in cervical cancer. Cell Death Dis. 2020;11:531.
Lin J, Liu Z, Liao S, Li E, Wu X, Zeng W. Elevation of long non-coding RNA GAS5 and knockdown of microRNA-21 up-regulate RECK expression to enhance esophageal squamous cell carcinoma cell radio-sensitivity after radiotherapy. Genomics. 2020;112:2173–85.
Sharma NS, Gnamlin P, Durden B, Gupta VK, Kesh K, Garrido VT, et al. Long non-coding RNA GAS5 acts as proliferation “brakes” in CD133+ cells responsible for tumor recurrence. Oncogenesis. 2019;8:68.
Li C, Lv Y, Shao C, Chen C, Zhang T, Wei Y, et al. Tumor-derived exosomal lncRNA GAS5 as a biomarker for early-stage non-small-cell lung cancer diagnosis. J Cell Physiol. 2019;234:20721–7.
Li G, Cai Y, Wang C, Huang M, Chen J. LncRNA GAS5 regulates the proliferation, migration, invasion and apoptosis of brain glioma cells through targeting GSTM3 expression. The effect of LncRNA GAS5 on glioma cells. J Neurooncol. 2019;143:525–36.
Wang J, Yokoyama Y, Hirose H, Shimomura Y, Bonkobara S, Itakura H, et al. Functional assessment of miR1291 in colon cancer cells. Int J Oncol. 2022;60:13.
Ma Y, Gao J, Guo H. miR-23a-3p regulates Runx2 to inhibit the proliferation and metastasis of oral squamous cell carcinoma. J Oncol. 2022;2022:8719542.
Fan X, Tao S, Li Q, Deng B, Tan QY, Jin H. The miR-23a/27a/24-2 cluster promotes postoperative progression of early-stage non-small cell lung cancer. Mol Ther Oncolytics. 2022;24:205–17.
Hu X, Wang Y, Liang H, Fan Q, Zhu R, Cui J, et al. miR-23a/b promote tumor growth and suppress apoptosis by targeting PDCD4 in gastric cancer. Cell Death Dis. 2017;8:e3059.
Shang M, Weng L, Wu S, Liu B, Yin X, Wang Z, et al. HP1BP3 promotes tumor growth and metastasis by upregulating miR-23a to target TRAF5 in esophageal squamous cell carcinoma. Am J Cancer Res. 2021;11:2928–43.
Zhu L, Wang F, Fan W, Jin Z, Teng C, Zhang J. lncRNA NEAT1 promotes the Taxol resistance of breast cancer via sponging the miR-23a-3p-FOXA1 axis. Acta Biochim Biophys Sin. 2021;53:1198–206.
He Z, Long J, Yang C, Gong B, Cheng M, Wang Q, et al. LncRNA DGCR5 plays a tumor-suppressive role in glioma via the miR-21/Smad7 and miR-23a/PTEN axes. Aging. 2020;12:20285–307.
Lin YT, Wu KJ. Epigenetic regulation of epithelial-mesenchymal transition: focusing on hypoxia and TGF-beta signaling. J Biomed Sci. 2020;27:39.
Vos EL, Salo-Mullen EE, Tang LH, Schattner M, Yoon SS, Gerdes H, et al. Indications for total gastrectomy in CDH1 mutation carriers and outcomes of risk-reducing minimally invasive and open gastrectomies. JAMA Surg. 2020;155:1050–7.
Zhao X, Sun Q, Dou C, Chen Q, Liu B. BMP4 inhibits glioblastoma invasion by promoting E-cadherin and claudin expression. Front Biosci. 2019;24:1060–70.
Malgulwar PB, Nambirajan A, Pathak P, Rajeshwari M, Suri V, Sarkar C, et al. Epithelial-to-mesenchymal transition-related transcription factors are up-regulated in ependymomas and correlate with a poor prognosis. Hum Pathol. 2018;82:149–57.
Acknowledgements
This study was supported by grants from the agricultural and social development project of Hangzhou (grant no. 20211231Y025; grant no. 202204B11).
Author information
Authors and Affiliations
Contributions
QD is responsible for the design of the study; HW is responsible for experimental implementation; DW is responsible for collection and analysis of data; YS and CS are responsible for the writing the manuscript; QH and LJ are responsible for reviewing the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Zhejiang University School of Medicine.
Patient consent for publication
The patient informed consent was obtained.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, H., Wang, D., Shen, Y. et al. GAS5 attenuates the malignant progression of glioma stem-like cells by promoting E-cadherin. Cancer Gene Ther 30, 450–461 (2023). https://doi.org/10.1038/s41417-022-00566-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-022-00566-y
- Springer Nature America, Inc.
This article is cited by
-
LncRNA GAS5 restrains ISO-induced cardiac fibrosis by modulating mir-217 regulation of SIRT1
Scientific Reports (2024)