
Abstract
Osteoarthritis (OA), a major cause of pain and disability, is a serious public issue worldwide. Some microRNAs (miRNAs) and SOX9 have been found to be expressed in OA. Therefore, the aim of this study is to investigate effects of microRNA-384-5p (miR-384-5p) on cartilage cell proliferation and apoptosis in mice with OA by targeting SOX9 through the NF-κB signaling pathway. First, bioinformatics was used to predict the SOX9-mediated miRNA (miR-384-5p), and dual luciferase reporter gene assay was conducted to further verify the relationship between miR-384-5p and SOX9. Then, the expression of miR-384-5p, SOX9, and NF-kB in mice modeled with OA was detected. To investigate the specific mechanism of miR-384-5p in OA, mimic and inhibitor of miR-384-5p and siRNA against SOX9 were used to transfect cartilage cells. Finally, proliferation, cell cycle, and cell apoptosis were detected using MTT assay and flow cytometry, respectively. Our results indicated that OA mice exhibited decreased expression of SOX9 and NF-kB but higher miR-384-5p expression. In addition, over-expressed miR-384-5p or silenced SOX9 could inhibit cell proliferation, and block cell cycle entry and induces apoptosis. SOX9 was a target gene of miR-384-5p. The NF-kB signaling pathway was inactivated after overexpression of miR-384-5p. Furthermore, we also observed that the effect of miR-384-5p inhibition was rescued when SOX9 was knocked down. The results support the view that inhibition of miR-384-5p could impede apoptosis and promote proliferation of cartilage cells through activating the NF-κB signaling pathway by promoting SOX9, thereby preventing the development of OA.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is one of the most common rheumatic disorders, whose main characteristics are cartilage breakdown and synovial inflammation. Clinical symptoms include joint swelling, synovitis, and inflammatory pain [1]. Pain is the prime symptom of OA and sensitization may contribute to the pain severity [2]. The known causes of OA include many aspects such as age, adiposity, injury, overuse, and infection [3]. Recently, the incidence of OA is steadily rising due to the aging population and the growing epidemic of obesity [4]. OA is the most frequent form of arthritis worldwide affecting about 40% people over the age of 70 years [5]. At present, treatment regimens for articular injury, such as physical therapy or anti-inflammatory drugs, aim to cure the surface symptoms, but have minute effects on the underlying causes [6]. Due to the susceptibility and high incidence of OA, it is trivial to detect early diagnosis markers of OA to improve treatment efficacy [7].
microRNA (miRs) is one of the small non-coding classes of RNAs that post-transcriptionally regulate gene expressions by combining the 3′ UTR of their target gene messenger RNAs (mRNAs), leading to mRNA degradation or translation inhibition [8]. miR-382-5p is one of the members of miRNAs that only recently proved to have great effects on the function of the neural system [9]. miR-382-5p/DLC-1 axis is reported to play an important role in the mediation of hepatitis B virus core protein-enhanced hepatocellular carcinoma motility [10]. SOX9 is one of the well-known signs for the skeleton before the appearance of cartilage blastemas [11], which have been found expressed in OA [12]. Besides, nucleus factor-kappa B (NF-κB) proteins, including a group of transcription factors, contribute to the formation of homo- and heterodimers [13], which has also been found in OA [14]. Liu et al. demonstrated melatonin alleviated inflammasome-induced pyroptosis by blocking NF-κB/GSDMD signaling pathway in adipose tissue of mice [15]. Wu et al. found that novel therapeutic strategies have been developed and are under investigation to selectively modulate miRNAs, which could potentially enable personalized OA therapy [16]. Nevertheless, the literature investigating the association between miR-384-5p and OA remains to be limited in number.
Henceforth, the present study aims to evaluate the effects of miR-384-5p on cartilage cell proliferation and apoptosis in mice with OA by targeting the SOX9 gene through the NF-κB signaling pathway.
Materials and methods
Ethical statement
All animal experimentation in the study was approved by the Institutional Animal Research Committee of Tongji Medical College. The animal experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee.
Chip variation analysis
Using the GEO database (https://www.ncbi.nlm.nih.gov/geo/), the related chips of OA were retrieved, and the GSE16464 expression chip was obtained. The chip included OA samples and normal samples. The difference between gene expression in OA samples and normal samples was analyzed using the R limma package, and |logFC| > 2 and p value < 0.05 were used as screening criteria for differentially expressed genes. Through the R language “pheatmap” package, the infrared images of the differentially expressed genes were constructed.
Genetic association analysis
Using the MalaCards database (http://www.malacards.org/), “osteoarthritis” was used as the key word in order to obtain the known genes of OA, and the 10 highest score genes were selected for correlation analysis. In the STRING database (https://string-db.org/), the 10 known genes of OA and the first 30 differentially expressed genes in GSE16464 were analyzed to find the most core gene.
SOX9-regulated miRNA prediction
SOX9-regulated miRNAs in mice were predicted in databases DIANA (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index), miRDB (http://mirdb.org/miRDB/index.html), miRSearch (https://www.exiqon.com/miRSearch), microRNA.org (http://34.236.212.39/microrna/home.do), and TargetScan (http://www.targetscan.org/vert_71/). All prediction results of DIANA, miRDB, miRSearch, and microRNA.org and Conserved sites of TargetScan were selected in the current study. The web (http://bioinformatics.psb.ugent.be/webtools/Venn/) was established using a Venn diagram to find the intersection of the prediction results of five databases. In the microRNA.org, mirSVR score of miRNA in the intersection was retrieved to determine the miRNA to be investigated.
Establishment of mice model
A total of 30 clean and healthy male mice (weighing about 20 g) were purchased from the Experimental Animal Center of Tongji Medical College (Wuhan, China). The mice were raised in the following feeding conditions: free access to food and water, under natural light, at room temperature (18–22 °C), relative humidity (40–70%), and under noise (≤50 dB). The mice were raised in the aforementioned conditions and subsequently divided into the following two groups: the control group and the OA group, with 15 mice in each group. OA models were established after destabilization of the medial meniscus (DMM) as follows: in the OA group, after mice were fixed and disinfected, the medial patellar ligament was sliced using a microblade, exposing the articular cavity; the fat pad was separated with a blunt dissection to expose the area within the femoral intercondylar in order to observe the medial meniscus tibial ligament; the medial meniscus tibial ligament was sliced using a microblade, and then the overlying skin and tissues were sutured. Injuries of the articular cartilage injury were avoided with maximal care during surgery. The mice in the OA group were not fixed after surgery, and they could move freely in the cage. The mice were administered an intraperitoneal injection of penicillin (80,000 unit/rat, 0.1 ml, Sigma, San Francisco, USA) to prevent infection; no treatment was performed in the control group. During the observation, in the OA group, the articular surface of mice appeared to be rough or anabrotic, synovial hyperplasia occurred in intra-articular and the amount of synovial fluid increased significantly, the degree of cartilage deficiency was serious, and the structure of cartilage was disordered. Henceforth, using the aforementioned observations, we could conclude that OA mice models were well established.
Hematoxylin–eosin (HE) staining
After 12 weeks, mice in two groups were sacrificed by neck dislocation, fresh knee cartilage tissues were collected and then put in the 40 g/L paraformaldehyde solution with phosphate buffer saline (PBS) for 20 h. Fixed specimens were put into 0.5 mol/L ethylene diamine tetraacetic acid (EDTA) solution with PBS to decalcify for 30 days, the solution was replaced one time during the process, then a vertical incision was made to the articular cartilage with a scalpel blade for the area of 1.0 cm × 0.5 cm × 0.2 cm, dried by automatic dehydrator (ASP6025, Lecia, Germany), embedded by paraffin, conventional histopathological slides were collected with 3 μm consecutive section. After conventional dewaxing and dehydration of sections, HE staining was conducted as follows: the sections were placed on a heating plate at 60 °C for 30 min. Dimethylbenzene (12868603-3, 500 ml, Xinchuyang Chemical Industry Co., Ltd., Wuhan, Hubei, China) was rinsed two times using absolute alcohol for 5 min. The sections were soaked in 95%, 90%, 80%, 70% alcohol for 5 min respectively, then rinsed with distilled water for 5 min. After hematoxylin staining (AR0005, BOSTER Biological Technology Co., Ltd., Wuhan, Hubei, China) for 5 min, the sections were washed with tap-water for 5 min. Sections were immersed in 1% hydrochloric acid–ethanol for 20 s to differentiate unsecured hematoxylin. Then, the sections were left until they turned blue, using lithium carbonate for 30–60 s, and washed with distilled water for 30 s. Sections were dried in 75% and 85% ethanol for 30 s, respectively; in 95% I and II ethanol for 3 min, respectively; in 100% I and II ethanol for 10 min, respectively, and dried naturally. After clearing by dimethylbenzene I and II for 10 min, respectively, the sections were blotted and mounted in neutral resin, and differences in cellular morphology and tissue staining were observed under a microscope at a magnification of ×100.
Immunohistochemistry
After the cartilage of mice were conventionally sectioned and dewaxed, immunohistochemistry was conducted as follows: after heating three times in a microwave with 3% H2O2 at room temperature for 10 min, the antigen retrieval solution was added to the cartilage tissue at room temperature for 10 min. Cartilage tissue was rinsed three times with 0.15 mol/L PBS for 2 min; normal goat serum was not washed but kept at room temperature for 20 min. Primary antibody of SOX9 (SOX9 antibody of Rabbit anti-Mice, 1 µg/ml, Cell Signaling Technology) was added to the cartilage tissue at 4 °C overnight and rinsed with 0.15 mol/L PBS (two times) for 3 min. Then, goat anti-rabbit IgG antiserum (diluted at a ratio of 1:1000, Abcam Trading Co., Ltd., Shanghai, China) was added to the cartilage tissue at 37 °C for 20 min and rinsed with 0.15 mol/L PBS for 3 min with two times. Afterwards, the Strept Avidin–Biotin Complex (SABC) solution was added to the cells at 37 °C for 20 min and rinsed with 0.15 mol/L PBS for 4 min with three times. Sections were colored using diaminobenzidine (DAB) for 5–10 min (Sigma, San Francisco, USA) and uniformly washed with distilled water. Subsequently, the sections were stained with hematoxylin slightly (Sigma, San Francisco, USA), dehydrated, and sealed. The protein expression of SOX9 was observed under a microscope, counted, and photographed. Criteria for positive expression of the protein were as follows: five visual fields were collected continuously, completely unstained as negative; positive index <10% as low expression; 10–50% as high expression; >50% as the significant high expression [17].
TUNEL assay
Sections were treated according to the instructions of the TUNEL kit (Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China). Paraffin section was used as the pretreatment before TUNEL assay. The operation steps of TUNEL staining were as follows: 10 μl reaction solution was added to the sections (mixture of 1 μl enzyme and 9 μl reaction buffer); and then cells were rinsed with Buffer A two times (each time for 1 min). A total of 50 μl blocking buffer was added to the cells and then cells were cultured at 37 °C for 30 min, and blocking buffer was removed. A total of 40 μl colored reaction buffer was added to the cells, then cells were cultured at 37 °C for 60 min, and rinsed two times with Buffer B (each time for 1 min). A total of 40 μl BUIP/NBT colored reaction buffer with sections was added to the cassette for 20 min, while staining was allowed to proceed continuously without any background; rinsed two times with Buffer B (each time for 1 min). Cells were counterstained with nucleus fast red for 20 s, washed by water, and dehydrated with ethanol of gradient concentration, until cleared and sealed. Nucleus of positive cell was yellow in color. Six non-overlapping high-power visual fields from each section were collected randomly (×200), and calculated for average percentage of positive cells according to the visual fields. Apoptosis Index (AI) = apoptotic nucleus number/total nucleus number × 100% [18].
Targeting relationship between miR-384-5p and SOX9
Targeted gene of miR-384-5p was analyzed using various databases including microRNA.org and miRbase, and TargetScan was used to verify if SOX9 is a direct target gene of miR-384-5p. To confirm this, we firstly established DNA recombinant plasmid pSOX9-Wt and pSOX9-Mut, which was inserted into the SOX9-3′-UTR wild sequence and mutation sequence. Gene amplification was performed for full length of 3′-UTR of SOX9, production of polymerase chain reaction (PCR) was cloned at multiple clone sites of downstream of pmirGLO Luciferase (Promega company, Madison, WI, USA), which was the binding site of miR-384-5p and its targeted gene was predicted with bioinformatics. Aimed at site-directed mutagenesis of this site, pRL-TK carrier (TaKaRa, Dalian, China), which express ranilla luciferase, was regarded as the internal reference, in order to adjust differences of transfection rate and cell number. miR-384-5p mimic and negative control (NC) of miR-384-5p were transfected with luciferase, respectively in order to enter in the cartilage cells. After transfection for 48 h, the former culture medium was cleaned, and rinsed with PBS two times. Passive lysis buffer (PLB) (100 μl) was added to the cells in each hole, after lightly shaken at room temperature for 15 min, the cell lysate was collected. Program was set to perform pre-reading for 2 s and reading for 10 s. LARII Stop &Glo® Reagent (100 μl) was added each time. Well-prepared LARII Stop & Glo® Reagent and luminescent tube or plate with cell lysate (20 μl for each sample were collected) were placed in the bioluminescence detector. Start program, and data were saved after reading the fluorescence. The calculation was performed according to the following formula: relative fluorescence value = activity value of firefly luciferase/activity value of ranilla luciferase.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Based on the published mRNA sequence of mice, primers of miR-384-5p, SOX9, NF-κB, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were detected using Primer Premier 5.0 (Taka, Dalian, China) (Table 1), and primers were synthetized by the TaKaRa company (Dalian, China). According to the instruction of Trizol (Invitrogen, Car, CA, USA), total RNA of the cells and cartilage tissues were extracted. Then, RNA was reverse transcribed to cDNA by PrimeScript RT kit (TaKaRa, Dalian, China) following the instructions of the kit, and the system of reverse transcription was 10 μl. The set reaction conditions were as follows: reverse transcription reaction at 37 °C for 45 min; reverse transcription of enzyme inactivation at 85 °C for 5 s. The reaction mixture was taken to operate RT-qPCR with the instruction of SYBR® Premix Ex TaqTM II kit (TaKaRa Company, Dalian, China). The reaction system was as follows: 25 μl SYBR® Premix Ex TaqTM II (2×), 2 μl PCR forward primer, 2 μl reverse primer, l μl ROX Reference Dye (50×), 4 μl DNA template, and 16 μl dH2O. RT-qPCR was conducted using the ABI PRISM® 7300 system (ABI, Massachusetts, USA), reverse transcription and amplification process were performed according to the instructions of kit. Reaction conditions were as follows: 40 cycles of pre-denaturation at 95 °C for 30 s; denaturation at 95 °C for 5 s; annealing extension at 60 °C for 30 s. U6 was regarded as the internal reference of miR-384-5p, while GAPDH was the internal reference of other genes. The differences in relative gene expression between two groups were analyzed by 2−△△Ct. All experiments were repeated three times (the method is also applicable for in vitro/cell experiments).
Western blot analysis
Cartilage tissue of mice was collected and added with liquid nitrogen, and ground to a homogeneous fine powder. Protein lysate (Solarbio Science & Technology Co., Ltd., Beijing, China) was added to the samples, and the samples were centrifuged at 4 °C for 20 min with 25,764g, and the supernatant was collected and stored, respectively for sparing. After lytic protein was immersed in the water bath at 100 °C for 10 min, samples were loaded to 10% SDS-polyacrylamide gel electrophoresis at 80 V for 30 min and 100 V for 90 min. After electrophoresis, the protein was transferred to 0.2 mol/L nitrocellulose filter (NC filter) for 120 min at 200 mA. The membrane was placed in 50 g/l 1× Tris-buffered saline and Tween 20 (TBST) in skimmed milk, lightly shaken at room temperature for 1 h in order to block the nonspecific binding site. And then, 1× TBST was used to rinse samples for 5 min, repeated three times. Primary antibody (rabbit-anti-mouse antibody of SOX9 for 1 µg/ml, rabbit-anti-mouse of NF-κB antibody for 0.5 µg/ml, Cell Signaling Technology) was added to the cleaved protein, and incubated in refrigerator at 4 °C overnight, then the primary antibody was removed, followed by the addition of 1× TBST for 5 min again, repeated three times. Then, the secondary antibody (goat anti-rabbit labeled by horseradish peroxidase (HRP), diluted at a ratio of 1:1000, Shanghai USEN Biological Technology Co., Ltd., Shanghai, China) was added to the cleaved protein and incubated at room temperature for 2 h, then the secondary antibody was removed, and washed with 1× TBST three times. The membrane was put in the chemiluminescence (ECL) reaction mixture at room temperature for 1 min. Surface moisture of the membrane was absorbed and covered with a preservative film, exposed, developed, and fixed in a dark room, and staining results were observed. GAPDH (diluted at a ratio of 1:1000, Sigma, San Francisco, USA) was regarded as the internal reference, and the ratio of gray value between the targeted brand and internal reference brand was expressed as the relative expression of protein (the method is also suitable for in vitro/cell experiments).
Cell culture and grouping
OA mice and normal mice were sacrificed using the neck dislocation method (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China). Bilateral slides of knee joint cartilage of mice were extracted after sterilization and rinsed with L-DMEM (Gibco, CA, USA) three times, followed by the addition of 1 g/L II collagenase (Sigma, San Francisco, USA) to another culture medium. The cartilage fragments were placed in the medium and then the medium was placed in an incubator (Heal Force HF90, Healthforce Biomedical Technology Company, Hong Kong, China) at 37 °C for 4 h. After digestion, the cell suspension was filtered with a 150 stainless steel mesh, centrifuged for 3–5 min at 179g, and the supernatant was discarded, then, double antibodies (100 U/ml streptomycin and 100 U/ml penicillin) (Procell Co., Ltd., Wuhan, China) and 12% fetal bovine serum (FBS) (HyClone, UT, USA) were added. Cell number was counted, and the cartilage cells were placed in the medium culture plate and cultured in an incubator with 5% CO2 and saturated humidity for 24 h. The culture medium was replaced through the degree of cell adhesion. After the first time of medium change, it was changed every 3 days. Cell growth was observed under an inverted microscope (XDM-200C, Puqian Optical Instrument Co., Ltd., Shanghai, China). When cartilage cells reached 90% confluency, 0.02% EDTA (Promega, WI, USA) and 2.5 g/L trypsin (Promega, WI, USA) were used to conduct cell-passage activity. Cells were rinsed with PBS solution and then digested with trypsin. Finally, cells were centrifuged and collected. Cell counter (Vi-CELL XR, Beckman Coulter, Brea, USA) was applied in order to count the number of cells, and then inoculated in a 30 mm culture medium for 6 × 106 cells/ml. The third generation of cartilage cells was inoculated in a 6-well plate at a density of 1 × 106 cells/well. Cell grouping and transfection was as follows: the normal group (cartilage cells from normal mice without any treatment), the blank group (cartilage cells from OA mice without any treatment), the NC group (cartilage cells from OA mice transfected with miR-382-5p negative sequence), the miR-384-5p mimic group (cartilage cells from OA mice transfected with miR-384-5p mimic sequence), the miR-384-5p inhibitor group (cartilage cells from OA mice transfected with miR-384-5p inhibitor sequence), the siRNA-SOX9 group (cartilage cells from OA mice transfected with siRNA sequence of SOX9), the miR-384-5p inhibitor + siRNA-SOX9 group (cartilage cells from OA mice transfected with inhibitor of miR-384-5p and siRNA sequence of SOX9). Cartilage cells in the logarithmic phase were inoculated in a 6-well plate, when cell density reached 30–50%, cells were transfected following the instructions of lipofectamin 2000 (Invitrogen, Car, CA, USA). In the miR-384-5p mimic, siRNA-SOX9 (transfected sequence of siRNA-SOX9 was 5′-AACGAGAGCGAGAAGACCC-3′), miR-384-5p inhibitor, miR-384-5p inhibitor + siRNA-SOX9 synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China) and NC lyophilized powder (Invitrogen, Car, CA, USA) were centrifuged and dissolved in water without RNase. The lipofectamin 2000 (5 μl) was diluted by 250 μl serum-free culture medium Opti-MEM (Gibco, CA, USA). The above two solutions were mixed and incubated at room temperature for 20 min. The mixture was added to the culture plate; after cultured at 37 °C with 5% CO2 for 6–8 h, the complete medium was replaced and incubated for 24–48 h to continue the follow-up experiments.
MTT assay
Cartilage cells in the logarithmic phase of growth were collected and monolayer cell culture was conducted with 0.25% trypsin. Cell suspension (cell density of 2.5 × 105 cells/ml) was prepared using RPMI 1640 culture medium (Shanghai Fusheng Industrial Co., Ltd., Shanghai, China) with 10% FBS, and cells were inoculated into a 96-well plate, 8 wells (100 μl each) in each group. Cells were cultured with 5% CO2 at 37 °C, and 10 μl of 5 mg/ml MTT solution (Sigma, San Francisco, USA) was added at the 24th hour, 48th hour, and 72nd hour time intervals, respectively, followed by continuous culture for 4 h. Optical density (OD) value at the wavelength of 570 nm in each well was measured using an automatic microplate reader (BIO-RAD, CA, USA). All experiments were repeated three times.
Flow cytometry
After transfection for 48 h, cartilage cells among different groups were collected, centrifuged at 1610g for 8 min in order to form a single cell suspension, and the supernatant was removed, followed by adding 1× 300 μl binding buffer and 5 μl Annexin V-FITC (Sigma, San Francisco, USA) for incubation at room temperature in dark conditions for 15 min. A total of 5 μl propidium iodide (PI) dye and 200 μl 1× Binding Buffer were added 5 min before detection, and cell apoptosis was detected using flow cytometry (FACSCalibur, Beckman Coulter, Brea, USA). The figure of cell apoptosis: the left lower quadrant presented the normal cells (An−PI−), the right lower quadrant presented apoptotic cells in the early stage (An+PI−), the right upper quadrant referred to apoptotic cells in the late stage (An+PI+), the left upper quadrant referred to apoptotic cells (An−PI+). Cell apoptosis rate was expressed by cell apoptosis percentage at an early stage in the total cell number, and all experiments were repeated three times.
After transfection or 48 h, cartilage cells in different groups were collected, centrifuged at 179g for 5 min, then the supernatant was removed, followed by rinsing with PBS two times, and the cells were resuspended. Cell concentration was adjusted to 1 × 106 cells/ml, and 1 ml of pre-cooled 75% ethanol (−20 °C) was added to fix cells at 4 °C for 1 h, and then cells were centrifuged to abandon absolute ethanol. Cells were rinsed two times in PBS and the supernatant was removed, followed by the addition of 100 ml RNase A (Sigma, San Francisco, USA) in dark conditions, and the cells were immersed in a water bath at 37 °C for 30 min, followed by adding 400 μl PI dye (Sigma, San Francisco, USA) at 4 °C in dark conditions. After 30 min, the cells were passed through a 200-mesh sieve (10,000 cells were counted), and cell cycle was detected using flow cytometry. Distribution curve of DNA concentration was obtained in each cell. All experiments were repeated three times. The figure of cell cycle: Y-axis: effective cell count;X-axis: DNA contention.
Statistical analysis
Statistical analysis was performed using the SPSS 21.0 software (IBM Corp., Armonk, NY, USA), and measurement data were expressed as mean ± standard deviation (SD) while enumeration data were expressed byt-test. Comparisons among groups were analyzed using one-way analysis of variance (ANOVA). p < 0.05 was considered to be statistically significant.
Results
miR-384-5p is predicted to affect OA by regulating SOX9
The GEO database was used to retrieve the related expression chip data of OA. The GSE16464 expression chip was obtained. The difference between OA samples and normal control samples was analyzed. Finally, 1331 differentially expressed genes were obtained, of which 717 genes were up-regulated in OA, and 614 genes were down-regulated in OA. Infrared images of the 30 most significantly different genes were constructed (Fig. 1a). In order to screen other OA-related genes, the MalaCards database was used to search for the known genes of OA and the top 10 genes were chosen for subsequent analysis (Table 2). In the STRING database, the correlation analysis of 30 different genes in GSE16464 and the retrieved 10 OA-related genes was conducted (Fig. 1b). It was found that COL2A1, GDF5, MATN3, and other nine genes were at the core position. Further analysis of the nine genes showed that SOX9 were not the known genes associated with OA, and it was found that SOX9 was related to the NF-kB signaling pathway [19,20,21]. In order to further understand the mechanism of SOX9 in OA, the miRNA databases, DIANA, miRDB, TargetScan, miRsearch, and microRNA.org, were used to predict the miRNA regulation of SOX9. In order to improve the accuracy of prediction results, predictions of the SOX9 gene were conducted in five databases of DIANA, miRDB, TargetScan, miRsearch, and microRNA.org, and extracted the intersection of five databases (Fig. 1c). A total of six miRNAs were in the intersection of five databases and the combination ability of the six miRNAs and SOX9 was consequently analyzed. The prediction results of microRNA.org database showed that mmu-miR-384-5p with the lowest miRSVR score had the strongest binding capacity with SOX9. These results imply that mmu-miR-384-5p can regulate the NF-kB signaling pathway and ultimately affect the development of OA by inhibiting the expression of the SOX9 gene.

Genetic variation analysis and miRNA prediction of osteoarthritis. a Variation analysis of osteoarthritis chip GSE16464; the abscissa indicates the sample number, and the ordinate represents the gene name; the upper tree diagram represents the sample clustering, the red and blue bar represents the sample type, the red represents the osteoarthritis disease sample, and the blue represents the normal control sample; the left side of the tree graph indicates clustering gene expression; the upper right color histogram represents color gradation, red indicates high expression, blue indicates low expression; each square represents the expression level of a gene in a sample. b Gene interaction association analysis; each circle represents a gene, word in the circle is GeneSymbol, the color and the size of that circle represents core degree of the gene, and the red and large circle indicates a high degree of core while blue and small circle represents degree of core; direct line of circles indicates a direct or indirect interaction between the two genes. c Venn analysis of SOX9-mediated miRNA prediction; blue represents DIANA database, red represents miRDB database, gray represents TargetScan database, yellow represents miRSearch database, and green represents microRNA.org database; the number represents the number of miRNA in the region and “6” in the center area means there are six miRNAs all found in the five databases; SOX9 SRY-related HMG box-9 (color figure online)
OA mice present with pathological features with cartilage exfoliation
In the control group, cartilage matrix was light pink under the microscope with homogenous staining, and cartilage cell was blue. Four-layer structures and tidal line of cartilage were clear. The surface layer appeared to be covered with flat cartilage cells, the middle level was covered with circular cartilage cells, and proliferating cartilage cells could be observed in the deep and calcified layers (Fig. 2a). In the OA group, cell number significantly decreased, arranged without sense of depth, and the cartilage matrix was stained unevenly, cell electron concentration significantly increased, the nuclei and cytoplasm were beyond identification, the surface layer of cartilage was incomplete or even disappeared, and subchondral bone revealed. All these conform to pathological characteristics of OA (Fig. 2b).

HE staining results of cartilage tissues of mice in the OA and control group (the original magnification is ×100). HE staining hematoxylin–eosin staining and OA osteoarthritis
OA mice have decreased positive expression rate of SOX9 in cartilage tissue
Results of immunohistochemistry (Fig. 3) indicate that the protein expression of SOX9 was largely observed in the nuclei and expressed as brown-yellow granules, and the positive expression rate of SOX9 in the control group was significantly higher compared to the OA group (p < 0.05), indicating low expression of SOX9 in OA mice.

Decreased positive expression rate of SOX9 in mice with OA detected by immunohistochemical staining (the original magnification is ×200). a Immunohistochemical staining of cartilage tissue of mice in the OA group and the control group (the original magnification is ×200). b Comparison of positive expression rate of SOX9 in cartilage tissue of mice. *p < 0.05 vs. the control group; OA osteoarthritis and SOX9 SRY-related HMG box-9
Cell apoptosis in cartilage tissue of OA mice is increased
Cell apoptosis in cartilage tissue was detected using TUNEL staining as follows: in normal cartilage cells, cells were well-distributed, and there was no evident staining of the cell nuclei, or variety in the size of cell body. There was weak positive expression in normal cartilage, primarily localized in the surface layer of cartilage (Fig. 4a). In apoptotic cells, TUNEL staining was expressed by positive expression, which infers that the cell nuclei were stained dense brown-yellow, with bright agglomeration in the chromatin, as well as decreased adhesion between the apoptotic cells and adjacent cells with oval or round shrunk cell. Positive expression was observed in the migrating layer and deep layer of cartilage in the OA group (Fig. 4b). Compared with the normal group, cell AI increased significantly in the OA group (p < 0.05) (Fig. 4c).

Induced cell apoptosis in cartilage tissue of mice with OA. a TUNEL staining of cartilage tissue of mice in OA and normal mice (the original magnification is ×200). b Comparison of cell apoptosis index of cartilage tissue of mice in OA and normal mice. *p < 0.05 vs. the control group; OA osteoarthritis
SOX9 is the target gene of miR-384-5p
According to the online analysis software (microRNA.org, miRbase, and TargetScan), there was a specific binding region between 3′-UTR on SOX9 gene and the miR-384-5p sequence, indicating that SOX9 was the targeted gene of miR-384-5p (Fig. 5a). A luciferase assay was performed in order to test the targeting relationship between SOX9 and miR-384-5p (Fig. 5b). The experimental results indicated that compared with NC group, the luciferase activity of SOX9-Wt was decreased significantly in the miR-384-5p mimic group (p < 0.05), and the luciferase activity of SOX9-Mut has no significantdifferences among the miR-384-5p mimic group and NC group (p > 0.05), suggesting that miR-384-5p could specifically bind to SOX9-3′-UTR and down-regulate the expression of SOX9 after transcription.

SOX9 is targeted by miR-384-5p. a Predicted binding site of miR-384-5p on SOX9.b Luciferase activity detection of luciferase reporter gene assay. *p < 0.05 vs. the negative control group; miR-384-5p microRNA-384-5p and SOX9 SRY-related HMG box-9
OA mice have decreased expression of SOX9 and NF-κB but increased expression of miR-384-5p in cartilage tissue
Results of RT-qPCR and Western blot analysis indicated (Fig. 6) that compared with the control group, the expression of miR-384-5p in cartilage tissue significantly increased, while mRNA and protein expression of SOX9 and NF-κB remarkably decreased in the OA group (all p < 0.05).

Mice with OA exhibit decreased expression of SOX9 and NF-κB and increased expression of miR-384-5p in cartilage tissue. a Expression of miR-384-5p is increased and mRNA expression of SOX9, NF-κB is decreased in cartilage tissue of mice with OA by RT-qPCR.b Western blot analysis found protein expression of SOX9 and NF-κB is decreased in cartilage tissue. *p < 0.05 vs. the control group; OA osteoarthritis, miR-384-5p microRNA-384-5p, SOX9 SRY-related HMG box-9, NF-κB cell nucleus factor-κB, GAPDH glyceraldehyde-3-phosphate dehydrogenase, and RT-qPCR reverse transcription quantitative polymerase chain reaction. The experiment is repeated for three times
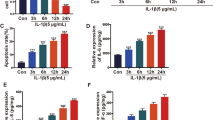
miR-384-5p inhibits cell proliferation after transfection
Results of MTT assay are shown in Fig. 7: within 24 h, cell proliferation among groups was essentially consistent (p > 0.05). After 48 h and 72 h, compared with the normal group, cell proliferation decreased remarkably in other groups (all p < 0.05). Compared with the blank group, there were no significant cell proliferation differences among the NC group and the miR-384-5p inhibitor + siRNA-SOX9 group (p > 0.05). Cell proliferation in the miR-384-5p mimic group and the siRNA-SOX9 group decreased evidently (bothp < 0.05), and cell proliferation in the miR-384-5p inhibitor group increased significantly (p < 0.05); compared with the miR-384-5p inhibitor group, cell proliferation in the miR-384-5p mimic group, siRNA-SOX9 group, and miR-384-5p inhibitor + siRNA-SOX9 group all decreased significantly (all p < 0.05).

Cells transfected with miR-384-5p mimic or siRNA-SOX9 show decreased proliferation rate detected by MTT assay. *p < 0.05 vs. the normal group; #p < 0.05 vs. the blank group; &p < 0.05 vs. the miR-384-5p inhibitor group; miR-384-5p microRNA-384-5p and SOX9 SRY-related HMG box-9
miR-384-5p promotes cell apoptosis after transfection
Cell apoptosis and cell cycle distribution are shown in Fig. 8. Compared with the normal group, cell apoptosis rate significantly increased in other groups, cell number also increased in G0/G1 phase while decreased in S phase (all p < 0.05). Compared with the blank group, in the NC group and the miR-384-5p inhibitor + siRNA-SOX9 group, there were no significant differences on apoptosis rate and cell number both in G0/G1 and S phases (all p > 0.05). Compared with the blank group, in the miR-384-5p mimic group and the siRNA-SOX9 group, cell apoptosis rate significantly increased, and cell number significantly increased in G0/G1 phase while decreased in S phase (p < 0.05); in the miR-384-5p inhibitor group, cell apoptosis rate significantly decreased, cell number obviously decreased in G0/G1 phase while increased S phase (all p < 0.05). Compared with the miR-384-5p inhibitor group, in the groups of miR-384-5p mimic, siRNA-SOX9 and miR-384-5p inhibitor + siRNA-SOX9, cell apoptosis rate and cell number in G0/G1 phase significantly increased, but cell number in S phase significantly decreased (all p < 0.05).

miR-384-5p mimic or siRNA-SOX9 block cell entry and induce cell apoptosis. a,b Cell apoptosis is promoted by miR-384-5p detected by flow cytometry.c,d Cell cycle is blocked by miR-384-5p. *p < 0.05 vs. the normal group; #p < 0.05 vs. the blank group; &p < 0.05 vs. the miR-384-5p inhibitor group; miR-384-5p microRNA-384-5p and SOX9 SRY-related HMG box-9
miR-384-5p or silencing of SOX9 inhibits the NF-κB signaling pathway
As shown in Fig. 9, compared with the normal group, in other groups, expression of miR-384-5p significantly increased, but mRNA and protein expression of SOX9 and NF-κB decreased (all p < 0.05). Compared with the blank group, there was no significant difference in the NC group (p > 0.05); in the miR-384-5p mimic group, expression of miR-384-5p significantly increased, but mRNA and protein expression of SOX9 and NF-κB decreased (all p < 0.05); in the miR-384-5p inhibitor group, expression of miR-384-5p significantly decreased, mRNA and protein expression of SOX9 and NF-κB increased significantly (all p < 0.05); in the siRNA-SOX9 group, there was no significant difference on expression of miR-384-5p (p > 0.05), mRNA and protein expression of SOX9 and NF-κB obviously decreased (all p < 0.05); in the miR-384-5p inhibitor + siRNA-SOX9 group, expression of miR-384-5p significantly decreased (p < 0.05), there was no significant difference on mRNA and protein expression of SOX9 and NF-κB (bothp > 0.05). Compared with the miR-384-5p inhibitor group, in the miR-384-5p mimic group and the siRNA-SOX9 group, expression of miR-384-5p significantly increased, but mRNA and protein expression of SOX9 and NF-κB was significantly decreased (all p < 0.05); in the miR-384-5p inhibitor + siRNA-SOX9 group, there was no significant difference on expression of miR-384-5p (p > 0.05), mRNA and protein of SOX9 and NF-κB significantly decreased (both p < 0.05).

miR-384-5p decreased expression of SOX9 and NF-κB in cartilage cells. a mRNA expression of SOX9 and NF-κB is decreased by miR-384-5p detected by RT-qPCR. b,c Western blot analysis found that protein expression of SOX9 and NF-κB is decreased by miR-384-5p. *p < 0.05 vs. the normal group; #p < 0.05 vs. the blank group; &p < 0.05 vs. the miR-384-5p inhibitor group. miR-384-5p microRNA-384-5p, SOX9 SRY-related HMG box-9, NF-κB cell nucleus factor-κB, GAPDH glyceraldehyde-3-phosphate dehydrogenase, and RT-qPCR reverse transcription quantitative polymerase chain reaction. The experiment is repeated for three times
Discussion
OA is a complex multifactorial disease, known to be caused by various genetic, mechanical, and environmental factors, and also prevails as one of the most common diseases of articulated joints [22]. miRs are an evolutionally conserved, small (18–25 nucleotides), non-coding class of RNAs exerting critical effects in post-transcriptional gene regulation [23]. Therefore, the present study aims to evaluate the effects of miR-384-5p on cartilage cell proliferation and apoptosis in OA mice by targeting the SOX9 gene through the NF-κB signaling pathway.
The findings of our study indicate that miR-384-5p is capable of regulating the expression of SOX9, which is the targeted gene of miR-384-5p, thereby inducing the occurrence of OA by the NF-κB signaling pathway. And in our experiment, high expression of miR-384-5p and low expression of SOX9 and NF-κB were found in cartilage tissues of OA mice, suggesting that upregulation of miR-384-5p and downregulation of SOX9 and NF-κB are involved in OA development. Recently, Hiroshi et al. found that miRs, regulate the expression of several targeted genes, such as SOX9 and SP1 [24]. Gu et al. found that activation of NF-κB signaling pathways would promote the release of pro-inflammatory cytokines, which play important roles in cartilage damage and bone metabolism homeostasis [25]. Interestingly, Yamagishi et al. demonstrated that Polycomb-mediated silencing of miR-31 was implicated in the activation of NF-κB signaling in tumors, including adult T cell leukemia/lymphoma (ATL) tumor cells [26]. SOX9 is a special major transcription factor owing to its crucial role in regulating various developmental pathways like chondrogenesis, gonadogenesis, and the pancreas [27]. Zhong et al. noted that the mRNA and protein expression of SOX9 in OA cartilage decreased, and OA was initiated by differential regulation of signaling pathway, such as WNT, which regulates the protein activity of SOX9 [28]. Henceforth, we inferred that miR-384-5p potentially holds great influence on OA by targeting the SOX9 gene through the NF-κB signaling pathway.
In addition, from comparisons of experimental data, we detected that compared with the normal group, apoptosis rate was higher in the OA group, and cell proliferation was lower in other groups transfected with miR-384-5p mimic, indicating that miR-384-5p could promote cell apoptosis and inhibit cell proliferation. Recently, Jovanovic et al. showed the potential and important role of miRs in regulating apoptosis at various levels, and in several organisms by regulating gene expression [29]. For example, Chen et al. found that miR-200a regulates hepatic cellular cancer (HCC) cell proliferation, migration, and invasion using its targeted gene Foxa2 [30]. Moreover, Zhao et al. found that miR-365 suppressed cell cycle progression and promoted colon cancer cell apoptosis by targeted gene Cyclin D1 and Bcl-2 [31]. Based on these aforementioned reasons and previous studies, we could infer that miR-384-5p could regulate cell proliferation and apoptosis in cartilage tissue. So, understanding the regulation of miR-384-5p may inspire alternative therapeutic methods for OA.
In conclusion, this study showed that miR-384-5p could regulate the expression of SOX9 negatively and inactivate the NF-κB signaling pathway in order to induce the appearance of OA, and further understanding the regulation of miR-384-5p may yield alternative treatments OA methods. However, due to the limited sample size and time, the specific mechanism of miR-384-5p in OA still lacks adequate clinical data to be used in a clinical setting. Therefore, a well-designed, prospective, randomized, multi-center clinical trial will be needed in the future.
Change history
29 July 2020
The original version of this Article contained inaccuracies in Figure 6B (“Sox9” band) and Figure 8A (“mir-384-5p inhibitor” and “8 mir-384-5p inhibitor + sirna-sox9” groups). Both figures have been corrected in the PDF and HTML versions of the Article.
29 July 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–35.
Arendt-Nielsen L, Nie H, Laursen MB, Laursen BS, Madeleine P, Simonsen OH, et al. Sensitization in patients with painful knee osteoarthritis. Pain. 2010;149:573–81.
Wang CJ, Cheng JH, Chou WY, Hsu SL, Chen JH, Huang CY. Changes of articular cartilage and subchondral bone after extracorporeal shockwave therapy in osteoarthritis of the knee. Int J Med Sci. 2017;14:213–23.
Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377:2115–26.
arcOGEN Consortium, arcOGEN Collaborators.Zeggini E,Panoutsopoulou K,Southam L,Rayner NW, et al. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380:815–23.
Tang Y, Pan ZY, Zou Y, He Y, Yang PY, Tang QQ, et al. A comparative assessment of adipose-derived stem cells from subcutaneous and visceral fat as a potential cell source for knee osteoarthritis treatment. J Cell Mol Med. 2017;21:2153–62.
Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta-analysis. Osteoarthr Cartil. 2010;18:24–33.
Chen X, Zhao Y, Wang F, Bei Y, Xiao J, Yang C. MicroRNAs in liver regeneration. Cell Physiol Biochem. 2015;37:615–28.
Wang B, Zhong Y, Huang D, Li J. Macrophage autophagy regulated by miR-384-5p-mediated control of Beclin-1 plays a role in the development of atherosclerosis. Am J Transl Res. 2016;8:606–14.
Du J, Bai F, Zhao P, Li X, Li X, Gao L, et al. Hepatitis B core protein promotes liver cancer metastasis through miR-382-5p/DLC-1 axis. Biochim Biophys Acta. 2018;1865:1–11.
Montero JA, Lorda-Diez CI, Francisco-Morcillo J, Chimal-Monroy J, Garcia-Porrero JA, Hurle JM. Sox9 expression in amniotes: species-specific differences in the formation of digits. Front Cell Dev Biol. 2017;5:23.
Ko JY, Sun YC, Li WC, Wang FS. Chaperonin 60 regulation of SOX9 ubiquitination mitigates the development of knee osteoarthritis. J Mol Med (Berl). 2016;94:755–69.
Tanaka T, Maekawa N, Kashio T, Izawa K, Ishiba R, Shirakura K, et al. Tumor necrosis factor alpha induces the expression of the endothelial cell-specific receptor Roundabout4 through the nuclear factor-kappaB pathway. Biol Pharm Bull. 2017;40:504–9.
Rigoglou S, Papavassiliou AG. The NF-kappaB signalling pathway in osteoarthritis. Int J Biochem Cell Biol. 2013;45:2580–4.
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal Res. 2017; 63 https://doi.org/10.1111/jpi.12414.
Wu C, Tian B, Qu X, Liu F, Tang T, Qin A, et al. MicroRNAs play a role in chondrogenesis and osteoarthritis (review). Int J Mol Med. 2014;34:13–23.
Singh V, Manalang M, Singh M, Apte U. A Brief report of immunohistochemical markers to identify aggressive hepatoblastoma. Appl Immunohistochem Mol Morphol. 2017 https://doi.org/10.1097/PAI.0000000000000492.
Cosan D, Soyocak A, Basaran A, Degirmenci I, Gunes HV. The effects of resveratrol and tannic acid on apoptosis in colon adenocarcinoma cell line. Saudi Med J. 2009;30:191–5.
Zheng W, Feng Z, Lou Y, Chen C, Zhang C, Tao Z, et al. Silibinin protects against osteoarthritis through inhibiting the inflammatory response and cartilage matrix degradation in vitro and in vivo. Oncotarget. 2017;8:99649–65.
Chen C, Zhang C, Cai L, Xie H, Hu W, Wang T, et al. Baicalin suppresses IL-1beta-induced expression of inflammatory cytokines via blocking NF-kappaB in human osteoarthritis chondrocytes and shows protective effect in mice osteoarthritis models. Int Immunopharmacol. 2017;52:218–26.
Pan T, Chen R, Wu D, Cai N, Shi X, Li B, et al. Alpha-Mangostin suppresses interleukin-1beta-induced apoptosis in rat chondrocytes by inhibiting the NF-kappaB signaling pathway and delays the progression of osteoarthritis in a rat model. Int Immunopharmacol. 2017;52:156–62.
Kopanska M, Szala D, Czech J, Gablo N, Gargasz K, Trzeciak M, et al. MiRNA expression in the cartilage of patients with osteoarthritis. J Orthop Surg Res. 2017;12:51.
Yu J, Wang JG, Zhang L, Yang HP, Wang L, Ding D, et al. MicroRNA-320a inhibits breast cancer metastasis by targeting metadherin. Oncotarget. 2016;7:38612–25.
Miyaki S, Asahara H. Macro view of microRNA function in osteoarthritis. Nat Rev Rheumatol. 2012;8:543–52.
Gu R, Liu N, Luo S, Huang W, Zha Z, Yang J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine (Baltimore). 2016;95:e4315.
Uribesalgo I, Ballare C, Di Croce L. Polycomb regulates NF-kappaB signaling in cancer through miRNA. Cancer Cell. 2012;21:5–7.
Egunsola AT, Bae Y, Jiang MM, Liu DS, Chen-Evenson Y, Bertin T, et al. Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J Clin Invest. 2017;127:1475–84.
Zhong L, Huang X, Karperien M, Post JN. Correlation between gene expression and osteoarthritis progression in human. Int J Mol Sci. 2016;17:1126.
Jovanovic M, Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25:6176–87.
Chen SY, Ma DN, Chen QD, Zhang JJ, Tian YR, Wang ZC, et al. MicroRNA-200a inhibits cell growth and metastasis by targeting Foxa2 in hepatocellular carcinoma. J Cancer. 2017;8:617–25.
Zhao W, Shen WW, Cao XM, Ding WY, Yan LP, Gao LJ, et al. Novel mechanism of miRNA-365-regulated trophoblast apoptosis in recurrent miscarriage. J Cell Mol Med. 2017;21:2412–25.
Acknowledgments
This study was supported by Natural Science Foundation of Xinjiang Uygur Autonomous Regions Province of China (Project: 2014211C093).We would like to acknowledge the helpful comments on this paper received from our reviewers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhang, W., Cheng, P., Hu, W. et al. Inhibition of microRNA-384-5p alleviates osteoarthritis through its effects on inhibiting apoptosis of cartilage cells via the NF-κB signaling pathway by targeting SOX9. Cancer Gene Ther 25, 326–338 (2018). https://doi.org/10.1038/s41417-018-0029-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-018-0029-y
- Springer Nature America, Inc.
This article is cited by
-
MicroRNA-22-3p ameliorates Alzheimer’s disease by targeting SOX9 through the NF-κB signaling pathway in the hippocampus
Journal of Neuroinflammation (2022)
-
Early life and adult stress promote sex dependent changes in hypothalamic miRNAs and environmental enrichment prevents stress-induced miRNA and gene expression changes in rats
BMC Genomics (2021)
-
Coding the noncoding: 2 years of advances in the field of microRNAs and long noncoding RNAs
Cancer Gene Therapy (2021)
-
A novel natural antisense transcript at human SOX9 locus is down-regulated in cancer and stem cells
Biotechnology Letters (2020)